
Abstract
Traumatic brain injury (TBI) often causes raised intracranial pressure (ICP), with >50% of all TBI- related deaths being associated with this increase in ICP. To date, there is no effective pharmacological treatment for TBI, partly because widely used animal models of TBI may not replicate many of the pathophysiological responses observed in humans, and particularly the ICP response. Generally, rodents are the animal of choice in neurotrauma research, and edema formation has been demonstrated in rat models; however, few studies in rats have specifically explored the effects of TBI on ICP. The aim of the current study was to investigate the ICP response of rats in two different, focal and diffuse, injury models of TBI. Adult male Sprague-Dawley rats were subjected to brain trauma by either lateral fluid percussion or impact-acceleration induced injury, in the presence or absence of secondary hypoxia. ICP, mean arterial blood pressure (MABP), and cerebral perfusion pressure (CPP) were monitored for 4 h after TBI. TBI alone or coupled with hypoxia did not result in any significant increase of ICP in rats unless there was an intracranial hemorrhage. At all other times, changes in CPP were the result of changes in MABP and not ICP. Our results suggest that rats may be able to compensate for the intracranial expansion associated with cerebral edema after TBI, and that they only develop a consistent post-traumatic increase in ICP in the presence of a mass lesion. Therefore, they are an inappropriate model for the investigation of ICP changes after TBI, and for the development of therapies targeting ICP.
Introduction
T
The pathophysiological changes that follow TBI are a complex network of structural, physiological, and functional secondary changes that are initiated by the primary mechanical event. Whereas a number of injury factors have been indentified in this secondary cascade, brain edema development, blood–brain barrier (BBB) breakdown, and decreased brain tissue perfusion, in particular, have been identified as critical to outcome (Bareyre et al., 1997; Unterberg et al., 2004; Vink and Nimmo, 2009). Brain tissue edema is a critical factor, as it causes an expansion of brain volume and thus increases intracranial pressure, consequently reducing cerebral perfusion and oxygenation (Marmarou 2007; McIntosh et al., 1990).
Traumatic brain edema is basically classified as either vasogenic or cytotoxic (Donkin and Vink, 2010; Klatzo 1967). Vasogenic edema is the result of the BBB disruption and accumulation of extracellular fluid, rich in protein, thus increasing the volume of the interstitial space without cell swelling (Gardenfors et al., 2002; Papadopoulos et al., 2004). In contrast, cytotoxic edema results from the sustained intracellular water accumulation, rich in electrolytes, mainly caused by active ion pump failure (e.g., Na+/K+-ATPase), resulting in cell swelling and decreased interstitial space (Liang et al., 2007). In reality, the edema that occurs after TBI is more often a mix of the two types with several subtypes having been suggested to differentiate between the pathogenic origins (Katayama and Kawamata, 2003; Unterberg et al., 2004). In any case, various mediators, such as glutamate, lactate, nitric oxide, arachidonic acid, substance P, and kinins are being released, which also contribute to the further development of both vasogenic and cytotoxic edema (Schilling and Wahl, 1999; Schneider et al., 1992), thus perpetuating the deleterious effects of brain water accumulation.
More than half of all deaths following TBI result from unresolved secondary brain edema that causes increased intracranial pressure (ICP) and reduced cerebral perfusion pressure (Feickert et al 1999; Rowland and Morris, 2007; Stahl et al., 2001). This makes management of raised ICP the single most important treatment strategy in TBI today (Miller et al., 1977; Marmarou, 1992; Wahlstrom et al., 2005; Steiner and Andrews, 2006). Although ICP has been the focus of intensive care management in TBI survivors for a number of decades, the mechanisms associated with brain water accumulation are largely unknown (Marmarou et al., 1991; Soustiel and Larisch, 2010) and, consequently, no effective therapeutic intervention currently exists for secondary cerebral edema and raised ICP (Beauchamp et al., 2008; Tolias and Bullock, 2004; Vink and Nimmo, 2009).
The development of novel pharmacological treatments requires extensive animal research. Whereas a number of effective animal models have been developed that recreate many of the pathophysiological responses observed in humans, the cost-effective and highly reproducible nature of rat models has made them the model of choice in neurotrauma research (Cernak, 2005; Lighthall et al., 1989). Although a few studies have looked at ICP changes after TBI in rats, no study had adequately investigated the effects of TBI on ICP in different models of TBI in rats. A thorough characterization of this effect is of paramount importance to the future development of ICP treatment, and is the primary focus of this study. In addition, we will investigate the effects of superimposed secondary hypoxia on ICP. In clinical scenarios, TBI is frequently accompanied by a reduction in the oxygen concentration of arterial blood (Bullock et al., 2000). This hypoxemia results from the apnea that accompanies the traumatic event, and is caused by failure or dysfunction of the respiratory response (Atkinson et al., 1998; Kemp et al., 2003). Hypoxemia leads to hypoxic or ischemic brain injury, which is known to significantly contribute to morbidity and mortality following TBI (Stocchetti et al., 1996).
Methods
Subjects
A total of 48 adult male Sprague-Dawley rats weighing 400–500 g were used in this study. All animals were housed in a conventional rodent facility at 24°C on a 12-h light/dark cycle with access to standard rodent food and water ad libitum. All experimental protocols were conducted according to the guidelines established by the Australian National Health and Medical Research Council and were approved by the animal ethics committees of the Institute of Medical and Veterinary Science and the University of Adelaide.
Experimental design
All animals were randomly assigned to the following six experimental groups: sham group for both models (uninjured, no hypoxia, n=9); lateral fluid percussion (LFP) injury only (n=8); LFP injury with secondary hypoxia (n=9); weight drop (WD) injury only (n=7); WD injury with secondary hypoxia (n=7); and hypoxia only for both models, as a control for the hypoxic episode (n=8). Sham group animals underwent all pre-trauma surgical preparation for both models, but were not subject to TBI. In two of the injury groups, secondary hypoxia (10% oxygen; 90% nitrogen) was induced for 15 min immediately following either LFP or WD in order to replicate the secondary hypoxia often experienced in clinical scenarios through transient apnea. The parameters for hypoxic ventilation were adapted during preliminary experiments from the methods of Rolett and colleagues (Rolett et al., 2000) to achieve a blood PO2 of ∼ 45–50 mmHg. Following the 15 min hypoxic episode, animals were ventilated with a normoxic mixture (30% O2, 70% N2) for the remainder of the monitoring period.
At the conclusion of the monitoring period, animals were killed by decapitation under pentobarbital anesthesia and their brains subjected to gross neuropathological examination to detect mass lesions. To understand the impact of bleeding on the variables under investigation, data from animals with significant intracranial hemorrhage were pooled and subject to separate analysis. A subgroup of animals without brain hemorrhage (6 shams, 6 trauma, and 6 trauma plus hypoxia) were used for determination of edema.
LFP injury
LFP was induced as described in detail elsewhere (McIntosh et al., 1989, Rogatsky et al., 1996, Thompson et al., 2005). Briefly, anesthesia was induced with 4% isoflurane in a normoxic mixture of 70% N2O and 30% O2 for 3–5 min. Anesthesia was then maintained at 2% of isoflurane via a nose cone while a tracheostomy was performed and the animals subsequently ventilated at ∼ 225 mL/min. The animal's right femoral artery and vein were cannulated using a polyethylene cannula (PE 50; inside diameter=0.58 mm, outside diameter=0.97 mm), with the arterial cannula used for monitoring of blood pressure and sampling of arterial blood for gas analysis and the venous cannula used for urethane administration after switching from isoflurane to urethane for maintenance anesthesia. Unlike isoflurane, urethane produces a long-lasting steady level of surgical anesthesia with minimal effects on the autonomic, respiratory, and cardiovascular systems (Fish, 1997; Maggi and Meli, 1986). The animal's core temperature was monitored and maintained at 37.5±0.5°C using a thermostatically heating pad. A sagittal incision (2 cm) was made on the dorsal scalp of the head and the temporal muscles were reflected before a 5 mm in diameter craniectomy was trephined into the skull centered 3 mm right of the sagittal suture and midway between the bregma and lambda. The dura was kept intact at the opening and a specially designed steel-bolt (4 mm inside diameter and 5 mm outside) was screwed and secured into the craniectomy with cyanoacrylate adhesive.
Immediately prior to trauma, animals were placed in a prone position onto a foam block and the steel bolt was filled with isotonic saline. Then the animal was attached to the LFP device (McIntosh et al., 1989) and a moderate trauma was induced with a force of 2.6–2.8 atm. After injury, an ICP probe (Codman ICP Express) was inserted to a depth of 6 mm below the dura through the steel bolt and into the cerebral parenchyma, and then sealed with sterile bone wax (Ethicon, W810). Arterial blood gases were analyzed periodically and arterial blood pressure was monitored continuously.
WD injury
Animals were injured using the Marmarou impact-acceleration model of diffuse TBI, which has been described extensively elsewhere (Marmarou et al., 1994). Briefly, rats were anesthetized, intubated, ventilated, and their core temperature was maintained at 37.5±0.5°C, as described for the LFP animals. The skull was exposed after midline sagittal incision (2 cm) and soft tissue retraction. A drop of cyanoacrylate adhesive was then applied to the skull along the midline, midway between the lambda and the bregma sutures, and a protective stainless steel disc (10 mm diameter x 3 mm thick) was fixed centrally upon the skull. The stainless steel disc acted as a helmet during trauma to reduce the incidence of skull fractures. Immediately prior to trauma, animals were secured in a prone position onto a foam block of uniform density (depth 11.5 cm) and placed under the injury apparatus, ensuring that the brass weight was centrally aligned above the protective steel disc. A TBI was induced by releasing the brass weight (450 g) from a height of 2 m via a cylindrical PVC conduit and allowing it to impact onto the steel disc attached to the animal's skull. The animal was rapidly relocated after the initial impact to prevent any further contact from the rebounding weight. The stainless steel disc was removed and the animal placed in a stereotactic frame (David Kopf Instruments, Tujunga, CA). For insertion of the ICP probe, a burr hole (2 mm diameter) was made in the animal's left parietal bone at the stereotactic coordinate, 0.5 mm posterior to the bregma and 4 mm lateral to the midline suture. After puncturing the dura mater, the ICP probe was directly inserted 6 mm below the dura into the cerebral parenchyma.
Edema measurement
Brain water content was assessed by the wet weight–dry weight method (O'Connor et al., 2006; Donkin et al., 2009) at 5 h after focal or diffuse trauma. Rats (n=6 per group) were decapitated under pentobarbital anesthesia and their brains removed. The cerebellum and brainstem were then rapidly removed and the cerebral hemispheres placed in glass vials. After weighing, brains were dried at 100°C for 24 h. The dry weight was then obtained and brain water content calculated according to the formula
Blood gas analysis
Arterial blood samples (0.2 mL) were obtained via the femoral cannula and analyzed using an Osmetech OPTI cassette system with an Osmetech OPTI blood gas analyzer (CCA, Helena Laboratories Pty Ltd., Australia). After TBI, blood gas analysis was conducted twice in each animal, once at 10 min and again at 210 min post-trauma.
ICP monitoring
ICP was measured with a CODMAN® microsensor probe, which consisted of a nylon catheter with a silicon strain-gauge type sensor mounted at the tip (1.2 mm diameter; Codman & Shurtleff, Inc., DePuySpine™). The ICP probe was connected to a Codman Express monitor (DePuySpine™) and measurements of ICP were recorded continuously by a MacLab data acquisition system (MacLab 2e) following insertion of the probe. Probe insertion was complete by 60 min post-trauma.
Blood pressure monitoring
Mean arterial blood pressure (MABP) was monitored using a MacLab data acquisition system (MacLab 2e). A Statham-type pressure transducer was connected via a polyethylene tube to the animal's arterial cannula. The pressure trace was relayed from the transducer to the MacLab via a bridge amp and all data were recorded and stored using a personal computer.
Determination of cerebral perfusion pressure
Cerebral perfusion pressure (CPP) at individual time points was calculated by the equation:
and stored using a MacLab data acquisition system (MacLab 2e) linked to a personal computer.
Statistical analysis
All data are shown as means and SEMs and were analyzed by two-way, repeated measures analysis of variance (ANOVA), followed by individual Student Newman Keuls tests. A p value of <0.05 was considered significant.
Results
Mortality and hemorrhage
The overall mortality rate for this investigation was 8% (2 FPI; 2 WD), which is consistent with of <10% mortality previous reports at this severity of injury (McIntosh et al., 1989; Marmarou et al., 1994). Deaths were caused by skull fracture or respiratory failure. Mass hemorrhagic lesions were found in WD animals that were excluded on the basis of skull fracture but not in animals that did not sustain a skull fracture. Following FPI, 5 animals were found to have mass hemorrhagic lesions and were analyzed separately.
Edema
Brain water content in sham (uninjured animals) was 78.5±0.3%. At 5 h after TBI, brain water content had increased to 79.8±0.1%, whereas in injured animals exposed to a hypoxic episode, it had increased to 81.4±0.4%. These results are consistent with previous reports that have shown that these rodent TBI models typically cause between 1 and 4% edema formation (Donkin et al., 2009; Nida et al., 1995; O'Connor et al., 2006; Soares et al., 1992), which may be exacerbated by hypoxia (Ishige et al., 1987; Van Putten et al., 2005).
Blood gas analysis
Blood oxygen saturation (SO2) was significantly diminished in animals subjected to TBI with hypoxia or hypoxia only (Table 1) confirming the effectiveness of the hypoxic period. The observed decrease in SO2 was physiologically as well as statistically significant, as the initial reading for each group fell below the clinical reference limit of 90%. By 210 min following trauma, SO2 had returned to sham levels in all groups.
Denotes significance (p<0.05) between an experimental group and sham animals at that time point (** p<0.01; *** p<0.001).
Denotes significance (p<0.05) between time points within an experimental group (†† p<0.01; ††† p<0.001).
Arterial blood pressure
MABP in sham and injured animals, with and without hypoxia, is shown in Figure 1. Prior to injury, the MABP across all animals was 94±3 mm Hg. During the hypoxic episode, MABP decreased to <60 mm Hg in both injured and non-injured animals, and returned to near sham levels soon after the resumption of normoxia. The return to near normal MABP after hypoxia was more gradual in the WD injured animals than in the LFP injured animals. After TBI, mild hypotension was apparent over the post-traumatic monitoring period. Such hypotension is typical of TBI, albeit that it was less apparent after a hypoxic episode.

Changes in rat mean arterial blood pressure (MABP) following traumatic brain injury (TBI) induced by lateral fluid percussion (LFP) injury
ICP analysis
Sham group animals in both models of TBI demonstrated an average ICP of ∼ 4–5 mm Hg during the 4 h post-injury. In the groups receiving TBI only, there was no significant increase in ICP compared to sham groups in either FPI or WD injury (Fig. 2), with ICP typically changing by <2 mm Hg. Similarly, animals exposed to hypoxia (with no TBI) showed a slight increase of 1–2 mm Hg in ICP compared to sham group animals, which was also insignificant. However, when TBI was combined with hypoxia, there was a slight increase in ICP in both models. In the LFP model, this increase never exceeded a mean of 8 mm Hg, which was insignificant. However, when secondary hypoxia was combined with WD injury, there was a small but significant rise in ICP (p<0.05). ICP gradually increased over time from a mean of 8 mm Hg to a mean of 11 mm Hg at 4 h after injury. This increase in ICP, although statistically significant compared to shams, was only 2–4 mm Hg above that of transient hypoxia alone (p>0.05), which is unlikely to be particularly deleterious.

Changes in rat intracranial pressure (ICP) following traumatic brain injury (TBI) induced by lateral fluid percussion injury
No previous study has explored the importance of brain hemorrhage on ICP changes after TBI in rats. We had retrospectively separated animals that had intracranial bleeding and excluded them from the above analysis. When pooling these animals with an intracranial hemorrhage and analyzing their ICP changes, it was noted that ICP was significantly increased in this group when compared to animals with no intracranial bleeding (Fig. 3). Indeed, ICP approached 20 mm Hg by the conclusion of the 4 h monitoring period. Moreover, the scale of the increase in ICP varied and seemed related to the level of intracranial bleeding. Therefore, the presence of a mass lesion facilitated the development of increased ICP in rats following TBI.
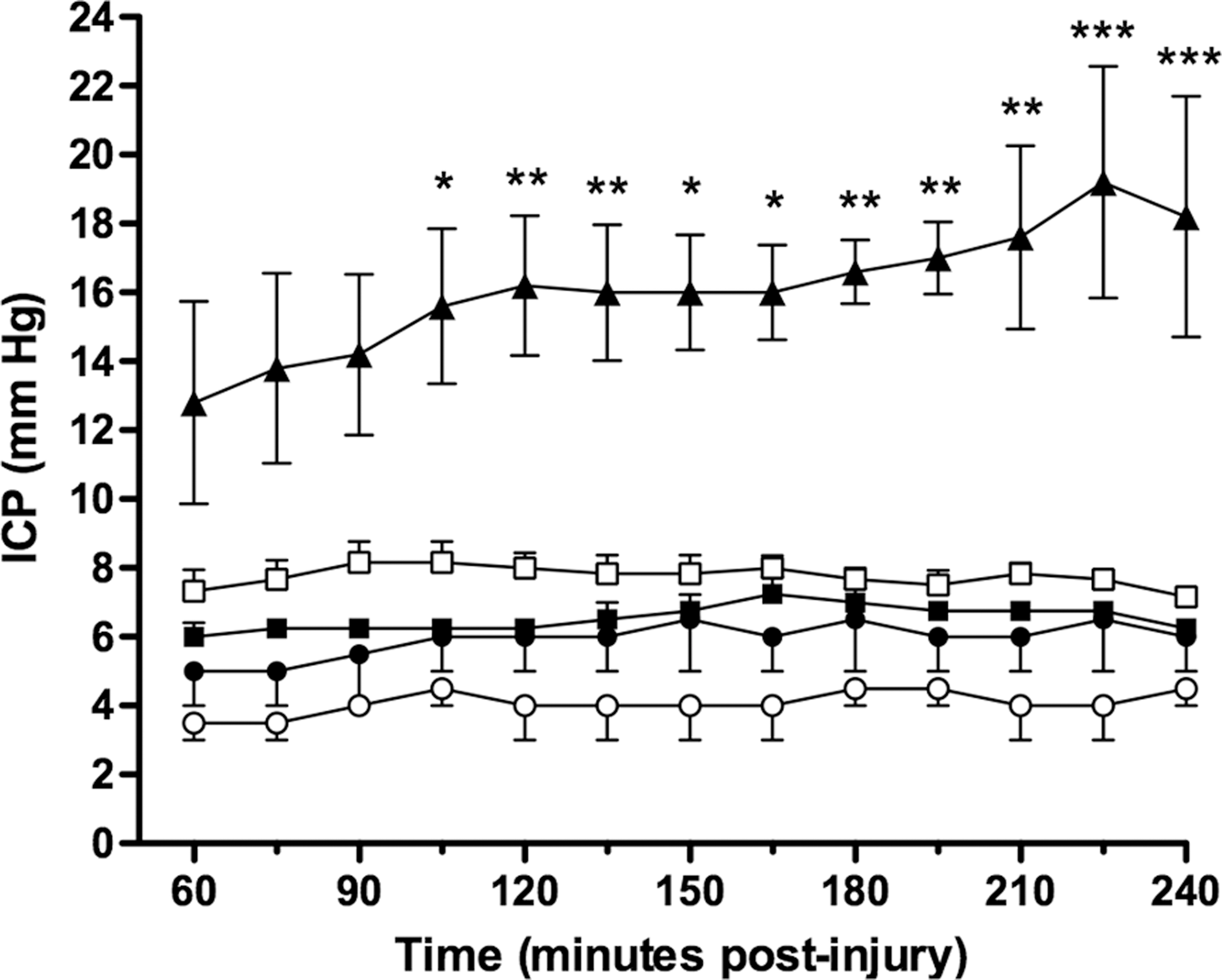
Effects of mass lesions on rat intracranial pressure (ICP) following fluid percussion induced traumatic brain injury (TBI). ICP was significantly greater in TBI animals with mass lesions than in all other without an intracranial hemorrhage. *=p<0.05; **=p<0.01; ***=p<0.001 versus TBI alone. ▴=TBI with intracranial hemorrhage present (n=5); ▪=TBI alone (n=4); □=TBI plus hypoxia (n=6); •=hypoxia alone (n=4); ○=shams (n=4).
CPP analysis
With respect to CPP, sham group animals demonstrated pressures of 100 mm Hg over the 4 h period post-injury (Fig. 4). Following both FPI and WD injury alone, there was a significant decline in CPP of >20 mm Hg in both injury groups relative to shams (p<0.05). Surprisingly, the decline in CPP between 1 and 4 h post-trauma was attenuated by the earlier exposure to secondary hypoxia (p>0.05 versus shams). The increased CPP in animals previously exposed to hypoxia was most likely related to the effects of MABP to a prior hypoxic episode, given that the CPP changes closely resembled changes in MABP. Whether this increase in MABP after the hypoxic episode is a rebound effect in an attempt to restore tissue oxygenation is unknown.

Changes in rat cerebral perfusion pressure (CPP) following traumatic brain injury (TBI) induced by lateral fluid percussion injury
The relationship between MABP and CPP was even more evident during the 15 min hypoxic period. Given that the ICP probe could be simply inserted through the existing craniotomy in FPI animals, very early ICP/CPP measurements could be obtained in this subgroup of animals (Fig. 5). During the hypoxic episode immediately after TBI, ICP did not change significantly at any stage (mean=10±1 mm Hg). In contrast, CPP decreased from 100±3 mm Hg to 42±6 mm Hg (Fig. 5A). This profound decrease in CPP was associated with a decrease in MABP during the hypoxic episode (see Fig. 1). CPP stayed low throughout the entire period of secondary hypoxia (Fig. 5B), but when the animals were returned to a normoxic gas mixture, CPP rapidly returned to normal values, in association with the increase in MABP (see Fig. 1). Such falls in CPP and MABP were not observed in trauma-only animals, suggesting that any decline in MABP during periods of hypoxia are highly deleterious to CPP.

Changes in rat cerebral perfusion pressure (CPP) and intracranial pressure (ICP) during and following the 15 min hypoxic episode induced immediately after FPI.
Discussion
The current study examines ICP changes in two different and well-characterized models of TBI in the rat: the focal fluid percussion injury model and the diffuse impact-acceleration model, with a particular focus on the effects of secondary hypoxia and mass lesions on post-traumatic ICP. Our results demonstrate that TBI alone or in combination with secondary hypoxia does not result in any sustained increase in ICP in the rat unless there is a mass lesion associated with a hemorrhage. In the absence of ICP changes, reductions in CPP that were noted in TBI were closely associated with changes in MABP.
Apnea is frequently observed in rats following experimental TBI, however hypoxemia is typically avoided by rapid ventilation (Prins and Hovda, 1998). To simulate the effect of post-traumatic apnea in these studies, animals received a 15 min hypoxic episode immediately following TBI. The hypoxic episode reduced arterial PO2 to ∼ 50 mm Hg. This fall in oxygen saturation to 78.4±1.2 % would correlate to a significant increase in mortality and morbidity for human TBI survivors (Stocchetti et al., 1996).
Animals exposed to secondary hypoxia in addition to TBI showed some increase in ICP compared to the sham groups, albeit that this increase was very modest and never achieved statistical significance in the LFP group. Statistical significance was achieved in the WD model when compared to sham animals; however again, this increase of ICP was modest (2–4 mm Hg over hypoxia alone) and would not be considered clinically relevant. Nonetheless, the increase in the WD model compared to the LFP model may reflect the differential nature of edema formation in the two models of TBI. Early edema is typically considered vasogenic in the WDI model, although an element of cytotoxic edema is anticipated because of delayed secondary injury factors, as previously discussed (Unterberg et al., 2004). However, introduction of a hypoxic insult to this model may have resulted in a more rapid and substantial cytotoxic edema which, combined with the vasogenic edema, may have resulted in an increase in the brain water content over and above the 2–4% typically reported (O'Connor et al., 2006). This is in contrast with LFP where cytotoxic edema has been reported to dominate in the early phase after trauma (Albensi et al., 2000; Bareyre et al., 1997).
In both models of TBI, cytotoxic swelling of neurons and glia does not occur in isolation but in conjunction with vasogenic accumulation of fluid in the extracellular space. Intracellular swelling is therefore not balanced by a decrease in extracellular fluid. On the contrary, intracellular expansion will occur in concert with expansion of the extracellular space, ultimately resulting in substantial enlargement of cerebral tissue. The extracellular space only constitutes 12–19% of brain volume (Go, 1997), therefore swelling of the intracellular space along with the introduction of new intravascular constituents (i.e., water and plasma solutes) provides greater potential for tissue expansion than does vasogenic edema in isolation.
A number of previous studies have reported ICP values in rodents after TBI, with some reporting increases after TBI (Liu et al., 2002; Wei et al., 2010; Zweckberger et al., 2009) and others reporting no increase (Goren et al., 2001; Kahveci et al., 2001; Rogatsky et al., 2003; Thomas et al., 1998). Notably, those that report increases have usually been associated with a severe focal or penetrating injury that causes extensive hemorrhage. Although the reasons for such contradictions are unclear, the findings may well be explained by anatomical differences in relation to the tentorium cerebelli in the central nervous system of rats compared to other mammals such as humans, sheep, pigs, cats, or dogs. The tentorium cerebelli is an arched lamina of dura mater separating the supratentorial compartment (containing the cerebral hemispheres) from the infratentorial compartment, or posterior fossa, which contains the cerebellar hemispheres and brainstem (Klintworth, 1968). In mice, rats, hamsters, and gerbils, it consists of delicate meningeal folds that only separate the lateral portions of the cerebellum and cerebral hemispheres. In other mammals including goats, sheep, cats, dogs, and humans, the bilateral folds have joined to form a diaphragm, a structure sufficient to prevent expansion or collapse of the supratentorial matter into the infratentorial compartment (Bull, 1969). Such movement through the tentorial aperture is a feature of transtentorial herniation. Given that the tentorium cerebelli of the rat is quite rudimentary and underdeveloped, any pressure associated with brain swelling could potentially be readily transferred to the infratentorial compartment and beyond, thus reducing the supratentorial ICP. No post-traumatic increase in ICP would be noted. In the presence of hemorrhage, however, the mass lesion will overcome the volume expansion allowed by the absence of the tentorium (reduced compliance) and an increase in ICP will result. Therefore the inconsistencies in the post-traumatic ICP reported in rodents may well be related to the presence or absence of intracranial hemorrhage. Moreover, the presence or absence of increased ICP would have a critical impact on the development of effective neuroprotective therapies designed for translation into the human condition where increases in ICP are common.
Conclusions
Following TBI in rats, an elevation of ICP was only observed in animals that developed mass lesions. TBI alone or coupled with secondary hypoxia did not result in any relevant increase of ICP in both models of TBI in the absence of a mass lesion. In the absence of any change in ICP, CPP was driven by changes in MABP. Although the reasons for the lack of ICP change are unknown, we speculate that rats may be able to compensate for the intracranial expansion associated with cerebral edema along their craniospinal axis, partially because of an underdeveloped tentorium cerebelli. The development of neuroprotective treatments targeting the effects of increased ICP might therefore be best pursued in other species that may more consistently produced sustained increases in post-traumatic ICP.
Footnotes
Acknowledgments
We would thank Dr. Emma Thornton for her helpful comments during the preparation of this manuscript. This research was funded, in part, by a grant from the National Health and Medical Research Council of Australia.
Author Disclosure Statement
No competing financial interests exist.