
Abstract
A number of studies have established a deleterious role for inflammatory molecules and reactive oxygen species (ROS) in the pathology of traumatic brain injury (TBI). Caffeic acid phenethyl ester (CAPE) has been shown to exert both antioxidant and anti-inflammatory effects. The primary objective of the present study was to examine if CAPE could be used to reduce some of the pathological consequences of TBI using rodent models. Male Sprague-Dawley rats and C57BL/6 mice were subjected to controlled cortical impact (CCI) injury. Blood–brain barrier (BBB) integrity was assessed by examining claudin-5 expression and the extravasation of Evans blue dye. The effect of post-injury CAPE administration on neurobehavioral function was assessed using vestibulomotor, motor, and two hippocampus-dependent learning and memory tasks. We report that post-TBI administration of CAPE reduces Evans blue extravasation both in rats and mice. This improvement was associated with preservation of the levels of the tight junction protein claudin-5. CAPE treatment did not improve performance in either vestibulomotor/motor function (tested using beam balance and foot-fault tests), or in learning and memory function (tested using the Morris water maze and associative fear memory tasks). However, animals treated with CAPE were found to have significantly less cortical tissue loss than vehicle-treated controls. These findings suggest that CAPE may provide benefit in the treatment of vascular compromise following central nervous system injury.
Introduction
T
Caffeic acid (3,4-dihydroxycinnamic acid) is a phenolic compound found in fruits and vegetables (Sondheimer, 1958) that has been shown to block the production of ROS, increase glutathione levels, and reduce inflammatory cytokine levels following exposure to various toxic agents (Kono et al., 1997; Stewart et al., 2001; Uz et al., 1998). CAPE is a naturally-occurring derivative of caffeic acid that is highly cell permeable due to the presence of an ester linkage. Once inside the cell, the ester bond is cleaved by intracellular esterases to release caffeic acid, the active moiety. Similarly to caffeic acid, CAPE has also been shown to decrease inflammatory processes, brain lipid peroxidation (Irmak et al., 2003), and free radical damage. It has been reported that these effects of CAPE are due to its influences on xanthine/xanthine oxidase, nuclear factor-κB (NF-κB), cyclooxygenase-2 (COX-2), 5-lipoxygenase (5-LOX), the production of inflammatory cytokines, and the release of cytochrome c from mitochondria (Chan et al., 1995; Koshihara et al., 1984; Michaluart et al., 1999; Mirzoeva and Calder, 1996; Natarajan et al., 1996; Sud'ina et al., 1993; Uz et al., 1998).
Since TBI triggers many of the pathological cascades targeted by CAPE, we hypothesized that CAPE treatment would lead to decreased brain pathology and improved neurological outcomes. In the present study, we examined if post-injury CAPE administration can reduce TBI-induced enhanced blood–brain barrier (BBB) permeability, tissue loss, and neurological dysfunction in animals with TBI. Our results show that while post-injury CAPE administration reduced BBB permeability and cortical tissue loss, it failed to offer significant improvements in either motor or memory tasks.
Methods
Materials
Male Sprague-Dawley rats (250–275 g) and C57BL/6 mice (23–25 g) were purchased from Harlan Laboratories (Indianapolis, IN). CAPE was purchased from Biomol International, LP (Plymouth Meeting, PA). Antibodies for von Willebrand factor (vWF) and claudin-5 were purchased from AbD Serotec (Raleigh, NC) and Invitrogen (Carlsbad, CA), respectively. Antibodies for neuronal nuclei (NeuN) and glial fibrillary acidic protein (GFAP) were purchased from Millipore (Billerica, MA).
Rat controlled cortical impact injury
All experimental procedures were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the recommendations provided in the Guide for the Care and Use of Laboratory Animals. A controlled cortical impact (CCI) model was used to cause brain trauma as previously described (Dixon et al., 1991). Animals were initially anesthetized using 5% isoflurane with a 1:1 N2O:O2 mixture, mounted on a stereotaxic frame, and then maintained with 2.5% isoflurane with 1:1 N2O:O2 mixture via a face mask. For BBB studies, a midline incision was made and a 6-mm-diameter craniotomy was performed on the right side midway between the bregma and the lambda, with the medial edge of the craniotomy 1.0 mm lateral to the midline. An electromagnetic impact device (Custom Design and Fabrication, Richmond, VA) was used to deliver an impact at an angle of 10° from the vertical plane at 6.0 m/sec with 3.3 mm deformation using an impactor tip 5 mm in diameter. Based on our observations, this deformation caused an injury comparable to that seen using a depth of 1.8 mm in the pneumatic CCI device. For behavioral studies, the aforementioned craniotomy was made on both sides of the skull and the contusion was delivered only to the right parietal cortex at a velocity of 4.0 m/sec and a deformation of 2.7 mm (equivalent to 1.5 mm in the pneumatic CCI device). After injury, the incision was closed with wound clips. Core body temperature was maintained at 37°C during the surgery using a rectal thermometer coupled to a heating pad. All animals were allowed to completely recover from the anesthesia in a warm chamber before being returned to their home cages. Sham-operated animals received all the aforementioned surgical procedures except for the craniotomy and the impact.
CAPE administration
CAPE was prepared in 50% dimethylsulfoxide (DMSO) in saline and was administered by intraperitoneal (IP) injection with a dose of 10 mg/kg at designed time points following brain injury. The total injection volume for each animal was 1 mL for rat studies and 500 μL for mouse studies. Vehicle-treated animals received an identical volume of 50% DMSO by IP injection at corresponding time points. CAPE was administered 30 min following injury for immunohistochemistry assays and Evans blue studies, and was administered 30 min following injury, then daily for the next 4 days, for the behavioral studies.
Immunohistochemistry and fluorescence intensity quantification
Twenty-four hours after injury, the rats were decapitated and the brains were removed and quickly frozen in −80°C isopentane. Twenty-micron-thick coronal cryosections were prepared and mounted directly on slides. After air drying, the sections were fixed with 100% methanol for 20 min at −20°C, and then rinsed in Tris-buffered saline, pH 8.0 (TBS). The sections were blocked in 5% goat serum in TBS with 0.25% Triton X-100 (TBST) at room temperature for 1 h, followed by incubation with primary antibodies (1 μg/mL) in 2.5% goat serum in TBST at 4°C for 48 h. Following extensive washing in TBST, species-specific secondary antibodies conjugated to Alexa-Fluor (in 2.5% goat serum in TBST) were added and allowed to incubate for 3 h at room temperature. Images of immunofluorescence were captured using a Zeiss Axiovert S100 microscope and a MicroFire (Optronics, Goleta, CA) camera. The parameters used for image acquisition were preset to minimize the background and optimize the signal using a tissue section from an injured animal. These parameters were kept constant across all subsequent groups. Two non-overlapping regions in the ipsilateral cortex from each section and two sections from each animal were used for imaging. The fluorescence intensities, measured using Image J software (from the National Institutes of Health [NIH]), were averaged for each section, and the two sections were averaged for each animal.
Measurement of BBB permeability
At the indicated time points following injury, 3% Evans blue (EB) dye in saline was injected slowly through the jugular vein (4 mL/kg) and allowed to circulate for 1.5 h for both rats and mice. Following the circulation time, the animals were transcardially perfused with 1× phosphate-buffered saline (PBS). The brains were removed, and the cerebral hemispheres were separated and homogenized in 50% trichloroacetic acid (TCA). The homogenate was incubated at 4°C for 1 h and then centrifuged at 20,000g for 30 min. The supernatant solution was collected and 2× volume of ethanol was added to each sample followed by thorough vortexing. The optical density of the resultant solution was measured at 620 nm and used to determine the relative amount of EB dye in each sample.
Assessment of motor function
All behavioral tests were conducted by an experimenter blind to the treatment groups. A vestibulomotor (beam balance) and a motor skill task (paw placement) were used to determine the animals' motor performance following injury on days 1–4 and day 7 post-CCI. For the beam balance, three daily trials were given during which the length of time spent on a narrow wooden beam (1.5 cm wide) was recorded for up to 60 sec. Paw placement was evaluated by placing the rat on a wire grid (opening size of 2×2 cm) and counting the number of foot faults out of a total of 50 steps. A foot fault was defined as when a front paw misses and appears below the plane of the grid. Paw placement was repeated three times to give an average daily score.
Assessment of cognitive function
The rats were tested for their cognitive performance using the standard hidden platform version of the Morris water maze (MWM; Dash et al., 1995,2002; Hamm et al., 1992; Royo et al., 2007). All animals had recovered from the TBI-associated motor dysfunction prior to performing the cognitive testing. The animals were given 4 consecutive training trials per day for 9 days, with an inter-trial interval (iti) of 4 min (Hamm et al., 1992). If the animal failed to locate the platform within 60 sec on any given trial, it was led there by the experimenter. Movement within the maze was monitored using a video camera linked to tracking software (Ethovision; Noldus Information Technology, Leesbury, VA). The time to locate the platform during training was used as a measure of learning and memory. A probe trial in which the platform was removed from the water maze was given 24 h after training to assess measures of platform localization. The rats were allowed to search for the hidden platform for a period of 60 sec, during which the latency to first platform crossing and the number of platform crossings were recorded.
Contusion volume measurement
Following the completion of the behavioral studies, the animals were deeply anesthetized with sodium pentobarbital (100 mg/kg) and transcardially perfused with PBS followed by 4% paraformaldehyde. The brains were removed, post-fixed overnight in perfusant, then cryoprotected in a 30% sucrose solution. Cortical tissue loss was estimated by experimenters kept blind with respect to the treatment groups. In brief, cryosections (40-μm thick) spanning the rostral-caudal extent of the injured cortex were selected and stained with cresyl violet by an experimenter given only the animal's identifier code. Images of the resultant slides were then used for tissue loss measurement by a second experimenter. The area of cortical tissue loss for each section was carefully outlined and quantified by Image J software. Contusion volume was calculated using the equation A1(0.5X1)+A2(0.5X1+0.5X2)+An−1(0.5Xn−1+0.5Xn)+An(0.5Xn), where A is the area (mm2) of the contusion for each slice, and X is the distance (mm) between two sequential slices. Once the contusion volume had been calculated for each animal, the blind code was broken and group differences were assessed.
Statistical analysis
For evaluation of EB extravasation, immunofluorescence intensity, and contusion volume, two-tailed Student's t-tests for unpaired variables were used to determine differences between the vehicle-treated and CAPE-treated groups. For evaluation of behavioral data, a two-way repeated-measures analysis of variance (ANOVA) was utilized to determine group main effects or interactions. A Holm-Sidak method for multiple comparisons post-hoc test was used to determine the data points with significant differences. All data were tested for normality using a Shapiro-Wilk normality test. Any data found not to have a normal distribution or equal variance were analyzed using the appropriate non-parametric test. Data were considered significant at p<0.05 and presented as mean±standard error of the mean (SEM).
Results
Post-injury CAPE administration reduces TBI-induced loss of claudin-5
TBI disrupts cerebral autoregulation and causes breakdown of the BBB, allowing infiltration of circulating cells, blood products, and fluid into the brain (Baldwin et al., 1996; Golding et al., 1999; Lotocki et al., 2009). This can cause further brain damage by enhancing inflammation, edema, free radical production, and neurovascular dysfunction. Capillary endothelial cells and tight junctions are two major components of the BBB. We therefore examined if CAPE can reduce endothelial cell loss, preserve tight junction protein levels, or both after TBI. To address these possibilities, brain sections were prepared 24 h post-injury from rats injected IP with either CAPE (10 mg/kg; n=4) or vehicle (n=3) for the evaluation of vWF (a marker of endothelial cells), and claudin-5 (a marker of tight junctions) immunoreactivity. A group of sham-operated animals (n=4) were used as uninjured controls. This dose of CAPE was chosen based on its reported effectiveness in reducing infarct size in a rodent model of ischemia (Khan et al., 2007).
Figure 1A shows representative photographs of a blood vessel labeled with vWF and claudin-5, counterstained with the nuclear stain To-Pro-3. Consistent with it being a secreted endothelial cell protein, vWF immunoreactivity appears punctate and defines the location of the vessel. Claudin-5 localizes to the plasma membrane and can be seen as a continuous strip-like staining pattern along the vascular tube. The overlay image demonstrates the localization of vWF and claudin-5 relative to the nuclei of the endothelial cells. As To-Pro-3 is not selective for endothelial cells, nuclei belonging to presumed pericytes (overlaying the vessel) and neurons (distal to the vessel) can be observed. As we and others have previously described, TBI causes the loss of both vWF and claudin-5 immunoreactivity, although regions of vessels immunopositive for vWF but claudin-5 negative can be seen (TBI+vehicle; Fig. 1B; Khan et al., 2009; Zhao et al., 2007). CAPE treatment appears to protect both the levels and cellular localization of claudin-5 (TBI+CAPE; Fig. 1B), suggesting a preservation of vascular integrity. To quantify these changes, low-magnification images of vWF and claudin-5 immunoreactivity in the ipsilateral cerebral cortex from sham, TBI+vehicle, and TBI+CAPE animals (Fig. 1C) were used for fluorescence intensity measurements. Although the loss of vWF immunoreactivity was not influenced by post-injury CAPE treatment (TBI+vehicle=45.37±6.05%; TBI+CAPE=54.31±6.62%; p=0.382; Fig. 1D), a significant increase in claudin-5 levels was detected, suggesting preservation of tight junctions in the surviving endothelial cells (TBI+vehicle=17.65±1.11%; TBI+CAPE=35.06±3.47%; p=0.009; Fig. 1D).

Post-injury caffeic acid phenethyl ester (CAPE) administration reduces the loss of claudin-5 and improves vascular integrity following traumatic brain injury (TBI). (
Post-injury CAPE administration reduces TBI-associated vascular dysfunction
Several studies have shown that CCI injury causes an increase in vascular permeability (Baldwin et al., 1996; Baskaya et al., 1997; DeWitt and Prough, 2003; Whalen et al., 1998). Based on the preservation of claudin-5 levels we observed, we questioned if this was associated with improved BBB integrity. Rats were injured, then treated with CAPE (10 mg/kg IP; n=10) or vehicle (n=10) 30 min following injury. Permeability was assessed 24 h later by measuring the extravasation of EB into the brain following its intravenous administration. The results presented in Figure 1E show that brain injury increases EB extravasation compared to sham-operated controls, and that post-injury administration of CAPE significantly reduced these levels compared to those seen in vehicle-treated, injured controls (TBI+vehicle: 340.96±32.34%; TBI+CAPE: 242.07±20.02%; p=0.018). This protective effect of CAPE on BBB integrity following TBI was confirmed using a second species, C57BL/6 mice (Supplementary Fig. 1; see online supplementary material at
CAPE administration does not improve motor or cognitive function following TBI
Encouraged by the protection offered by CAPE on vascular permeability, we tested if CAPE can improve motor and cognitive functions in brain-injured animals. Rats (n=10/group) were injured, injected with either 10 mg/kg CAPE or an equal volume of vehicle 30 min later, and then daily for the first 4 days post-injury. Vestibulomotor and motor performance was assessed using the beam balance and paw placement tasks, respectively, on days 1–4 and day 7 post-injury (Fig. 2A). A separate group of animals received sham surgery and were tested on the beam balance and paw placement tasks to demonstrate normal performance in these tasks. Figure 2B–D show that CAPE treatment did not improve performance in either the beam balance (repeated measures two-way ANOVA: F(1,18)=0.310, p=0.584; Fig. 2B), nor the paw placement task (repeated measures two-way ANOVA: ipsilateral F(1,18)=0.410, p=0.530, Fig. 2C; contralateral F(1,18)=0.991, p=0.332; Fig. 2D) compared to vehicle-treated animals.
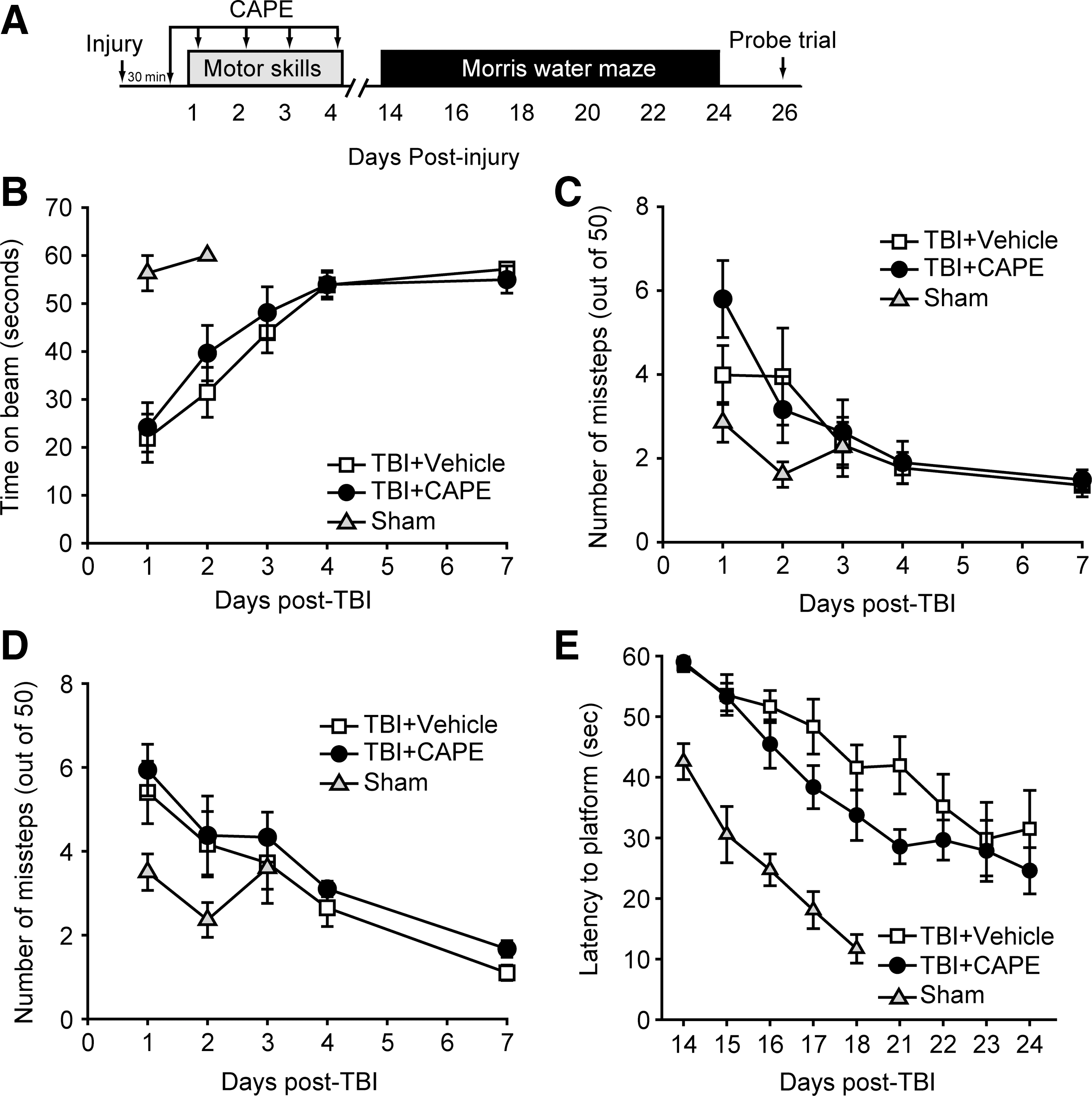
Caffeic acid phenethyl ester (CAPE) does not improve neurobehavioral outcome in injured rats. (
Spatial learning and memory was tested using the hidden platform version of the MWM task (Dixon et al., 1999; Hamm et al., 1992). Training in the task was carried out on days 14–18 post-injury, then following a 2-day rest period, the animals were given 4 more days of training. A group of sham animals were identically trained and used to demonstrate baseline performance. Figure 2E shows that when the latencies to find the hidden platform were analyzed, no significant difference was observed between vehicle and CAPE-treated animals (repeated measures two-way ANOVA: F(1,18)=1.870, p=0.188). When latency and number of platform crossings were assessed during a probe trial given 24 h following the completion of training, no differences were detected between the two groups (data not shown). In addition, when tested using a contextual fear memory task, no difference in memory retention was observed between injured animals treated with CAPE and those receiving vehicle (Supplementary Fig. 2; see online supplementary material at
CAPE administration reduces cortical contusion volume
After completion of the behavioral tests, the animals were perfused with paraformaldehyde and the brains were processed for immunohistochemistry using antibodies against NeuN (a neuron-specific marker) and GFAP (a glial-specific marker). Representative images of ipsilateral hippocampi immunostained for NeuN and GFAP from a sham, a vehicle-treated, and a CAPE-treated animal are shown in Figure 3A–C. The boxes in Figure 3A indicate the relative positions of the GFAP images represented in Figure 3B and C. Visual inspection of NeuN-immunostained sections revealed a moderate degree of cell layer thinning in the CA1 and CA3 subfields of the hippocampus after TBI. However, no overt difference in hippocampal cell loss was observed between vehicle-treated and CAPE-treated animals (Fig. 3A). Consistent with previous reports, TBI increased GFAP immunoreactivity in the CA3 (Fig. 3B) and dentate gyrus (Fig. 3C) subfields, an effect that was modestly reduced in CAPE-treated animals.

Administration of caffeic acid phenethyl ester (CAPE) after traumatic brain injury (TBI) reduces cortical tissue loss. (
As we observed that CAPE reduced claudin-5 loss and improved vascular integrity in the cerebral cortex, we questioned if these observations were associated with a reduction in cortical tissue loss. Figure 3D shows representative images of cresyl violet-stained tissue sections from a vehicle-treated and a CAPE-treated injured rat. Quantification of cortical tissue loss throughout the rostral-caudal extent of the injury revealed that CAPE significantly reduced both ipsilateral (TBI+vehicle: 24.82±4.27 mm3; TBI+CAPE: 12.32±2.28 mm3; p=0.017), and contralateral (TBI+vehicle: 15.74±3.59 mm3; TBI+CAPE: 3.93±1.46 mm3; p=0.006) contusion volumes compared to injured animals receiving vehicle (Fig. 3E).
Discussion
A number of studies have shown that TBI pathophysiology is complex and results from the combined action of multiple cellular and neurochemical mechanisms (Bramlett and Dietrich, 2007; Giza et al., 2002; Mechoulam and Shohami, 2007; Smith et al., 2010). Thus, it is thought that drugs that selectively target a single mechanism may not be as effective as drugs that attenuate more than one mechanism (Loane and Faden, 2010; Margulies and Hicks, 2009; Sayeed and Stein, 2009; Stoica and Faden, 2010). CAPE has been shown to reduce damage arising both from oxidative stress and inflammation, two important mechanisms in the progression of TBI pathology (Irmak et al., 2003). The present study revealed three major findings: (1) post-injury administration of CAPE reduces TBI-induced vascular permeability and (2) cortical tissue loss, but (3) had no effect on post-injury motor or cognitive performance.
CAPE has been reported to have anti-inflammatory, antioxidant, neuroprotective, antiviral, antitumoral, antiatherosclerotic, and immunomodulatory properties in a variety of systems (Fesen et al., 1993; Hishikawa et al., 2005; Ilhan et al., 2004; Liao et al., 2003; Orban et al., 2000; Park and Kahng, 1999; Wei et al., 2004). These diverse effects of CAPE may be attributed to its ability to readily permeate cells, where it inhibits enzymes such as lipoxygenases, cyclooxygenases, glutathione S-transferase, and xanthine oxidase (Chan et al., 1995; Koshihara et al., 1984; Michaluart et al., 1999; Mirzoeva and Calder, 1996; Orsolic et al., 2005). In addition, CAPE has been reported to be a potent inhibitor of NF-κB activation (Natarajan et al., 1996). Consistent with this, we found that CAPE reduces both the basal and tumor necrosis factor-α–induced expression of a NF-κB reporter gene in HT22 mouse hippocampal cells (unpublished data).
BBB breakdown is one of the early pathological events triggered by TBI that contributes to the infiltration of circulating cells and molecules, and facilitates the entry of fluid into the brain, all of which can exacerbate brain damage and tissue loss. A number of studies have reported that ROS and inflammatory molecules can compromise BBB integrity following TBI (Adelson et al., 1998). Consistent with this, it has been reported that either exogenous administration of free radical scavengers, or exogenous activation of endogenous antioxidant pathways, reduces loss of tight junction proteins and improves BBB integrity (Povlishock and Kontos, 1992; Smith et al., 1994; Zhao et al., 2007). We found that post-injury administration of CAPE effectively reduces BBB permeability, an effect seen in both rat and mouse models of TBI. Although the mechanisms by which CAPE reduces BBB permeability are not known at present, we observed that this improvement was associated with an increase in the levels of the tight junction protein claudin-5. Unlike endothelial barriers in other organs, the expression of high levels of claudin-5 in brain microvessels has been shown to give rise to the high trans-endothelial electrical resistance that is critical for maintaining brain homeostasis (Honda et al., 2006). Our finding that CAPE may improve BBB integrity by preserving claudin-5 levels is consistent with other studies that reported that activation of endogenous antioxidant proteins restores claudin-5 levels and markedly protects against TBI-induced enhanced permeability (Zhao et al., 2007).
A number of in vitro and in vivo studies have demonstrated CAPE to be neuroprotective. For example, CAPE/caffeic acid has been reported to protect the brain against induced diabetes, quinolinic acid-induced brain damage, aluminum-induced toxicity, neonatal hypoxia, and transient focal ischemia (Celik and Erdogan, 2008; Kalonia et al., 2009; Khan et al., 2007; Wei et al., 2004; Yang et al., 2008; Zhou et al., 2006). Caffeic acid has also been reported to reduce lesion area and glial scar formation following cryoinjury in mice (Zhang et al., 2007). Our finding that CAPE reduces cortical tissue loss is consistent with these studies. As TBI has been shown to cause oxidative damage, apoptosis, and necrosis of cortical cells (Colicos et al., 1996; Hall, 1993; Raghupathi, 2004; Stoica and Faden, 2010), the inhibition of one or more of these pathways may have contributed to the reduction of cortical contusion volume we observed.
Based on CAPE's effectiveness in reducing vascular permeability after injury, we anticipated that it would improve the function of the injured brain. Although we observed that CAPE reduced parietal cortex tissue loss, this was not accompanied by a reduction in motor dysfunction. The lack of motor skill improvement is likely due to the fact that the contusion caused by the CCI injury employed in the present study does not encompass the entire motor cortex (Supplementary Fig. 3; see online supplementary material at
Footnotes
Acknowledgments
The authors thank Cameron Jeter for her critical reading of the manuscript. The studies were made possible by funds from the NIH (NS049160).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.