
Abstract
Calcium influx into cells is responsible for initiating the cell death in neuronal tissue after hypoxic injury. Changes in intracellular calcium with subsequent increased expression of ryanodine receptor 2 (RyR2) are hypothesized to cause cell death after hypoxic injury. In the present study we have examined the time-dependent changes of RyR2 expression in hypoxic/reperfusion injury of spinal cord dorsal column. In this study we used western blotting, real time PCR (RT-PCR) and immunohistochemistry to examine changes in protein and gene expression of RyR2 after spinal cord injury (SCI) in the rat. Quantitative immunoblotting showed increase in the expression of RyR2 at 4 h during hypoxic/reperfusion injury of dorsal column. Moreover, RT-PCR showed 36-fold increases in mRNA of RyR2 after 4 h of hypoxic injury of white matter. By double immunofluorescence staining, RyR2 was localized on axons and astrocytes in the white matter of the spinal cord. After treatment with KN-62; (inhibitor of CaMKII) and SP600125 (inhibitor of JNK), there is a significant reduction in the expression of RyR2, indicating the role of these molecules in RyR2 regulation. Further removal of extracellular calcium does not have significant effect on RyR2 expression and phosphorylation of CaMKII, which was further confirmed by treatment with intracellular Ca++ chelator BAPTA-AM. Finally, bioassay with quantitative analysis showed that treatment with inhibitor significantly reduced the cellular oxidative stress suggesting RyR2 is responsible for increased cellular oxidative load. In summary, we provide evidence that RyR2 gene and protein expression in astrocyte and axons is markedly increased after hypoxic injury. Further CaMKII/JNK pathway upregulates RyR2 expression after hypoxic injury. Therefore we propose that inhibitors of CaMKII/JNK pathway would reduce the cellular oxidative load and thereby have a neuroprotective role.
Introduction
S
Intracellular Ca2+ channels are divided into two major classes: ryanodine receptor and inositol 1,4,5-triphosphate, both of these are present in endoplasmic reticulum. Three isoforms of ryanodine receptors (RyRs) are known: RyR1, RyR2, and RyR3, which are expressed in different degrees in the central nervous system (McPherson and Campbell, 1993). Interestingly, the intracellular distribution of RyRs is markedly different within cells and is variable among cells, which suggests their differential activity in response to injury (Llano et al., 2000; Martone et al., 1997; Ross et al., 1989). RyR1 and RyR2 have been shown to be involved in white matter traumatic and ischemic injury (Ouardouz et al., 2003; Thorell et al. 2002). Increased activity of RyRs has been found in axons and astrocytes during trauma and hypoxia (Aley et al., 2006).
In addition, RyR2 is also regulated by protein kinase A and Ca2+/calmodulin dependent protein kinase II (CaMKII) (Anthony et al., 2007). The RyR channels act as redox sensors as they are regulated by redox state of highly reactive cysteine residues (Eu et al., 2000; Pessah et al., 2002). RyRs form a macromolecular complex with regulatory proteins FKBP12.6, cytoskeletal proteins, adapter proteins, kinases, and phosphatases (Fill and Copello, 2002; Marx et al., 2000; Priori et al., 2001; Vest et al., 2005), which allows precise special regulation of RyRs activity in the microenvironment of the channel. The most common mechanism of intracellular Ca2+ signaling is through its association with calmodulin (CaM). The Ca2+/CaM complex binds to and modulates the functions of multiple key regulatory proteins, including a family of CaM kinases (CaMKs). CaMKs regulate several signaling molecules including mitogen-activated protein (MAP) kinase c-Jun N-terminal kinase (JNK) (Wu et al., 2009).
In the present study, we have shown that hypoxic/reperfusion injury is associated with an increase in RyR2 expression, which causes increased oxidative stress of dorsal column. Further CaMKII/JNK pathway upregulates RyR2 expression in hypoxic/reperfusion injury of white matter. We propose that use of pharmacological inhibitors of CaMKII/JNK pathway would reduce the cellular oxidative load and thereby have a neuroprotective role.
Methods
The composition of the Ringer's solution was: NaCl 124 mM, KCl 3 mM, NaH2PO4 1 mM, NaHCO3 26 mM, MgSO4 1.5 mM, CaCl2 1.5 mM and glucose 10 mM. The Ringer's solution was continuously bubbled with 95% O2 and 5% CO2.
Chemicals and reagents
HEPES, sucrose, EDTA, PMSF, BSA, Tween 20, thiobarbituric acid reactive substance (TBARS), trichloroacetic acid, sulphosalicylic acid, DTNB, H2O2, reduced glutathione (GSH), glutathione reductase (GR), NADPH, sodium azide, GSSG, Tris base, SDS, and APS were purchased from Sigma, USA. KN-62, SB202190, BAPTA-AM, FR180204, and SP600125 were purchased from Torcis Bioscience, Missouri USA. PVDF membrane, acrylamide, bis-acrylamide, TEMED, and real time PCR (RT-PCR) kit were purchased from BioRad. Chemiluminescence kit was purchased from GE Healthcare, NJ, USA and TRIzol® was purchased from Invitrogen, CA, USA. Primers were synthesized from Integrated DNA Technologies, (IDT), USA. Anti-RyR2 antibody was purchased from Affinity BioReagent Thermo Scientific and p-CaMKII, p-JNK, p-c-Jun, β-actin, and HRP conjugated goat anti-mouse and goat anti-rabbit were purchased from Santa Cruz Biotechnology, USA. Antibodies against Neurofilament 200 (NF-200) and glial fibrillary acidic protein (GFAP) were purchased from Sigma-Aldrich, St. Louis, MO, USA. Alexa Fluor 555 goat anti-rabbit and Alexa Fluor 488 goat anti- mouse SFX kits were purchased from Molecular Probes (Invitrogen), Oregon USA.
Animals
All the experiments were performed on male Wistar rats 8 weeks old, weighing 250–300 g. The animal procedures used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Nebraska Medical Center (UNMC).
Isolation of spinal cord dorsal column and induction of hypoxic/reperfusion injury
The method of dorsal column isolation and injury is described in our earlier publications (Agrawal and Fehlings, 1997; Thorell et al., 2002) and briefly discussed here. Rats were anesthetized with sodium pentobarbital (40 mg/kg intraperitoneal) and laminectomy was performed between T3 and T10 to expose the spinal cord. A 30-mm section of spinal cord was rapidly removed and placed in cold (2–4°C) Ringer's solution. A dorsal column segment was microdissected and was kept in Ringer's solution bubbled with 95% O2/5% CO2. Hypoxic condition was created by perfusing the ringer solution by 95% N2/5% CO2 for 1 h. After hypoxic injury reperfusion was performed for 2, 4, and 8 h. In another group, inhibitor was perfused during hypoxia and reperfusion for different time intervals. The removal of extracellular Ca2+ was done by replacing CaCl2 with 2mM MgCl2 in Ringer's solution during injury and reperfusion. n=3 for each time point in each experiment, and three independent samples were used to confirm the findings.
Western blotting
Spinal cord tissues were homogenized in ice cold lysis buffer (HEPES 5mM with sucrose 0.32M, MgCl2 1mM, ethylene glycol tetraacetic acid (EGTA) 2mM, PMSF 0.1mM, and protease inhibitors). Tissue lysates were centrifuged at 12000 rpm for 30 min. Protein quantification was done by Lowry's method. SDS-PAGE (7.5% for RyR2 and 10% for others) was done by Bio-Rad mini protein II electrophoresis cell. For each sample, 50 μg of protein was electrophoresed and electroblotted for 1 h at 20 V using semidry method. Membrane was blocked with 5% non fat dry milk in TBS-Tween (washing buffer) for 2 h at room temperature (RT) followed by three washings of 10 min each in washing buffer. After that, the blot was probed overnight with primary antibody followed by three washings of 10 min each and than incubated with HRP-labeled secondary antibody for 1 h at RT. Blots was washed with washing buffer three times for 10 min each. Lanes were illuminated using an electrochemiluminescence kit (Amersham) and exposure was taken on radiographic film.
Co-immunoprecipitation
Tissue lysates were centrifuged at 12000×g for 30 min at 4°C. Equal amounts of protein were incubated overnight with anti-p-JNK or IgG in a 500-μl total volume. Protein G-Sepharose beads (Oncogene Research, Boston, MA) were added to the lysate–antibody mix and incubated on a rotating platform for 2.5–3.5h at 4°C, followed by three to four washes with the lysis buffer. The immunoprecipitates were then immunoblotted with anti-p-CaMKII antibody and anti-mouse polyclonal antibody.
RNA isolation and quantitative PCR for miRNA of RyR
Total RNA was extracted from spinal cords using TRIzol regent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The concentration and quality of total RNA was measured by the UV absorbance at 260 and 280 nm (A260/280). An equal amount of RNA from each sample was used for quantitative PCR. One-step RT-PCR kit was purchased from QIAGEN. The PCR mixture (25 μl) will contain 0.5 μM of each primer, 12.5 μl of a commercial SYBR® green PCR master mixture, and 2.0 μg of mRNA and diethylpyrocarbonate (DEPC)- treated water. The samples were placed in eight-well strips that were sealed with an optical clear cap. The thermal cycling conditions were as follows: reverse transcription step 30 min at 50°C, initial PCR activation step (15 min at 95°C) and cycling step (denaturation for 30 sec at 94°C, annealing for 30 sec at 58°C, and then extension for 1 min at 72°C; 40 cycles) followed by melt curve analysis. An internal control, β-actin, was amplified in separate tubes. We have used comparative cycle threshold method 2ΔΔCT for relative quantitation of gene expression. Thus, all values for experimental samples have been expressed as differences (n-fold) between the sample mRNA and the sham mRNA. The data have been represented as the mean±standard deviations (SD) of data from three independent experiments. The primers for RT-PCR were:
RyR2:
Forward primer: 5’ GGAGAACTCAGAGACCAACAAGAGCA 3’
Reverse primer 5’ GGGTTTCAAAGCCATGCGGCAC 3’
β-actin:
Forward primer: 5’ TGGCCTCACTGTCCACCTTCCA 3’
Reverse primer: 5’ CGCAGCTCAGTAACAGTCCGCC 3’
Double immunofluorescence
Dorsal column was embedded in OCT compound (Fisher Scientific) medium and rapidly frozen in 2-methylbutane on dry ice. Cryostat sections (10 μm) were cut with a CM 1900 (Leica) at −18°C, mounted on superfrost slides and fixed for 15 min in cold acetone (-20°C). Permeablization was done with 0.5% Triton X-100 for 2 h at RT. Blocking was done with enhancer solution provided with the Aleax Fluor® SFX kit for 30 min at RT. Further sections were incubated with anti NF-200 (1:100 dilution in PBS) and RyR2 (1:100 dilution in PBS) or anti GFAP (1:100 dilution in PBS) and RyR2 (1:100 dilution in PBS) for overnight at 4°C. Slides were washed with PBS three times and incubated with Alexa fluor 488 anti-mouse (5 μg/mL) and Alexa Fluor 555 anti-rabbit (5 μg/mL) for 60 min at RT in the dark. Slides were washed three times with PBS. Slides were mounted with VECTASHIELD® mounting media and imaged with Zeiss 510 Meta Confocal Laser Scanning Microscope.
Bioassays
Lipid peroxidation
The level of lipid peroxoidase (LPO) in sample was measured as TBARS by the method of Buege and Aust (1978). Briefly, the reaction mixture contained sample (tissue lysate) (4 mg/mL) with an equal volume of Buege and Aust reagent (trichloroacetic acid, 15% w/v in 0.25 M HCl and thiobarbituric acid, 0.37% w/v in 0.25 M HCl) and kept in boiling water for 15 min. The samples were then centrifuged at 1000 x g for 10 min after cooling. The absorbance of the supernatant was measured at 532 nm. The concentration of TBARS was determined using an extinction coefficient of 1.56 x 105 and results were expressed as nmol of TBARS/mg of protein.
GSH
GSH was determined by the method of Jollow et al., 1974. Samples (tissue lysate) (0.3 mL) were mixed with 0.3 mL of sulphosalicylic acid (4%) and incubated at 4°C for 30 min. Thereafter, it was centrifuged at 1200×g for 15 min at 4°C and 0.1 mL of this supernatant was added to 1.7 mL phosphate buffer (0.1 M, pH 7.4) containing 0.2 mL DTNB (4 mg/ml) in a total volume of 2.0 mL. The yellow color developed was read immediately at 412 nm by spectrophotometer. The GSH content was calculated as nmol GSH/g tissue by using molar extinction coefficient of 13.6×103 M−1 cm−1.
Glutathione peroxidase (GPx)
Se-GPx (EC 1.11.1.9) activity was measured at 37°C by coupled assay system as described by Wheeler and associates (1990). The reaction mixture consisted of 0.01 mM H2O2, 0.05 mM GSH, 0.07 mM GR, 0.072 mM NADPH, 0.05 mM sodium azide, 0.1 M phosphate buffer (pH 7.4) and 0.1 mL sample in a total volume of 2 mL. The change in absorbance was recorded at 340 nm and the enzyme activity was calculated as nmol NADPH oxidized min−1 mg−1 protein using molar extinction coefficient of 6.22×103 M−1 cm−1.
GR
GR (EC 1.6.4.2) activity was measured according to the method of Carlberg and Mannervik, 1975. The assay mixture consisted of 0.025 mM EDTA, 0.025 mM oxidized glutathione, 0.005 mM NADPH, 0.1 M phosphate buffer (pH 7.6) and 0.1 mL sample in a total volume of 2.0 mL. The enzyme activity was quantitated at 25°C by measuring the disappearance of NADPH at 340 nm. The activity was calculated as nmol NADPH oxidized min−1 mg−1 protein using molar extinction coefficient of 6.22×103 M−1 cm−1.
Statistical analysis
Statistical analysis of data was done using analysis of variance (ANOVA) with post-hoc analysis. The Tukey–Kramer post-hoc test was applied to serve as significance among groups. The significance of results was ascertained at P<0.05. All the data are presented as means±SD of the means. Western blot radiographic images were scanned with CanoScan LiDE 25. Densiometric analysis was performed by using the public domain ImageJ program (developed at the National Institutes of Health and available at
Results
Increased expression of RyR2 protein after hypoxia
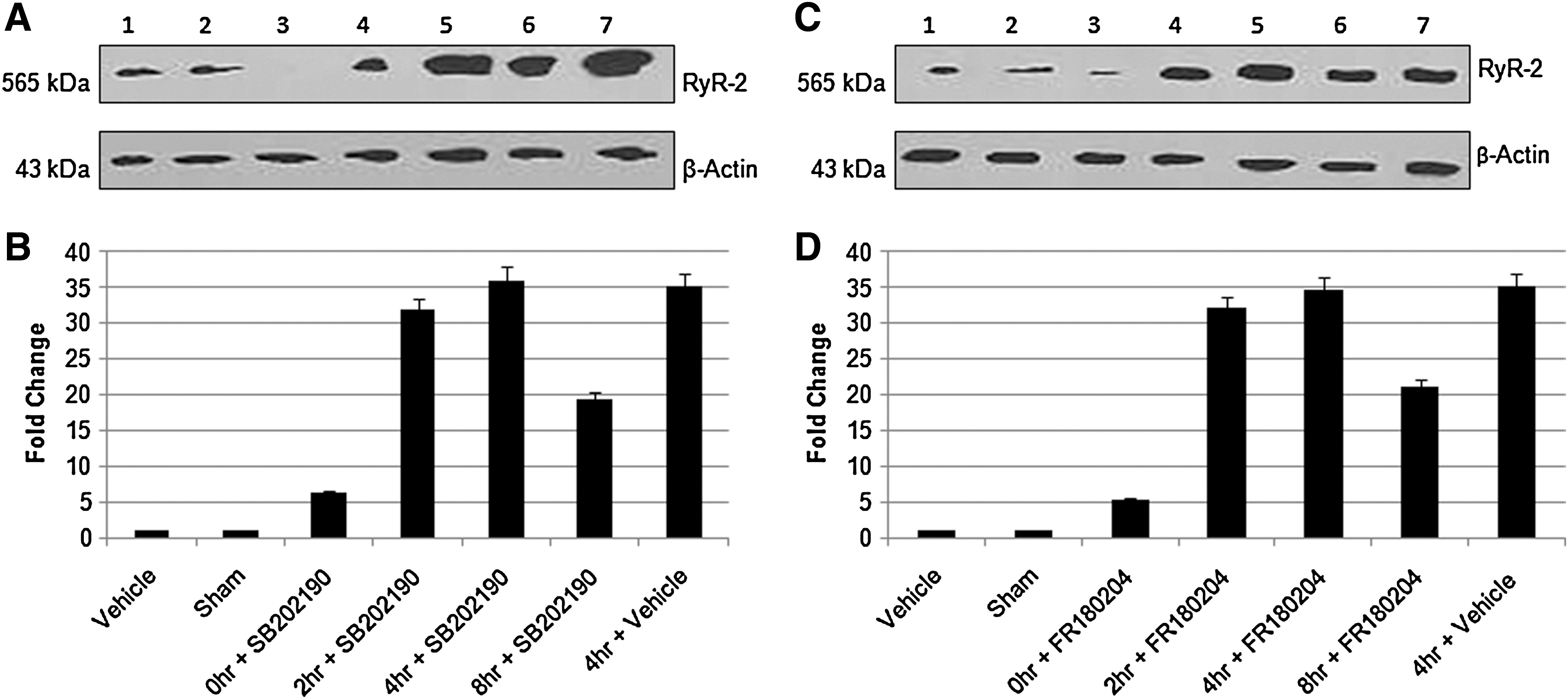
Western blot analysis revealed a single band (∼565 KDa) of immunoreactive protein (Fig. 1A), which is comparable to the predicted molecular weight of RyR2 (Priori and Napolitano, 2005). Figure 1A shows that RyR2 protein is visibly increased in the dorsal column at 4 h. Quantitative analysis was performed for sham, 0-h, 2-h, 4-h, and 8-h injured dorsal column (Fig. 1B). RyR2 protein was increased at 4 h as compared to sham. The transcriptional upregulation of RyR2 mRNA was determined using RT-PCR with quantification of fold changes as compared to sham. RT-PCR analysis also showed 35 fold increases in RyR2 mRNA expression (Fig. 1C).
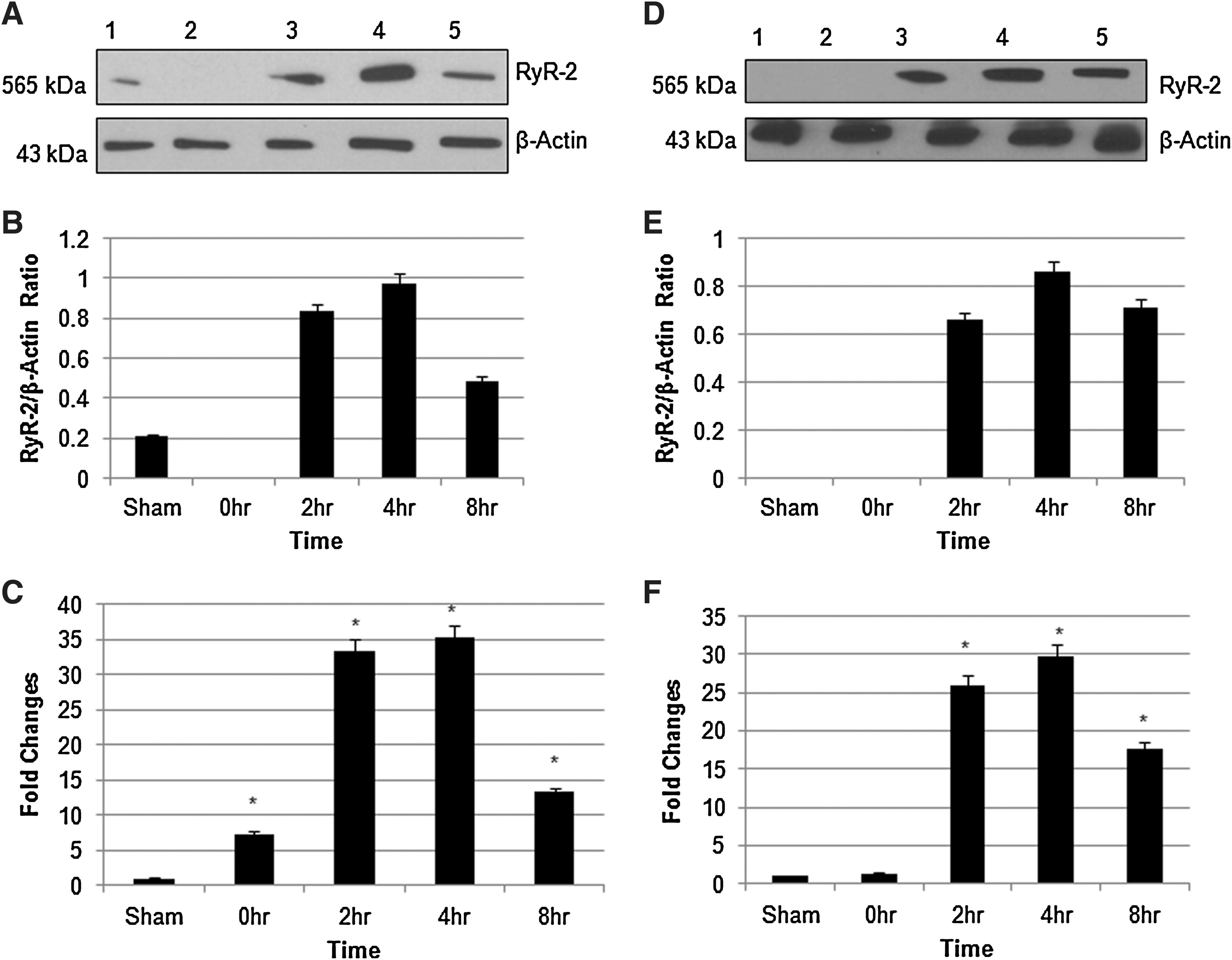
(
Role of extracellular Ca2+
To study the function of extracellular Ca2+ in regulation of RyR2, we replaced CaCl2 with equimolar MgCl2 in Ringer's solution. As shown in Figure 1D and E, there is no significant difference in RyR2 expression in the absence of extracellular Ca2+ as compared to its expression in the presence of Ca2+ (Fig. 1A and B). Similar effect was observed at the level of mRNA (Fig. 1F). This suggests that extracellular Ca2+ is not critical for RyR2 expression. Selective intracellular Ca2+ chelator BAPTA-AM (1μM) significantly reduced the expression of RyR2 protein and mRNA in the presence (Fig. 2A and B) or absence (Fig. 2C and D) of extracellular Ca2+, further signifying the importance of intracellular Ca2+ in RyR2 expression.
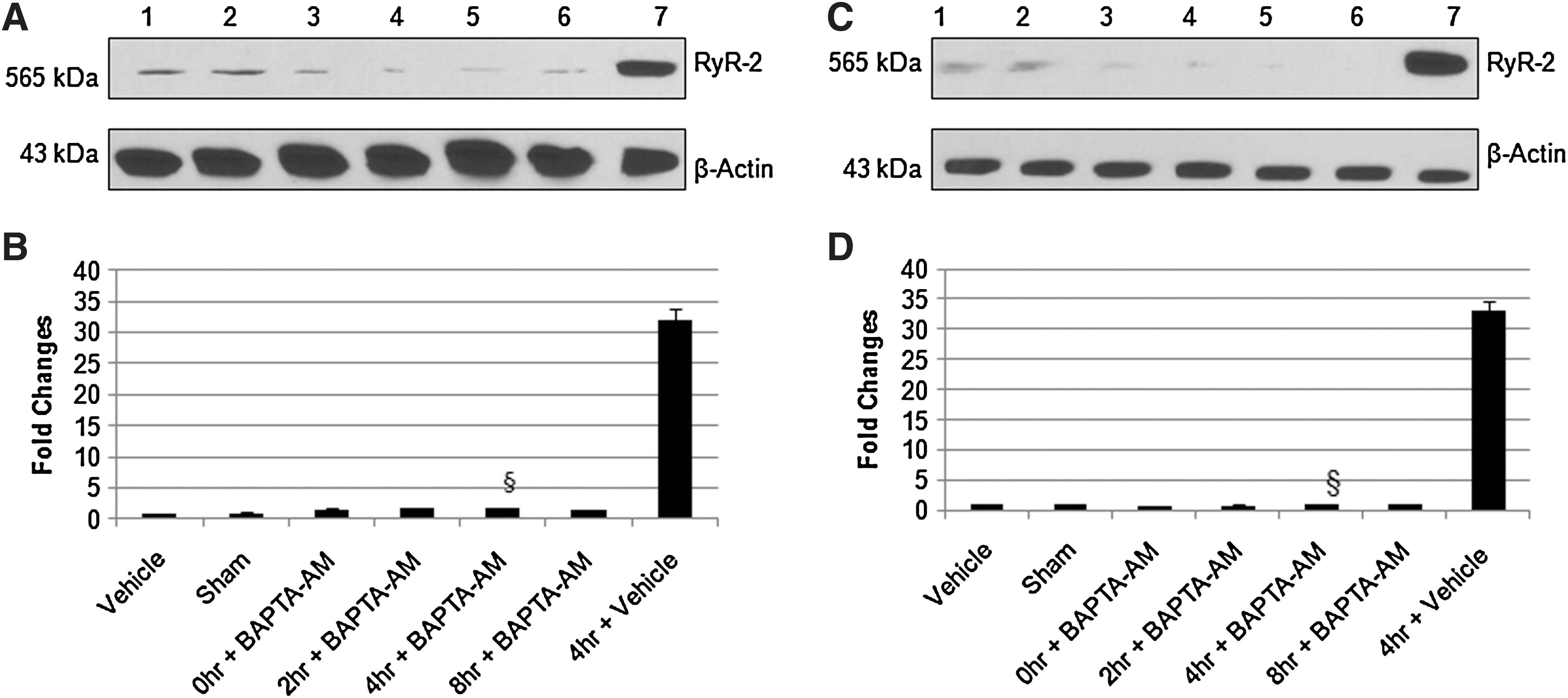
Cells expressing RyR2
In order to determine the cellular localization of RyR2 protein in the spinal cord white matter, we performed confocal immunohistochemistry with cell and subtype specific markers. Longitudinal sections of spinal cord were examined in sham control axons (Fig. 3A–C) and astrocytes (Fig. 4A–C) and 4 h following hypoxic injury to dorsal column (Figs. 3 D–F and 4 D–F). Double immunostaining revealed that RyR2 expression was increased in axons as well as in astrocytes when compared with sham. CaMKII inhibitor KN-62 (10 μM) was used during hypoxia and reperfusion and then double immunostaining was performed. We see a significant decrease in the expression of RyR2 in axons and astrocytes, respectively (Figs. 3 G–I and 4 G–I).

Double immunofluorescence staining of spinal cord dorsal column (original magnification x63) with neurofilament-200 and RyR2. Panel
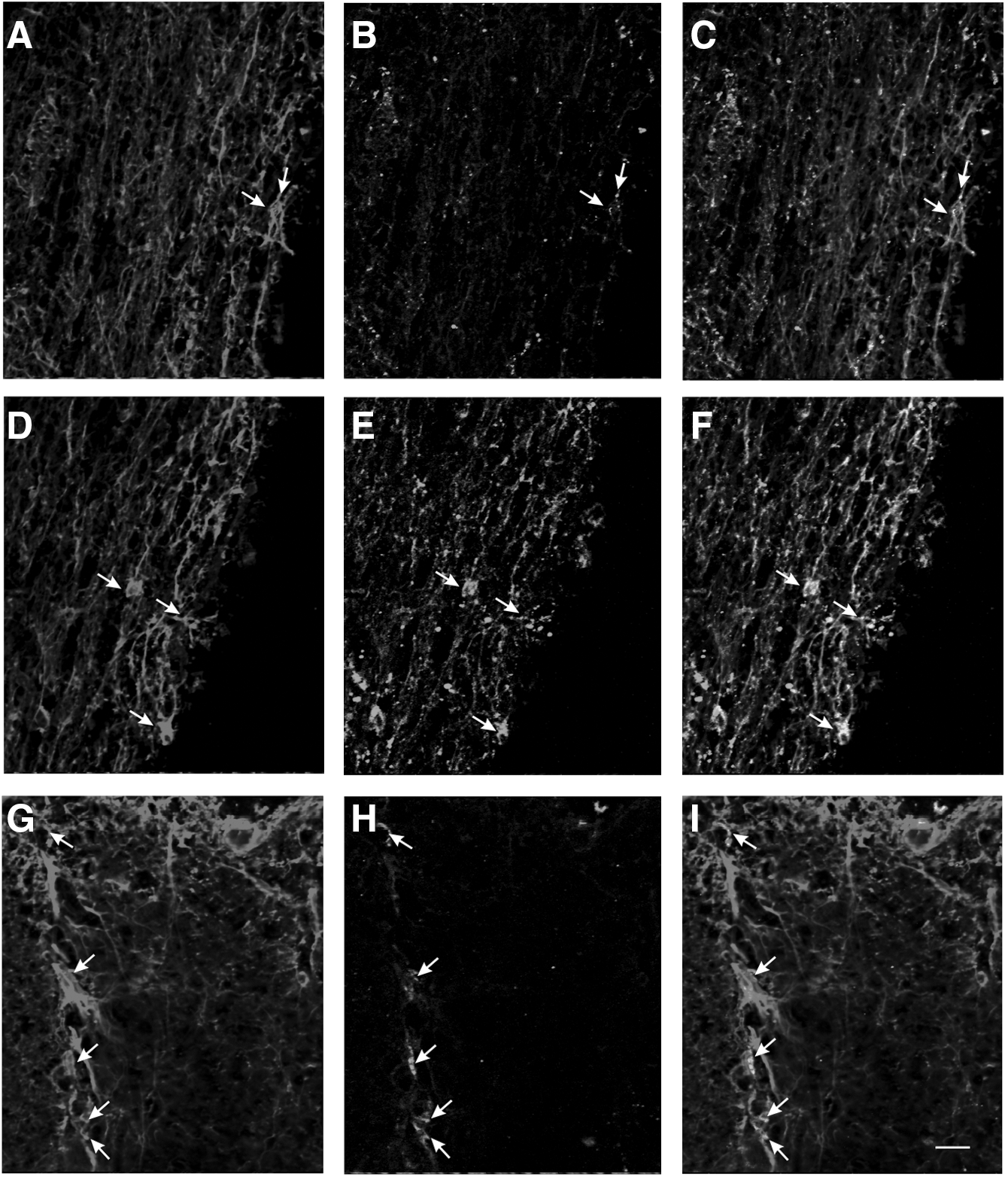
Double immunofluorescence staining of spinal cord dorsal column (original magnification x 20) with glial fibrillary acidic protein (GFAP) and RyR2. Panel
Involvement of CaMKII and JNK in hypoxic/reperfusion injury
To investigate the intracellular signaling molecules in RyR2 regulation, we used CaMKII inhibitor KN-62 (10 μM) and JNK inhibitor SP600125 (5 μM) during hypoxia and reperfusion. We observe a significant decrease in RyR2 protein and mRNA expression (Figs. 5A–D), which shows the involvement of CaMKII and JNK in RyR2 regulation in hypoxic/reperfusion injury of spinal cord dorsal column in vitro. MAP kinase, p38 inhibitor SB202190 (20 μM) and ERK inhibitor, FR180204 (20 μM) did not have any effect on RyR2 expression (Figs. 6A–D).
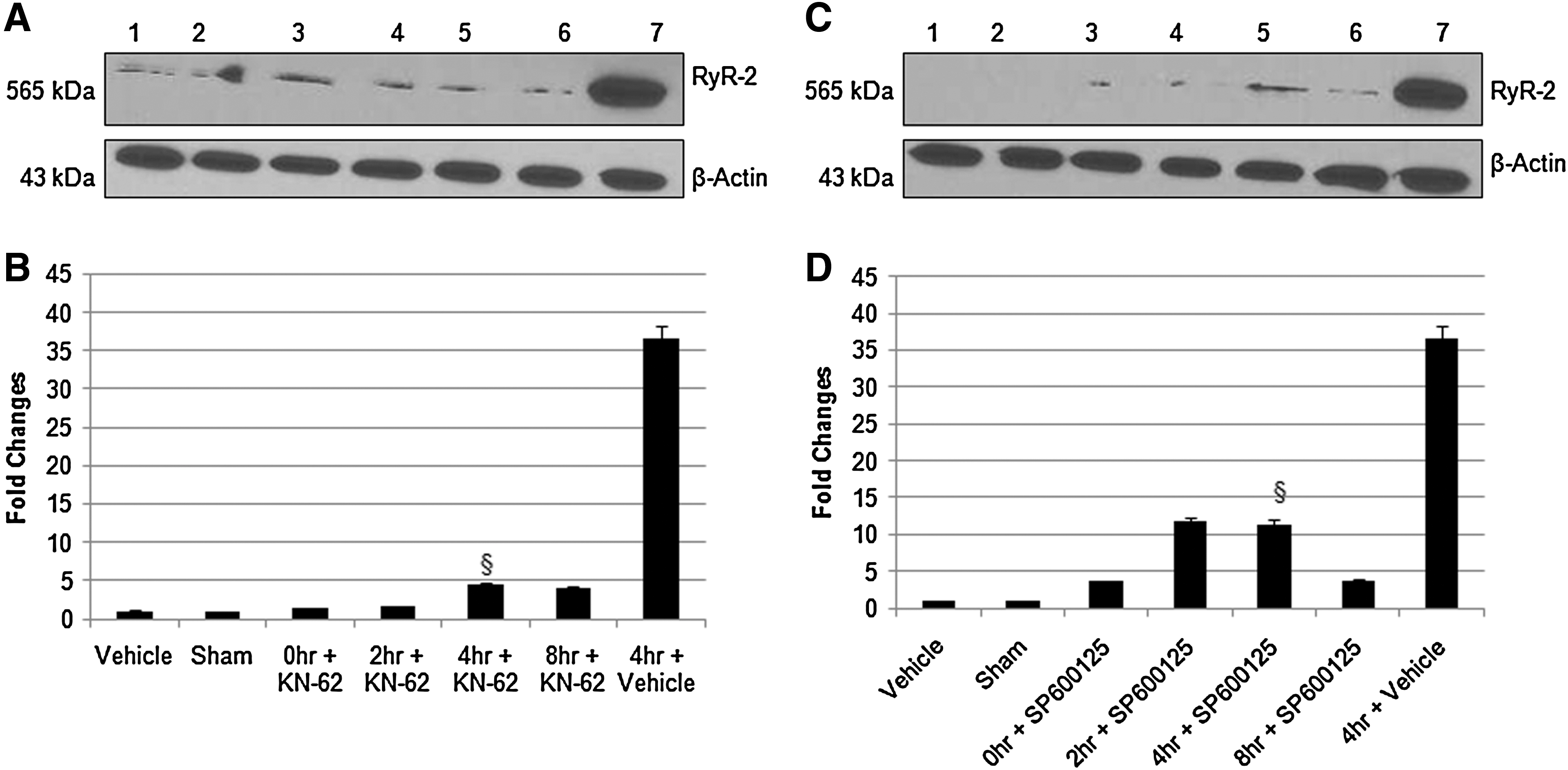
CaMKII activation in hypoxic/reperfusion injury
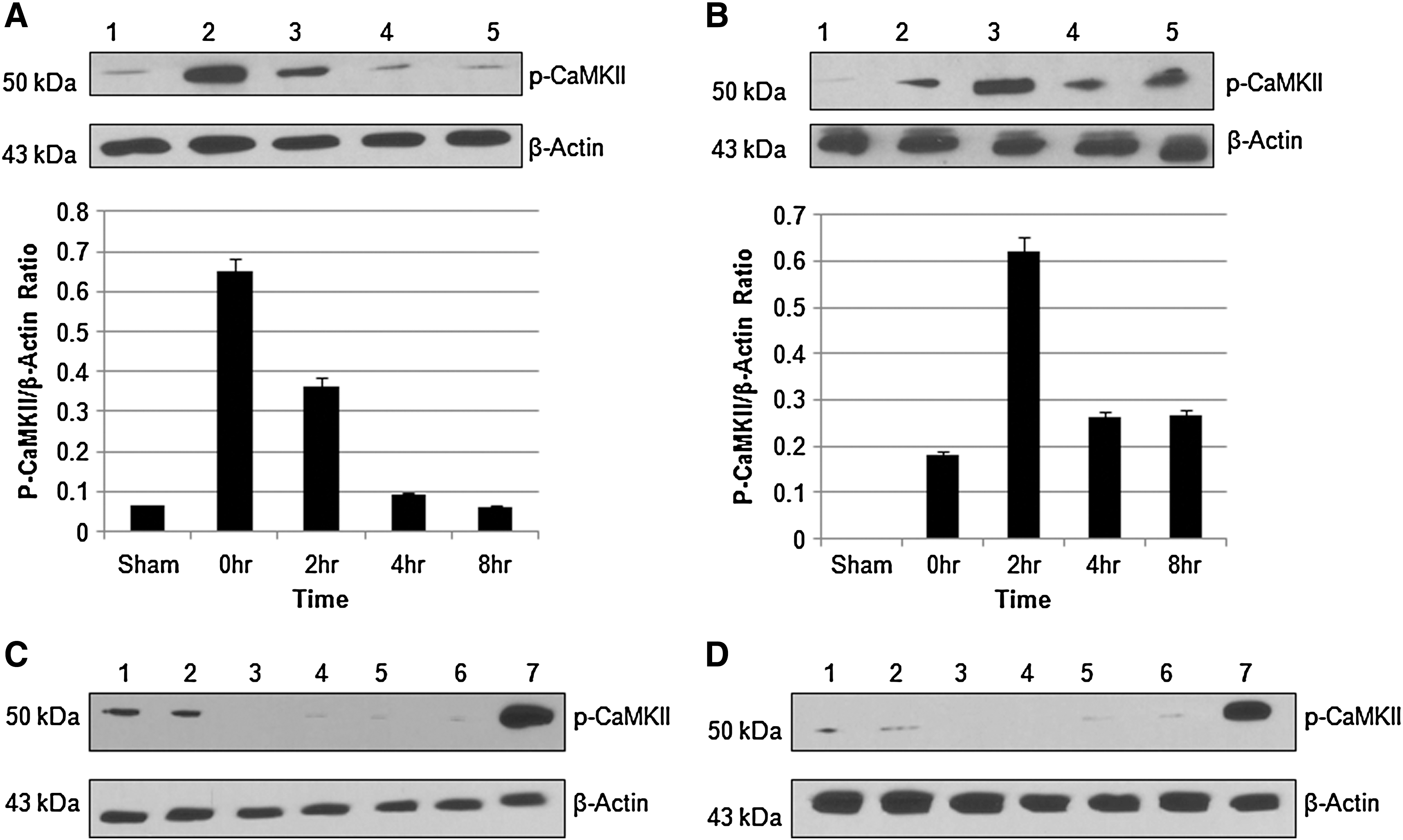
We studied the role of extracellular Ca2+ on CaMKII activation in hypoxic/reperfusion injury of spinal cord dorsal column. We observed time-dependent activation of CaMKII in the presence and absence of Ca2+. In the presence of Ca2+, the maximum activation (phosphorylation) of CaMKII was observed at 0 h. On removal of extracellular Ca2+, the maximum activation was observed at 2 h (Fig. 7A and B). We did not see any CaMKII activation in sham in the absence of Ca2+, which shows that extracellular Ca2+ participates in the basal level activation of CaMKII. Further BAPTA-AM (1 μM) reduced the activation of CaMKII in the presence (Fig. 7C) or absence (Fig. 7D) of extracellular calcium, which suggested the importance of intracellular Ca2+ in CaMKII activation in hypoxic/reperfusion injury of dorsal column.
Effect of CaMKII and JNK inhibitor
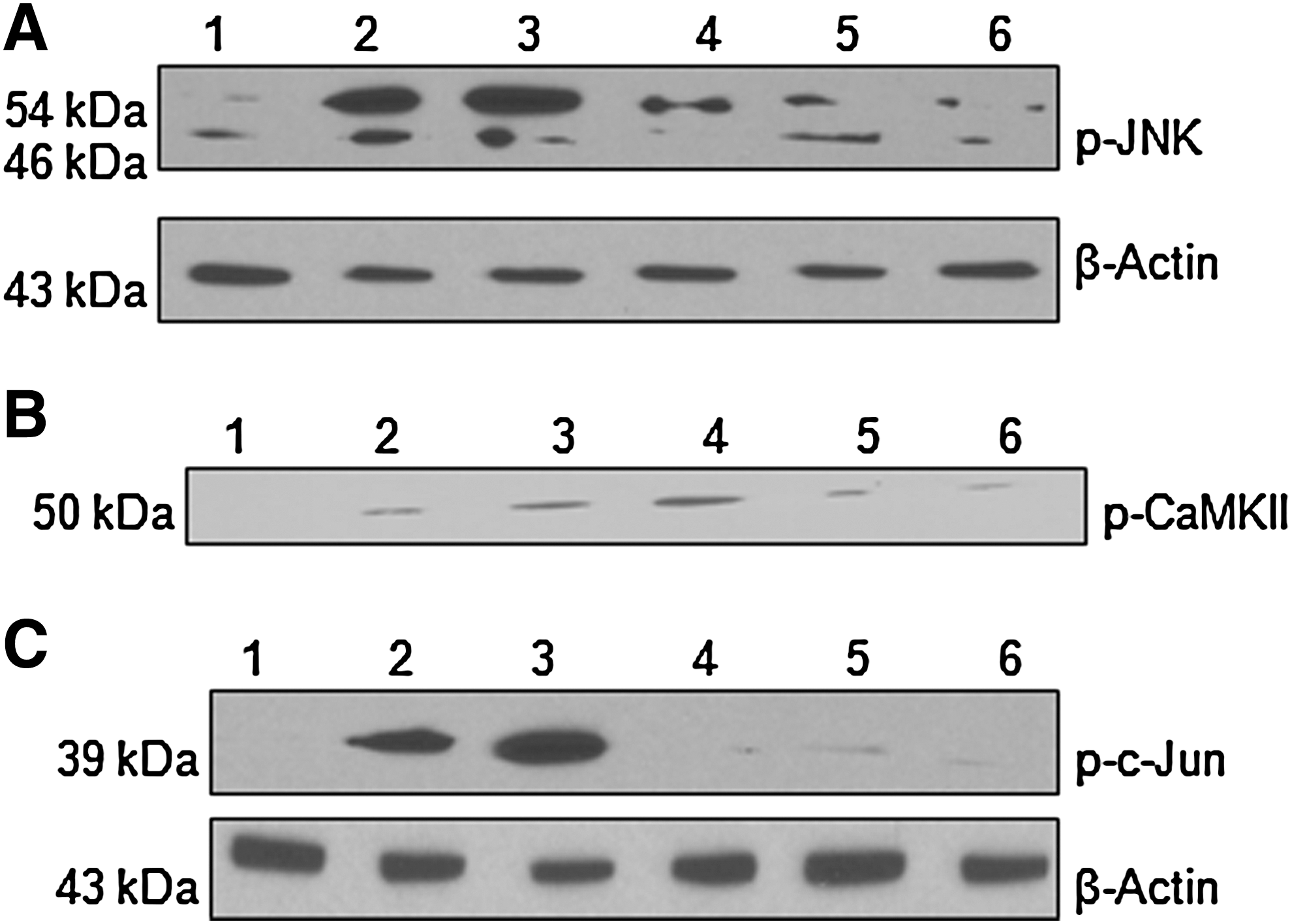
To establish the role of CaMKII/JNK pathway in injury, we studied the effect of KN-62 on JNK activation. We observed the maximum activation of JNK after 2 h of hypoxic/reperfusion injury and perfusion of KN-62 reduced the phosphorylation of JNK (Fig. 8A). This shows that CaMKII is upstream to JNK and that CaMKII/JNK pathway is involved in the regulation of RyR2 expression in hypoxic/reperfusion injury of spinal cord dorsal column in vitro. Co-immunoprecipiation of p-JNK and p-CaMKII also confirm the role of this pathway (Fig. 8B). The activation of c-Jun, a known transcription factor of JNK, was observed at its maximum 2 h after injury. Perfusion of SP600125 abolished the phosphorylation of c-Jun (Fig. 8C). This shows that c-Jun is activated in hypoxic/reperfusion injury of spinal cord white matter in vitro.
Effect of CaMKII and JNK inhibitor on cellular LPO, GSH content and GR and GPx activity
Treatment of dorsal column during hypoxic/reperfusion injury for 4 h with KN-62 or SP600125 significantly reduced the cellular oxidative stress as evident from the level of LPO and GSH and from the activity of GR and GPx (Table 1). This shows that RyR2 is responsible for increased cellular oxidative load, and that an inhibition of its expression can be neuroprotective.
Values are expressed as mean±SD. Significance was determined as p<0.05 when compared with *sham group or **hypoxic+reperfusion group.
Discussion
Traumatic or ischemic insult of white matter causes intracellular Ca2+ overload, and is considered to play a major role in SCI. In many cases removal of extracellular Ca2+ has been shown to be protective (Stys, 2004; Thorell et al., 2002). But axonal injury occurs even in the absence of extracellular Ca2+ because of Ca2+ overloading from intracellular stores (Nikolaeva et al., 2005; Ouardouz et al., 2003; Thorell et al., 2002). RyRs are localized on endoplasmic reticulum (ER) membrane and import Ca2+ from ER luminal space to the cytoplasm (Mackrill, 1999; Pozzan et al., 1994). Previous studies in our laboratory have shown the involvement of RyRs and IP3R in traumatic injury of spinal cord (Thorell et al., 2002). Ouardouz and associates (2003) have also reported the involvement of RyR1 and RyR2 in ischemic injury of white matter. As RyRs are important mediators of injury, the present study was undertaken to investigate the regulation and time dependency of RyR2 expression in hypoxic/reperfusion injury of spinal cord dorsal column in vitro.
Our study shows a change in the expression of RyR2 in hypoxic/reperfusion injury of dorsal column with time in vitro. Densiometric analysis shows maximum expression of RyR2 at 4 h, suggesting that reoxygenation upregulates the expression of RyR2. We observe a similar pattern in expression of mRNA. This transient increase in RyR2 expression might be caused by the to in vitro system, as there is no blood cell infusion that results in minimum secondary damage and fast switching off of the RyR2 expression. However, in our separate in vivo study, we found consistent increase in RyR2 expression until 8 h of study. A further double immunofluorescence study shows the localization RyR2 in axons and astrocytes in dorsal column. Studies have shown the localization of RyR2 in axonal fibers (Ouardouz et al., 2009). To find out the importance of extracellular Ca2+ in RyR2 regulation, we replaced CaCl2 with MgCl2. We did not observe any significant changes in the expression of RyR2, which suggest that extracellular Ca2+ is not critical for the regulation of RyR2 expression in hypoxic/reperfusion injury. We further confirm the importance of intracellular Ca2+ with BAPTA-AM.
Calmodulin is a well-known Ca2+-binding protein, which upon binding to calcium leads to activation of several signaling pathways in particular kinases (Wu et al., 2009). CaMKII is an abundant calmodulin-dependent kinase that is highly expressed in the central nervous system (CNS) (Yamauchi, 2007). Treatment of spinal tissue during hypoxic/reperfusion injury with KN-62, inhibitor of CaMKII, resulted in significant decrease in the expression of RyR2 both at mRNA and protein levels, suggesting the role of CaMKII in RyR2 gene regulation. We also observed that removing extracellular Ca2+ results in the shift in the time-dependent activation of CaMKII with maximum phosphorylation observed at 2 h as opposed to 0 h in the presence of Ca2+. Densiometric analysis showed that there is no significant change in the phosphorylation of CaMKII. This suggests that intracellular stores release enough calcium for the saturation of CaMKII activation. Treatment with BAPTA-AM reduced the activation of CaMKII, signifying the importance of Ca2+ in its activation.
JNK plays important role in stress, and its activation has been seen in SCI in mice (Esposito et al., 2009; Genovese et al., 2009; Yin et al., 2005). We found that JNK inhibitor SP600125 significantly inhibited the expression of RyR2 both at mRNA and protein levels, whereas other MAP kinase p38 and ERK did not show any effect. Activation of JNK causes its translocation to nucleus where it activates downstream transcription factors. Activation of c-Jun, a member of the AP-1 family of transcription factors, has been seen during spinal cord trauma and hypoxia/reoxygenation (Ravikumar et al., 2004; Xu et al., 2001). Our in silico analysis of RyR2 gene (NC_005116.2) promoter and further upstream regions show binding sites for AP-1. We found the maximum activation of c-Jun at 2 h after injury, and that treatment with SP600125 reduces its activation further, confirming our in silico findings.
There are reports that show the importance of CaMKII/JNK pathways in ER stress, similar to what we see in hypoxic injury of CNS (Timmins, et al., 2009). We observed that interaction of p-JNK and p-CaMKII by co-immunoprecipitation and treatment of KN-62 reduced the phosphorylation of JNK, which suggests that CaMKII/JNK pathway is involved in RyR2 regulation in hypoxic/reperfusion injury of dorsal column. It will be imperative to study the CaMKII/JNK pathway in specific cell types to understand its relative importance.
Our data show the operation of a positive feedback loop via Ca2+, which regulates the expression of RyR2 during hypoxic/reperfusion injury. Further, we speculate that after the removal of extracellular Ca2+, there is an increase in cytoplasmic Ca2+ because of the activation of RyR1 or IP3Rs, which are controlled by Cav1.2 voltage-gated Ca2+ channel and IP3 respectively. Voltage- gated Cav1.2 senses the stimulus caused by depolarization by Na+ and IP3 release through group I metabotropic glutamate receptors (mGluR-I) – phospholipase C (PLC) pathway (Pin and Duvoisin, 1995). Once RyR1 or IP3R become activated, they start to release Ca2+ from ER, which leads to the activation of RyR2 via Ca2+- induced Ca2+ release (CICR), resulting in Ca2+ overloading. This Ca2+ then further activates other signaling molecules. We have not looked at the regulation of RyR1 through this pathway, but we observe the involvement of CaMKII/JNK in the regulation of IP3R1 (data not shown).
Calcium overloading puts the cells in a state of oxidative stress (Csordas and Hajnoczky, 2009; Peng and Jou, 2010). To confirm the importance of downregulating the expression of RyR2 in neuroprotection, the effect of KN-62 and SP600125 was seen on important oxidative parameters. We found significant reduction in the cellular oxidative parameters and observed that inhibition of RyR2 expression is neuroprotective. It will be interesting to see the exact contribution of RyR2 downregulation on oxidative parameters, as there might be other pathways for RyR2 regulation, and CaMKII/JNK might affect cellular oxidative parameters by other means as well.
Conclusion
Our study shows the upregulation of RyR2 in axon and astrocytes in hypoxic/reperfusion injury of spinal cord dorsal column in vitro. We demonstrate the role of CaMKII/JNK pathway by the use of their specific inhibitors KN-62 and SP600125, respectively. Further, our studies suggest that extracellular Ca2+ is not critical for RyR2 expression in hypoxic/reperfusion injury of spinal cord dorsal column in vitro. We propose that agents blocking RyR2 expression would reduce the cellular oxidative stress, and have potent neuroprotective activity in SCI.
Footnotes
Acknowledgments
The authors thank the Department of Surgery, University of Nebraska Medical Center for providing financial support for this work.
Author Disclosure Statement
No competing financial interests exist.