
Abstract
Noise exposure leads to dramatic physiological and anatomical changes within the central auditory pathway in addition to the well-known cochlear damage. Our group previously described a significant loss of neurons in different central auditory structures upon acoustic overstimulation. The aim of the present study was to investigate if declined neuronal cell density is caused by apoptotic mechanisms. Mice were noise-exposed (3 h, 5–20 kHz) at 115 dB SPL under anesthesia and investigated immediately after, and at 6 h, 24 h, or 7 days after the exposure (n=16). Unexposed animals were used as controls (n=5). Apoptotic cells were detected by fluorescence microscopy after terminal deoxynucleotidyl transferase dUTP nick-end labeling assay (TUNEL). TUNEL-positive cells were compared to cell density (diamidino phenylindole, DAPI) within the dorsal and ventral cochlear nucleus (VCN), and the central nucleus of the inferior colliculus (ICC). In all investigated auditory areas, TUNEL-positive cells were significantly increased after acoustic overstimulation. In the acute, 6-h, and 24-h groups, their numbers were significantly increased in the VCN, as well as in the 6-h, 24-h, and 7-day groups in the dorsal cochlear nucleus (DCN). In the ICC, TUNEL-positive cells were significantly increased in all exposed mice. In the VCN, the number of TUNEL-positive cells of the same grid size was three times the numbers in the ICC. Our results show that noise exposure induces apoptosis-related pathophysiological changes within the central auditory pathway in a time-dependent manner. This may represent potential therapeutic targets, and helps clarify the complex psychoacoustic phenomena of noise-induced hearing loss.
Introduction
A
In the central auditory pathway, acoustic deprivation after cochlear ablation reduces dendritic spines at auditory nerve fibers on cochlear nucleus (CN) neurons (Kane, 1974), and diminishes their cell bodies (Benson et al., 1997). Recently, researchers have described increased synaptic plasticity in the ventral cochlear nucleus (VCN) due to elevated expression of neurotrophic proteins (Kraus et al., 2009; Suneja et al., 2005). Most experimental animal studies cover observation periods of a few days up to 1 month after acoustic deprivation, although the VCN is primarily affected by early neurodegenerative processes (Gil-Loyzaga et al., 2010). Evidence of apoptotic programmed cell death in the VCN in the first 2 days after acoustic deprivation due to cochlear removal was detected by terminal deoxynucleotidyl transferase dUTP nick-end labeling assay (TUNEL; Karnes et al., 2009; Mostafapour et al., 2000).
As the result of NIHL, axon degeneration occurs in the CN (Kim et al., 1997,2004), trapezoid body (Jean-Baptiste and Morest, 1975), and superior olivary complex (Aarnisalo et al., 2000). NIHL also significantly reduces cell density in higher auditory structures within all subdivisions of the medial geniculate body, the primary auditory cortex (layers IV–VI; Basta et al., 2005), and neuronal precursor cells in the hippocampus (Kraus et al., 2010b). Also, spontaneous neuronal activity in the inferior colliculus (IC) undergoes changes (Basta and Ernst 2005). Evidence of an apoptotic pathway was found during the first week after impulse noise by TUNEL within the temporal cortex (layers II–VI), and the cingulate and piriform cortices (Saljo et al., 2002).
Recently, our group showed that NIHL causes decreased cell density, both immediately after noise exposure in the VCN, and 7 days after noise exposure in all investigated central auditory areas (the VCN, dorsal cochlear nucleus [DCN], central nucleus of the inferior colliculus [ICC], the dorsal, ventral, and medial subdivision of the medial geniculate body, and the primary auditory cortex) (Gröschel et al., 2010).
Pathophysiological changes of the central auditory pathway are relevant from a therapeutic perspective. Long-term effects involve tinnitus and reduced speech intelligibility (Basta et al., 2005; Henderson et al., 2006; Hirose and Liberman, 2003; Yamashita et al., 2004). Early interventions may prevent these long-term effects, which can involve the cochlea as well as the central auditory pathway. Nevertheless, successful interventions for acute impairments of the cochlea have been described (Gedlicka et al., 2009).
The aim of the present study was therefore to investigate the time course of apoptotic signaling cascades that might contribute to the decline in neuronal cell density in central auditory structures during noise exposure.
Methods
This study investigated 21 young adult mice (NMRI strain) with normal hearing and of both sexes. The mice were 30–40 days old, and therefore had a mature central auditory pathway. This strain has been used to correlate NIHL with pathophysiological changes of the central nervous system (Basta and Ernst, 2005; Basta et al., 2005; Gröschel et al., 2010). The Governmental Commission for Animal Studies approved the experimental protocol. All efforts were made to minimize pain in the animals.
Sixteen mice were noise-exposed in a soundproof chamber (0.8 m×0.8 m×0.8 m, minimal attenuation 60 dB) for 3 h with broadband noise (5–20 kHz) at 115 dB sound pressure level (SPL) under anesthesia (ketamine/xylazine: 60/6 mg/kg IP), as described earlier (Gröschel et al., 2010). A video camera placed inside the lighted chamber was used to control the anesthesia (no visible muscle reflexes). Body temperature was maintained at 37°C by a heating pad (Thermolux CM 15W; Acculux, Murrhardt, Germany) placed below the animal. To verify normal hearing, we performed frequency-specific (4, 8, 12, 16, and 20 kHz) auditory brainstem responses in all mice (controls and noise-exposed animals) at the beginning of each anesthesia before the noise exposure was started. A measurement system (Viking IV; Viasys Healthcare, Conshohocken, PA) recorded auditory brainstem responses, and demonstrated that all animals had normal hearing. Thereafter, noise was delivered binaurally by high-tone loudspeakers (HTC 11.19; Visaton, Haan, Germany) placed above the animal's head. The speakers were connected to an audio amplifier (Tangent AMP-50; Tangent A/S, Aulum, Denmark), and a DVD player (DK DVD-438; DK Digital, Ratingen, Germany). Sound pressure levels were calibrated with a sound level meter (Voltcraft 329; Voltcraft, Hirschau, Germany) placed near the animal's ear.
Three mice were investigated immediately after noise exposure (the acute group), whereas the remaining mice were kept in cages for further investigations. Three mice were studied each after 6 h and 24 h (the 6-h and 24-h groups), and seven mice were studied after 7 days (the 7-day). Five unexposed animals served as the control group. All animals were perfused via the left heart chamber with 4% paraformaldehyde in order to fixate the neural tissue. The brains were removed, embedded in paraffin, and micro-sliced (10 μm) in the frontal plane using a microtome (model HM 325; Thermo Scientific, Walldorf, Germany). The tissue slices were fixed at 64°C overnight. Every fifth slice was stained with hematoxylin and eosin (H&E; Hahn and Netsky, 1977) in order to microscopically locate the appropriate layer of the murine VCN, DCN, and ICC (Imager.Z1; Carl Zeiss, Göttingen, Denmark). The brain areas were defined in accordance with the mouse brain atlas of Paxinos and Franklin (Paxinos and Franklin, 2001).
Eight slices of each central auditory area in each mouse were tested for apoptosis at the single-cell level, based on TUNEL (In Situ Cell Death Detection Kit; Roche, Mannheim, Denmark; Gavrieli et al., 1992). The four slices in between the two most central auditory areas detected with H&E, as well as the four slices adjacent to the most central areas were stained with TUNEL. The following protocol was used. Slices were incubated with xylene (2×5 min) and ethanol (100%, 95%, and 70%, each for 2 min) in order to deparaffinize the paraffin-embedded tissue, and were boiled with citrate buffer (0.01 M, pH 6, 5 min) in order to achieve antigen retrieval of the tissue. Each slice was incubated with 50 μL TUNEL reaction mixture (60 min at 37°C), and 100 μL of a solution containing diamidino phenylindole (DAPI; Morikawa and Yanagida, 1981), in a ratio 1:1000, and Tris-buffered saline (50 mM Tris-HCl and 150 mM NaCl, pH 7.4) for 5 min at room temperature. The tissue sections were then mounted.
The stained slices were magnified microscopically with fluorescence channels for TUNEL (540 nm) and DAPI (340 nm). The images were digitized with a digital camera (AxioCam MRC; Carl Zeiss) and converted to black and white (Adobe Photoshop CS4). The number of TUNEL-positive cells was counted in different grid sizes (DCN: 160 μm×100 μm, VCN: 220 μm×170 μm, and ICC: 450 μm×330 μm). A normal grid was defined according to the grid size of the ICC. The mean number of TUNEL-positive cells of each brain area was calculated using the results of 8 slices obtained from each animal, and set to the normal grid size. Data represent mean TUNEL-positive cells±standard deviation from the mean value (±standard error) after different exposure times compared to the control group.
Statistical analyses were performed using the Mann-Whitney U test (the data were not normally distributed). The control group and noise-exposed animals were compared, as well as, adjacent noise-exposed groups. Significance levels were calculated according to the Bonferroni procedure for multiple tests of significance (p<0.05 for one, p<0.0253 for two, p<0.0170 for three, and p<0.0127 for four).
Results
The lower central auditory areas, the VCN, the DCN, and the ICC, were stained with TUNEL and DAPI (Fig. 1). TUNEL-positive cells were found in all investigated areas. An example is shown in Figure 2.

Examples of the investigated lower central auditory areas. Fluorescence photomicrographs of the ventral (VCN) and dorsal (DCN) cochlear nuclei are shown at 200× magnification (left), and the central nucleus of the inferior colliculus (ICC) at 100× magnification (right). The slices were stained with TUNEL (light grey) and DAPI (dark grey) (TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling assay; DAPI, diamidino phenylindole).

Example of TUNEL-positive cells in the VCN. Fluorescence photomicrographs of the VCN at 400× magnification, from 24-h noise-exposed (right) and control animals (left). Arrows indicate TUNEL-positive cells (TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling assay; VCN, ventral cochlear nucleus).
The ventral cochlear nucleus
There was a significant increase in TUNEL-positive cells of noise-exposed mice in the acute, 6-h, and 24-h groups compared to controls (2.28±0.47). No significant difference was found between the 6-h and 24-h groups. The most TUNEL-positive cells were found after 24 h (8.14±0.50). The numbers in the 7-day group were not significantly different from those in controls (Fig. 3A).
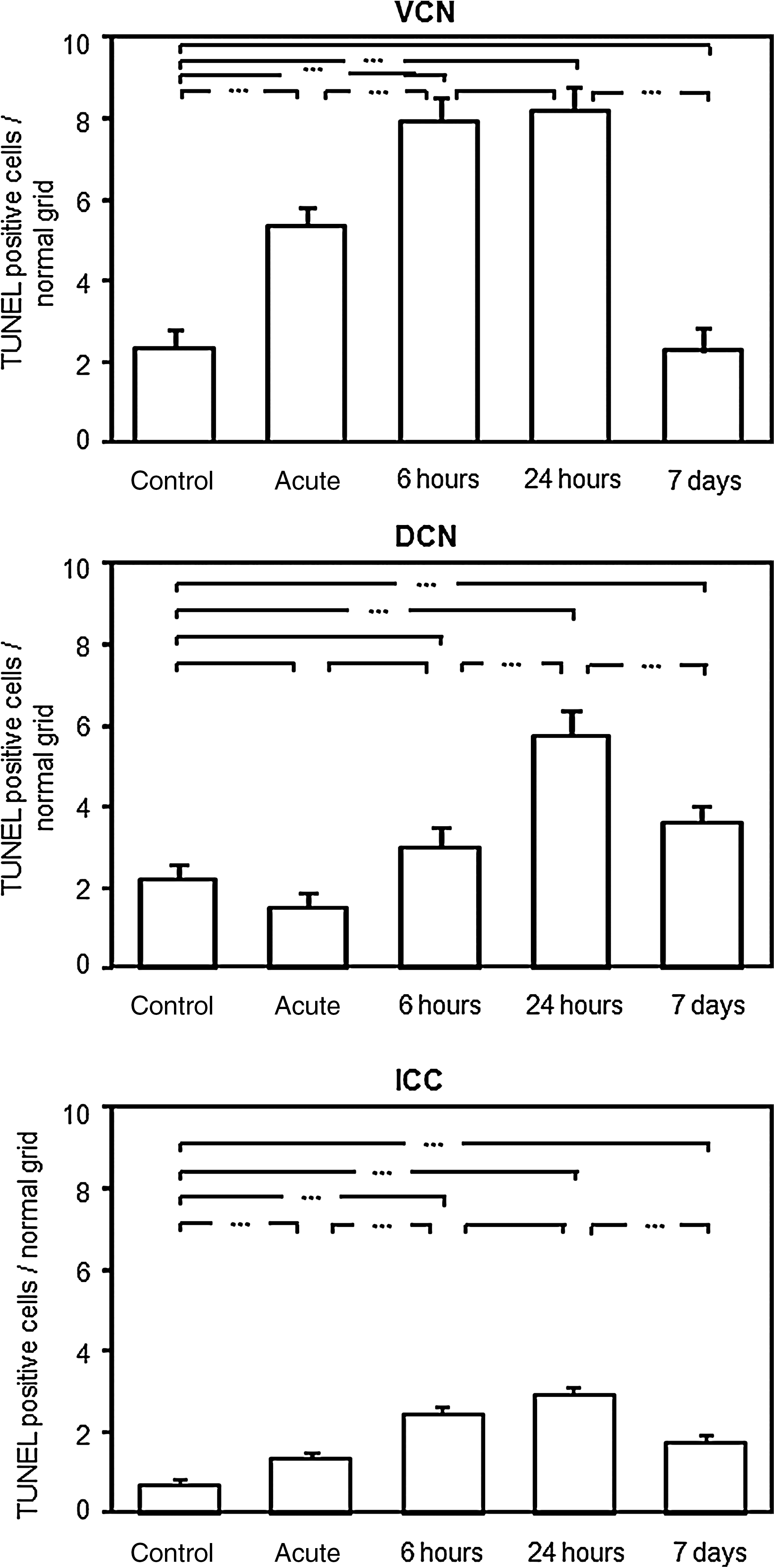
Mean numbers of TUNEL-positive cells after NIHL in the lower central auditory pathway (the VCN, DCN, and ICC). Data indicate mean TUNEL-positive cells±standard deviation from the mean value (±standard error), after different exposure times compared to the control group, and adapted to the standard grid size. Connecting lines with three asterisks indicate significant differences between groups (TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling assay; VCN, ventral cochlear nucleus; DCN, dorsal cochlear nucleus; ICC, central nucleus of the inferior colliculus; NIHL, noise-induced hearing loss).
The dorsal cochlear nucleus
TUNEL positive cells were significantly increased in the 24-h (5.74±0.60) and in the 7-day (3.61±0.41) groups compared to controls (2.19±0.36). No significant difference was found between the “control” group and acute or 6-h groups after noise exposure. The highest increase of TUNEL positive cells were found between 6 and 24 hours after noise exposure. Seven days after exposure, there was still a significant difference compared to controls (Fig. 3B).
The central nucleus of the inferior colliculus
There was a significant increase in TUNEL-positive cells of all noise-exposed mice compared to controls (0.72±0.07). Mean numbers of TUNEL-positive cells differed significantly at all investigated time points, except between 6 and 24 h. The most TUNEL-positive cells were found after 24 h (2.95±0.14). In the 7-day group, the mean number of TUNEL-positive cells (1.77±0.15) was significantly decreased compared to the 24-h group, but was significantly larger than in the control group (Fig. 3C).
Discussion
Acoustic overstimulation is well known as a major pathophysiological factor in sensory inner ear cells due to degeneration of the cochlear outer hair cells (Henderson et al., 2006; Nordmann et al., 2000; Ryan et al., 1992; Wagner et al., 2005). In recent years, studies have proven that the central components of the auditory pathway are affected both by acoustic deprivation (Benson et al., 1997; Gil-Loyzaga et al., 2010; Karnes et al., 2009; Kraus et al., 2009; Mostafapour et al., 2000; Suneja et al., 2005) and NIHL (Aarnisalo et al., 2000; Basta and Ernst, 2005; Basta et al., 2005; Jean-Baptiste and Morest, 1975; Kim et al., 1997,2004; Kraus et al., 2010b; Saljo et al., 2002). Our group recently demonstrated (Gröschel et al., 2010) that NIHL influences neuronal cell densities in key structures of the central auditory pathway. Cell density was significantly decreased in the VCN immediately after noise exposure, and in most investigated central auditory areas, 7 days after noise exposure. The present study proves for the first time that apoptotic mechanisms cause reduced neuronal cell density after NIHL in lower central auditory areas. TUNEL uses apoptotic cell detection. TUNEL-positive cells were significantly increased in all investigated lower central auditory areas (VCN, DCN, and ICC) after noise exposure. The most cells were detected after 6 and 24 h, while apoptotic cascades started immediately after noise exposure, especially in VCN neurons.
TUNEL of degraded DNA detects apoptosis at the single-cell level (Gavrieli et al., 1992). The immunofluorescence technique used in this study does not induce DNA strand breaks in paraffin-embedded tissues (Jackson et al., 2008).
The ventral cochlear nucleus
In the VCN, NIHL decreases cell density both immediately and 7 days after noise exposure (Gröschel et al., 2010). Other researchers have described a partly reversible destruction of sensory tissue after acute noise exposure (Henderson et al., 2006; Hirose and Liberman, 2003; Wagner et al., 2005), that contributes to audiologic symptoms. Acoustic overstimulation leads to excessive neuronal activation. Calcium influx accompanies acoustic overstimulation and leads directly to toxic neurodegenerative processes like apoptosis (Mattson, 2007; Yu et al., 2001). Intracellular calcium triggers neurotransmitter release into the synaptic cleft and can induce glutamate excitotoxicity (Greenwood and Connolly, 2007). Ischemia in activated brain regions follows excessive neuronal activation after acoustic overstimulation. Reactive oxygen species and free radicals, as well as mitochondrial dysfunction, induce cell death (Serrano and Klann, 2004). Other studies describe acute changes in cell density, mainly in the VCN, which has a stronger direct afferent input from the cochlea and higher excitotoxicity than the other lower central auditory areas (Shore et al., 2008).
We demonstrated in mice with NIHL a significant increase in TUNEL-positive cells in the VCN of the acute group compared to the control group, which is consistent with earlier studies that have shown lower cell density (Gröschel et al., 2010). Programmed cell death is relatively short (1–3 h from initiation to cell elimination), and appears in tissues in clusters (Kerr et al., 1972). The occurrence of this process in slowly-renewing tissues is very rare (Gavrieli et al., 1992). Another significant increase of TUNEL-positive cells in the VCN was found in the 6-h and 24-h groups compared to the control and the acute groups. No significant difference was found between 6 and 24 h after noise exposure. After 7 days, the number of TUNEL-positive cells was comparable to that of the control group. This lower cell density in the 7-day group (Gröschel et al., 2010) is the result of programmed cell death, which seems to be complete by this time. Other groups have found that decreased cell loss 2 weeks after cochlear removal was less than one-tenth that seen after 1 week (Mostafapour et al., 2000). The VCN is primarily affected by early neurodegenerative processes (Gil-Loyzaga et al., 2010).
Acoustic deprivation due to cochlear removal also causes apoptotic signaling cascades in the central auditory pathway. However, the kinetics of cell loss are delayed by 12 h compared to our results after NIHL. TUNEL-positive neurons were first detected 12 h after cochlear removal, and had their peak after approximately 48 h. No TUNEL-positive cells were found after 3 weeks (Mostafapour et al., 2000). Other groups had similar results (Karnes et al., 2009).
The central nucleus of the inferior colliculus
In the ICC, we showed a significant increase in TUNEL-positive cells after NIHL in all investigated groups compared to the control group. While reduced cell density in the ICC was lacking in the acute group (Gröschel et al., 2010), TUNEL was possible. The mean number of TUNEL-positive cells differed significantly between the acute and the 6-h groups, as well as between the acute and 24-h groups, similarly to the findings in the VCN. In the 7-day group, programmed cell death occurred, differing from the pattern seen in the VCN. A loss of cell density after 7 days has also been found in earlier studies (Basta and Ernst, 2005; Gröschel et al., 2010). TUNEL staining after NIHL has not been studied in the ICC. In the central auditory cortex, TUNEL-positive cells were detected 6 h to 7 days post-exposure (Saljo et al., 2002).
The number of TUNEL-positive cells in the ICC was one-third that seen in the VCN. One explanation for this finding is that inhibitory interneurons suppress cellular activity of neurons projecting from CN to higher structures (Ryan et al., 1992), and protect higher central auditory areas. Short-term plastic changes in post-synaptic AMPA-receptor densities is another mechanism limiting excitotoxic neuronal damage during noise exposure (Chen et al., 2009). Furthermore, a delayed time frame of apoptotic mechanisms in the ICC such as that shown in the present study may contribute to the earlier reported delayed reduction of cell density (Basta and Ernst, 2005; Gröschel et al., 2010).
The dorsal cochlear nucleus
In the DCN, mean TUNEL-positive cell numbers were significantly increased in the 24-h and the 7-day groups compared with controls. This is consistent with earlier studies that showed reduced cell density after 7 days (Gröschel et al., 2010). The number of TUNEL-positive cells was lower compared to that of the VCN. Other studies describe acute changes in cell density, mainly in the VCN, which has a stronger direct afferent input from the cochlea. The DCN has stronger multisensory innervations (Shore et al., 2008). Like in the ICC, programmed cell death was detectable in the 7-day group, and may continue beyond this time point.
Apoptotic cascades in central auditory structures contribute to the effects of long-term NIHL in humans. In patients with NIHL, a decrease in grey matter volume has been reported within the central auditory pathway (Landgrebe et al., 2009), and is combined with reduced speech intelligibility, despite good tonal hearing (Harris et al., 2009). The exact mechanisms that induce programmed cell death detectable by TUNEL are unknown. Rapid neuronal depolarization and a hypoxic state could lead to spreading depression and tissue damage (Somjen, 2001).
Acoustic deprivation induces long-lasting plastic changes. Different groups have recently demonstrated increased synaptic plasticity in central auditory structures. Cochlear ablation resulted in fiber growth and synaptogenesis in the VCN. These studies detected elevated expression of different neurotrophic proteins, such as brain-derived neurotrophic factor (Suneja et al., 2005), and GAP-43 (Kraus et al., 2009). Evidence for microglial reaction was shown in astrocytes, which undergo hypertrophy in deafferented cochlear nuclei (Campos Torres et al., 1999). Progressive neuropathologic disorders affect immunocompetent cells in the brain and the microglia, as shown by the proinflammatory and immunological responses (Kraus et al., 2010a).
Previous studies have shown the cytochrome c and active caspases-9 are markers for the activation of the intrinsic apoptotic pathway. Both markers were detected in both degenerating and surviving neurons in VCN (Wilkinson et al., 2003). Upstream apoptotic markers like caspase-3 may both exacerbate and modulate apoptotic death, while TUNEL staining of degraded DNA is restricted to subpopulations of deafferented neurons (Karnes et al., 2009). In the auditory cortex, c-Jun expression was found in neurons in twice the numbers seen with TUNEL-staining after acoustic exposure (Saljo et al., 2002). c-Jun belongs to activator protein 1, a transcription factor, which regulates cellular processes including differentiation, proliferation, and apoptosis (Ameyar et al., 2003).
In conclusion, our results show that apoptosis-related pathophysiological changes contribute to the noise-induced decline of neuronal cell density in lower central auditory areas. The reduced cell density due to programmed cell loss most likely contributes to the psychoacoustic phenomena of NIHL, such as tinnitus, acoustic discomfort, reduced speech intelligibility, and hyperacusis. Future investigations studying NIHL should focus on the specific cell types involved in degeneration and investigate mechanisms such as neuronal plasticity. The present results may help reveal potential therapeutic targets, and help us understand the complex psychoacoustic phenomena of noise-induced hearing loss.
Footnotes
Acknowledgments
This work was funded by a grant from ZIM/BMWiT (FKz. KF2227701). We are grateful to Ulrike Erben, Anja Kühn, and Katja Grollich, for allowing us to perform the experiments in their laboratory, as well as for instruction in immunohistological and fluorescence staining techniques.
Author Disclosure Statement
No competing financial interests exist.