
Abstract
Astrocyte activation contributes to the brain's response to disease and injury. Activated astrocytes generate harmful radicals that exacerbate brain damage including nitric oxide, peroxides and superoxides. Furthermore, reactive astrocytes hinder regeneration of damaged neural circuits by secreting neuro-developmental inhibitors and glycosaminoglycans (GAGs), which physically block growth cone extension. Therefore, targeted therapeutic strategies to limit astrocyte activation may enhance recovery from many neurodegenerative states. Previously, we demonstrated that the HIV-1 TAT cell-penetrating peptide, a short non-toxic peptide from the full-length TAT protein, delivered a protein cargo to astrocytes in a process dependent on cell-surface GAG. Since activated astrocytes produce GAG, in this study we tested whether TAT could transduce activated astrocytes, deliver a biologically active cargo, and produce a physiological effect. Astrocyte activation was induced by IL-1β, lipopolysaccharide (LPS), or mechanical stretch injury, and quantified by increased GAG and nitrite content. TAT-mediated delivery of a mock therapeutic protein, GFP, increased significantly after activation. Nitrite production, GAG expression, and GFP-TAT transduction were significantly attenuated by inhibitors of JNK, p38, or ERK. TAT fused to a peptide JNK inhibitor delivered the peptide inhibitor to activated astrocytes and significantly reduced activation. Our study is the first to report significant and direct modulation of astrocyte activation with a peptide JNK inhibitor. Our promising in vitro results warrant in vivo follow-up, as TAT-mediated protein delivery may have broad therapeutic potential for preventing astrocyte activation with the possibility of limiting off-target, negative side effects.
Introduction
I
We hypothesized that activated astrocytes could be targeted by the cell-penetrating peptide (CPP) from the transactivator of transcription (TAT) protein from the human immunodeficiency virus (HIV-1) through their increased expression of glycosaminoglycans (GAGs), since the first step in TAT transduction is an electrostatic interaction with the cell surface (Brooks et al., 2005; Tyagi et al., 2001). The CPP TAT is non-toxic and non-inflammatory (Vives et al., 1997) unlike the full-length TAT protein (Williams et al., 2009). Cultured astrocytes were activated with a well-characterized in vitro mechanical stretch injury model (Morrison et al., 2003), the proinflammatory cytokine interleukin (IL)-1β, or LPS. Activation was measured via functional changes, specifically production of nitrite, the oxidation product of NO (Murphy et al., 1993), and GAG content. We observed significant increases of nitrite and GAG content in mechanically- and chemically-stimulated astrocytes and an increase in green fluorescence protein (GFP)-TAT transduction. Inhibition of the JNK, p38 MAPK, or ERK pathways by small molecule inhibitors significantly reduced activation and significantly decreased GFP-TAT transduction. TAT-mediated delivery of a JNK peptide inhibitor to mechanically- or chemically-stimulated astrocytes significantly prevented activation. Our results highlight the potential of TAT-mediated cargo delivery to target activated astrocytes, to deliver a biologically active cargo, and to directly modify the gliotic response to activating stimuli. Such a strategy could hold therapeutic benefit for multiple CNS diseases and injury.
Methods
Cell culture
All protocols involving animals were approved by the Columbia University IACUC. Purified astrocyte cultures were prepared as previously reported (Simon et al., 2009). Briefly, cortices of 8- to 10-day-old Sprague-Dawley rat pups were digested with papain, triturated, and plated into T75 tissue culture flasks. After 1 h incubation, the flasks were vigorously shaken to remove non-adherent cells, and the media was discarded. The enriched astrocytes (>90%) were grown in DMEM (Sigma-Aldrich, St. Louis, MO) with 10% heat-inactivated newborn calf serum, 1 mM glutamine, 10 mM HEPES, and 10 mg/mL gentamicin for 2 weeks as previously described (Li et al., 2010; Simon et al., 2009). The astrocytes were then treated with trypsin/EDTA (Sigma-Aldrich) for 5 min and replated onto either uncoated tissue culture 24-well plates at a density of 30,000 cells per well, or onto highly stretchable silicone membranes (Specialty Manufacturing Inc., Saginaw, MI) coated with laminin, poly-L-lysine, and gelatin attached to custom-made stainless steel wells at a density of 5000 cells per cm2 (Morrison et al., 2003). The astrocytes were then cultured for an additional 2 days before experimentation.
Mechanical stretch injury of astrocytes
Astrocytes on silicone membranes were subjected to a single 30% equi-biaxial stretch injury at a strain rate of 20 sec−1 using a well-characterized stretch injury device (Morrison et al., 2003). These injury parameters were chosen to induce a moderate level of injury (Cater et al., 2006). Strain and strain rate were verified with high-speed video analysis (Cater et al., 2006). The cells were analyzed for nitrite, GAG, DNA, or GFP-TAT transduction 48 h following injury.
Chemical stimulation of astrocytes
Astrocytes were stimulated with 20 ng/mL murine IL-1β (Invitrogen, Carlsbad, CA) or 2 μg/mL LPS (Sigma-Aldrich) for 48 h. Cells were analyzed for nitrite, GAG, DNA, or GFP-TAT transduction.
Inhibition of mechanical activation
To verify the involvement of MAPK pathways in astrocyte activation in response to mechanical stretch injury, astrocytes were pretreated for 1 h (Hua et al., 2002) with either 20 μM (Nakajima et al., 2004; Yoo et al., 2008) SB203580 (Sigma-Aldrich), SP600125 (Sigma-Aldrich), or U0126 (Sigma-Aldrich) to inhibit specific MAPK according to previously published methods (Hua et al., 2002; Nakajima et al., 2004; Yoo et al., 2008). A pretreatment paradigm was utilized to confirm involvement of specific pathways and not to demonstrate therapeutic potential. For the TAT-JNK inhibitor delivery studies, astrocytes were treated with either 3 μM TAT-JNK inhibitor (Axxora, San Diego, CA), L-TAT control peptide (Axxora), GFP-TAT, or the TAT peptide utilized in our previous study (Gao et al., 2009) (hereafter referred to as mTAT) immediately following mechanical injury to more closely model a post-injury treatment paradigm. The half maximal inhibitory concentration (IC50) of the TAT-JNK inhibitor is 1 μM, as previously determined from JNK inhibition assays in vitro (Borsello et al., 2003). The concentration of the TAT-JNK inhibitor necessary for sufficient intracellular inhibition was based on previous delivery studies of GFP-TAT (Gao et al., 2009; Simon et al., 2009). The inhibitors then remained within the medium until nitrite, GAG, DNA, or GFP-TAT transduction was quantified at 48 h.
Inhibition of chemical activation
To verify the involvement of the MAPK pathways in astrocyte activation in response to chemical stimuli, astrocytes were pretreated for 1 h (Hua et al., 2002) with either 20 μM (Nakajima et al., 2004; Yoo et al., 2008) SB203580 (Sigma-Aldrich), SP600125 (Sigma-Aldrich), or U0126 (Sigma-Aldrich) to inhibit specific MAPK before chemical stimulation with either 20 ng/mL murine IL-1β (Kim et al., 2006) (Invitrogen), or 2 μg/mL LPS (Nakajima et al., 2004) (Sigma-Aldrich). These pretreatment paradigms have been used before to verify the involvement of MAPK cascades in chemically-mediated astrocyte activation (Hua et al., 2002; Kim et al., 2006; Nakajima et al., 2004; Yoo et al., 2008). For the TAT-JNK inhibitor delivery studies, astrocytes were chemically stimulated as above, and either 3 μM TAT-JNK inhibitor (Axxora), L-TAT control peptide (Axxora), GFP-TAT, or mTAT was added concurrently. The chemical stimulators and inhibitors then remained within the medium until nitrite, GAG, DNA, or GFP-TAT transduction was quantified at 48 h.
Quantification of nitrite
Nitrite production was chosen as a functional measure of astrocyte activation, specifically as a surrogate marker of NO overproduction as a result of iNOS expression, which is known to exacerbate brain injury (Wada et al., 1998b). Nitrite content was quantified 48 h after astrocyte activation because previous studies have shown that iNOS expression, NO production, and hence nitrite content are robustly increased at this time point (Yoo et al., 2008). Nitrite content was measured in triplicate for each well using the Griess reagent (Invitrogen). Equal volumes of N-(1-naphthyl)ethylenediamine and sulfanilic acid were mixed together to form the Griess reagent. Then 150 μL of cell supernatant or NaNO2 standard was mixed with 130 μL of deionized water and 20 μL of Griess reagent in a 96-well plate. After 30 min incubation in the dark at room temperature, sample absorbance was measured in a spectrophotometric microplate reader (Molecular Devices, Sunnyvale, CA) at 548 nm and converted to nitrite content with the standards. Nitrite content was normalized to total DNA content using the Quant-iT Picogreen dsDNA assay (Invitrogen). For the inhibitor studies, the average μmol nitrite/ng DNA was normalized to the average μmol nitrite/ng DNA in uninjured or unstimulated control cells to account for any basal effects of inhibitors on unactivated cells.
Quantification of GAG
Cellular GAG content was quantified 48 h after astrocyte activation, as previous studies have shown that GAG production is consistently increased at this time point after activation in vitro (Properzi et al., 2008; Smith-Thomas et al., 1994; Zhang et al., 2006). Increased GAG production as a result of injury has been shown to be inhibitory to neurite outgrowth and recovery (Smith-Thomas et al., 1994). To determine cellular GAG content, cells were incubated in 500 μL of lysis buffer (0.1 M sodium acetate buffer, pH 5, containing 5 mM EDTA, 5 mM cysteine HCl, and 0.6 U papain/mL) overnight at 37°C. The cells were sonicated and analyzed using the 1,9-dimethylmethylene blue (DMMB; Sigma-Aldrich) dye-binding assay, and compared to prepared GAG standards (Farndale et al., 1986). GAG content was normalized to total DNA content using the Quant-iT Picogreen dsDNA assay (Invitrogen). For the inhibitor studies, the average μg GAG/ng DNA was normalized to the average μg GAG/ng DNA in uninjured unstimulated control cells to account for basal effects of MAPK inhibitors on unactivated cells.
Quantification of protein transduction into primary astrocytes
The GFP-TAT construct has been described previously and consisted of an 11-amino acid peptide flanked by glycines (GYGRKKRRQRRRG) fused to the C-terminus of the GFP protein (Simon et al., 2009). Protein transduction of GFP or GFP-TAT was quantified in triplicate as previously reported (Gao et al., 2009; Simon et al., 2009). Briefly, astrocyte cultures were incubated with either 3 μM GFP or GFP-TAT for 4 h, as our previous studies have demonstrated that this duration and concentration yielded maximal uptake of the constructs (Gao et al., 2009). Cells were washed in PBS and treated with trypsin/EDTA (Sigma-Aldrich) for 5 min to remove protein bound to the surface of the cells, centrifuged, and resuspended in 250 μL PBS. Protein transduction was quantified with a FACSCanto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ) by calculating the percent increase in geometric mean fluorescence (excitation 488 nm, emission 530/30 nm) above that of cells treated with GFP only, to account for GFP and background autofluorescence. For the inhibitor studies, the average fold change of each injured or stimulated experimental group was normalized to the average fold change of uninjured or unstimulated groups to account for any basal effects of inhibitors on unactivated cells. A total of 10,000 events per sample were counted.
Results
Mechanical activation of astrocytes
Mechanical stretch significantly increased nitrite content after injury compared to uninjured controls (Fig. 1A). GAG content increased significantly after injury compared to uninjured controls (Fig. 1B). Mechanical injury increased GFP-TAT transduction more than twofold compared to uninjured controls (Fig. 1C). These results indicate that our in vitro model of stretch injury activated astrocytes, and that GFP-TAT preferentially transduced activated astrocytes.
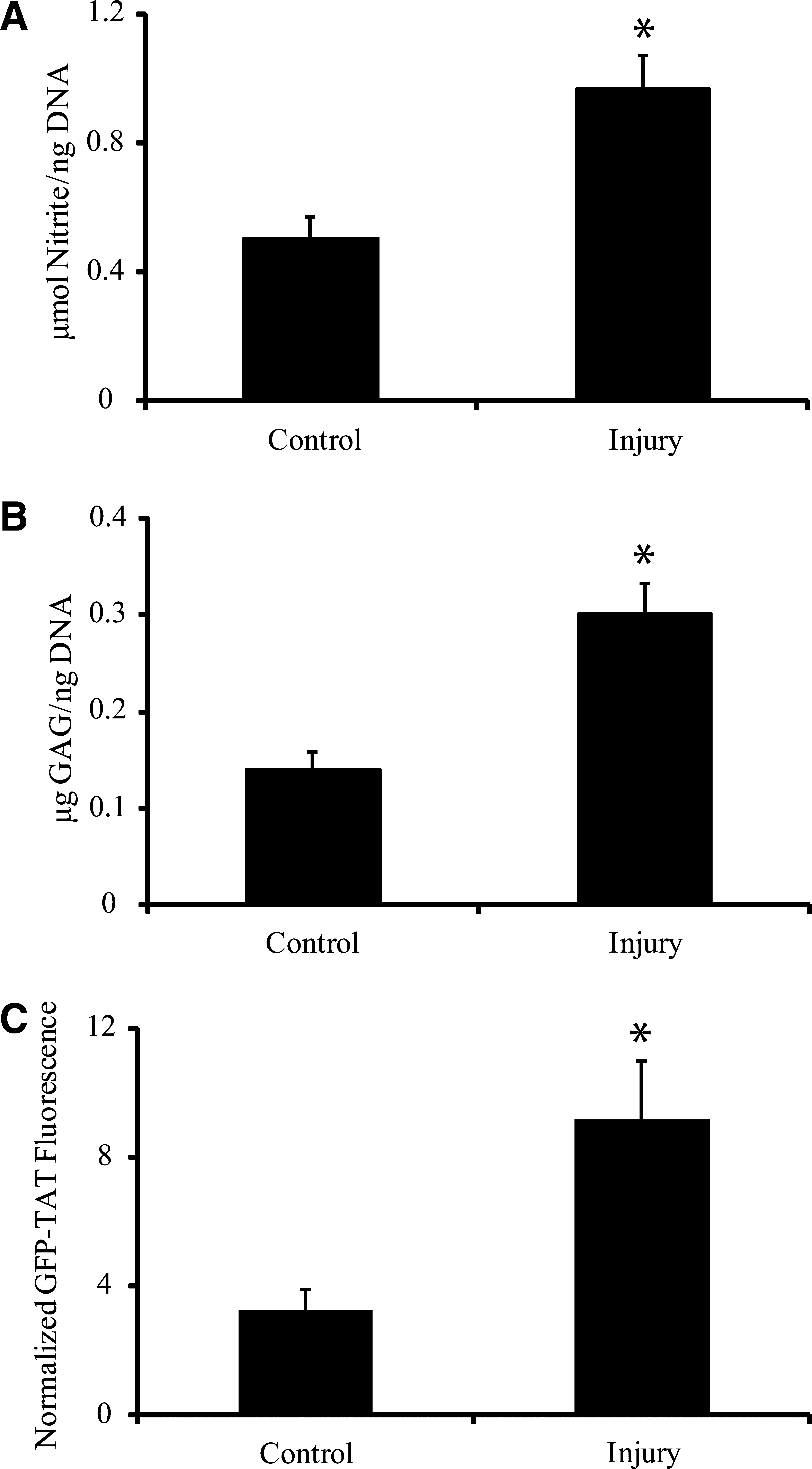
Effects of mechanical stretch injury. (
IL-1β and LPS activation of astrocytes
Both IL-1β and LPS treatment significantly increased nitrite content of astrocyte cultures compared to untreated controls (Fig. 2A). GAG content increased significantly in IL-1β- and LPS-treated astrocytes compared to untreated controls (Fig. 2B). In both IL-1β- and LPS-stimulated astrocytes, GFP-TAT transduction increased significantly compared to untreated controls (Fig. 2C), indicating that GFP-TAT preferentially transduced activated astrocytes.

Cytokine and LPS-mediated activation of astrocytes. (
Inhibition of mechanical activation via MAPK antagonism
Pretreatment of astrocytes with either 20 μM of SB203580, SP600125, or U0126 for 1 h before mechanical injury significantly attenuated nitrite production (Fig. 3A). GAG content in injured astrocytes was also decreased significantly after pretreatment with either SB203580, SP600125, or U0126 (Fig. 3B). GFP-TAT transduction was significantly reduced in injured astrocytes pretreated with MAPK inhibitors compared to untreated, injured astrocytes (Fig. 3C).
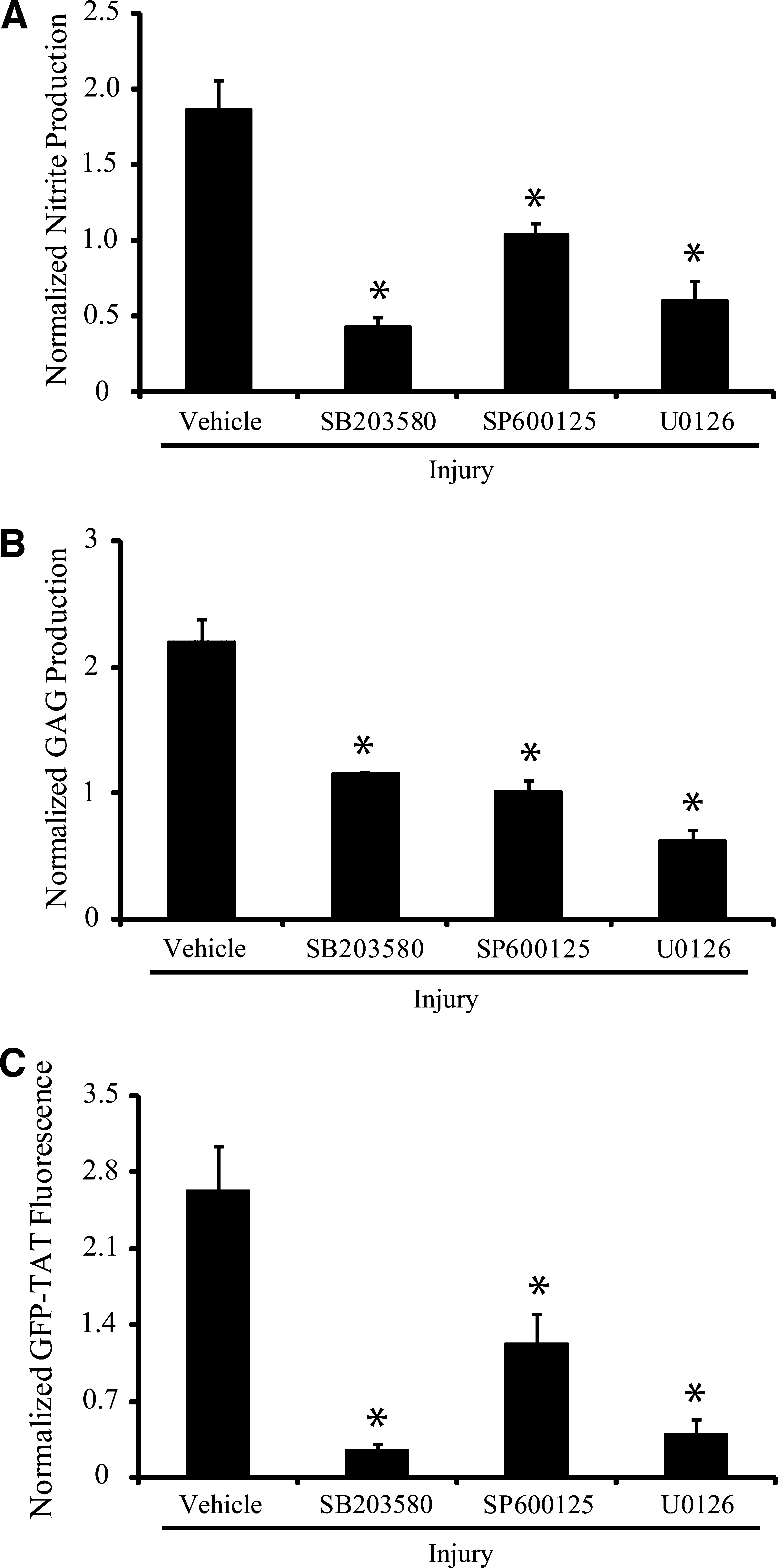
Effect of MAPK inhibition on mechanical activation. (
Inhibition of IL-1β and LPS activation via MAPK antagonism
After IL-1β or LPS stimulation, nitrite content was significantly decreased by MAPK inhibition with either SB203580, SP600125, or U0126, compared to stimulated cultures receiving vehicle (Fig. 4A and D). GAG content was significantly reduced in chemically-stimulated cultures pretreated with either SB203580, SP600125, or U0126, compared to vehicle-treated cultures (Fig. 4B and E). GFP-TAT transduction was significantly decreased in IL-1β- and LPS-stimulated astrocytes pretreated with either SB203580, SP600125, or U0126 (Fig. 4C and F).
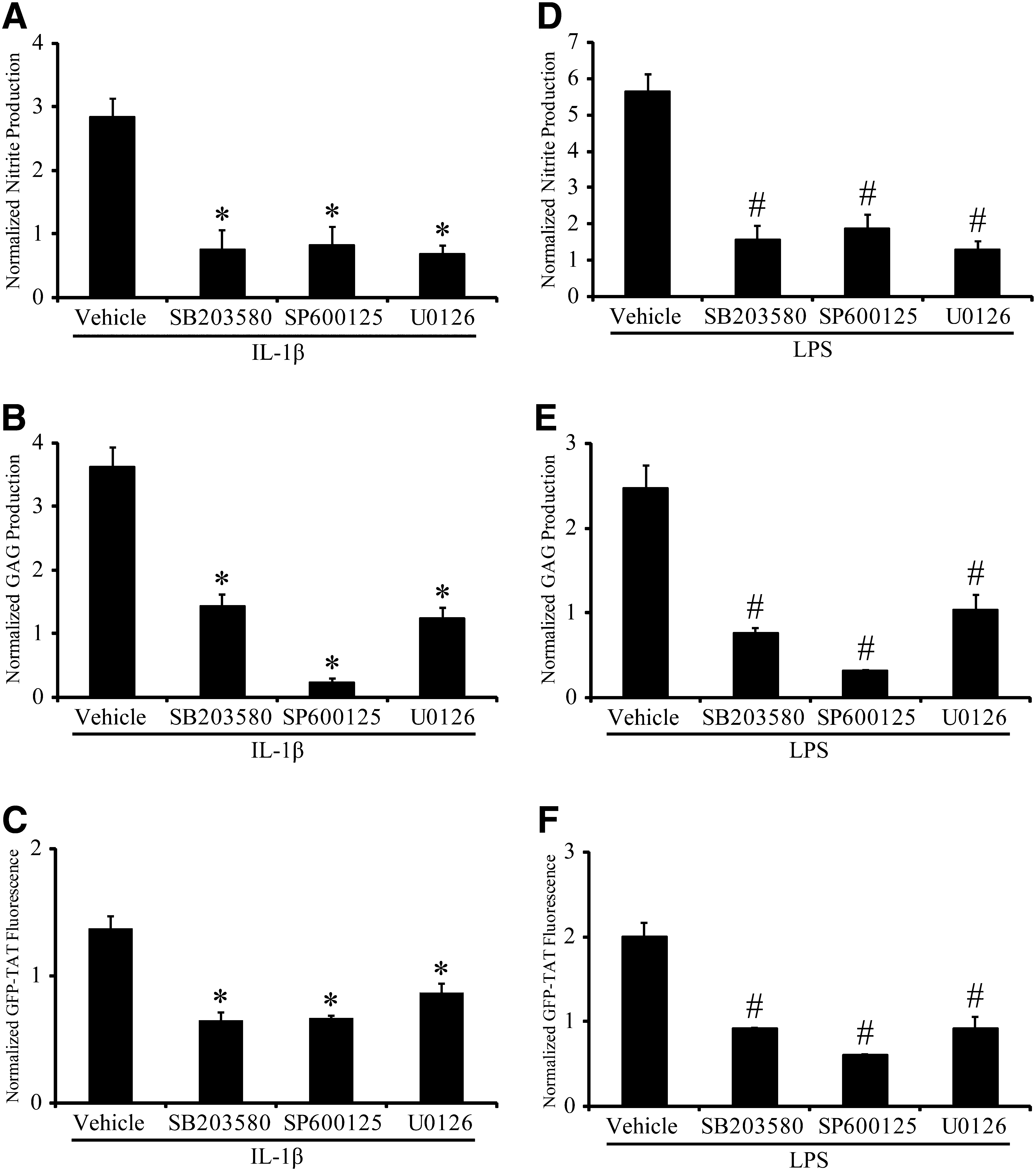
Effect of MAPK inhibition on IL-1β- and LPS-mediated activation. (
Inhibition of activation via a TAT-JNK peptide inhibitor
Treating cultures with the TAT-JNK peptide inhibitor immediately after mechanical stretch injury significantly prevented the injury-induced increase of nitrite (Fig. 5A), and GAG (Fig. 5B) content measured 48 h after injury. GFP-TAT, L-TAT control peptide, and mTAT had no effect on nitrite (Fig. 5A) or GAG (Fig. 5B) production after mechanical stretch injury. Treating cultures with the TAT-JNK inhibitor also prevented the IL-1β- and LPS-induced increase of nitrite (Fig. 5C and E), and GAG (Fig. 5D and F) content measured 48 h after stimulation. GFP-TAT, L-TAT control peptide, and mTAT did not significantly alter nitrite (Fig. 5C and E) or GAG (Fig. 5D and F) production after IL-1β or LPS stimulation.
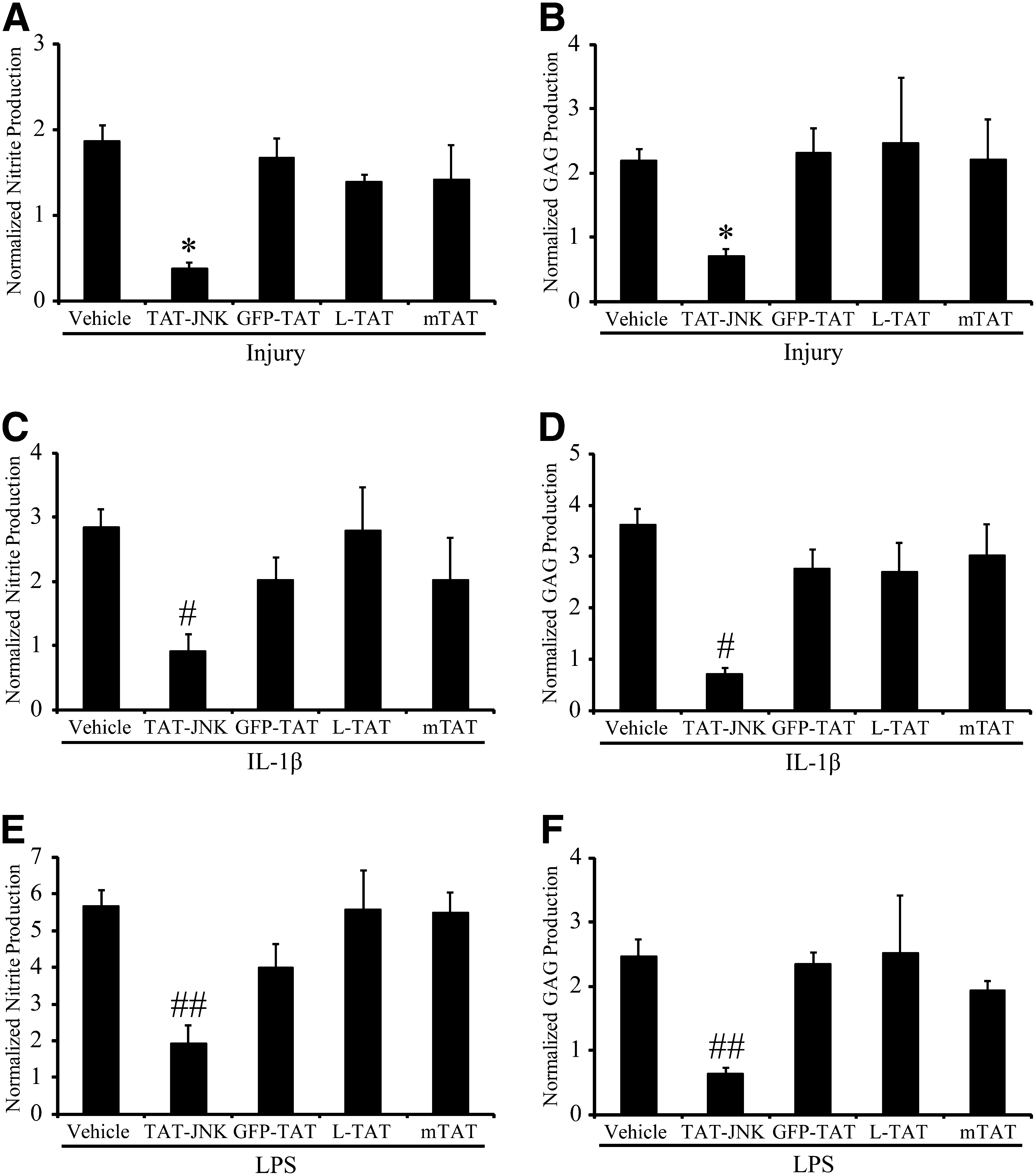
Effect of TAT-mediated delivery of a peptide JNK inhibitor on activation. (
Discussion
Astrocytes have been implicated to have both beneficial and harmful effects in the developing pathobiology after CNS insults (Morganti-Kossmann et al., 2002; Sofroniew, 2005). Many of their negative effects are associated with the process of reactive gliosis or astrocyte activation, in which astrocytes increase production of GAG and NO, among other changes in their physiology. In our study, NO and GAG production were chosen as functional measures of astrocyte activation because of their potential to detrimentally affect surrounding cells (Silver and Miller, 2004; Wada et al., 1998b). Although GFAP is often used as a histological marker of activation (Pekny and Nilsson, 2005), whether it plays a functional role in the injury cascade is unclear. However, it is well established that increased expression of iNOS (Lee et al., 1995), and the subsequent overproduction of NO (Wada et al., 1998a, 1998b) can cause cell death due to subsequent nitrosylation of proteins (Wada et al., 1998b). Furthermore, increased production of extracellular GAG is known to be inhibitory to neurite outgrowth (Smith-Thomas et al., 1994). GAGs form a significant component of the glial scar and cause growth cone collapse, preventing regeneration. Therefore, we chose to focus on these more relevant, functional measures of activation. In the present study, astrocytes were activated by very different factors, including mechanical stimulation with a well-characterized stretch injury model developed previously in our laboratory (Morrison et al., 2003), cytokine treatment (Kim et al., 2006), or LPS (Bhat et al., 1998), to determine if the activation process could be controlled by the intracellular delivery of a peptide TAT-JNK inhibitor.
Utilization of TAT to target activated astrocytes via their increased GAG content could be of significant therapeutic value. Whereas the 86-amino acid, full-length HIV-1 TAT has been implicated in neurodegeneration and neuroinflammation (Williams et al., 2009), the CPP TAT is an 11-amino acid peptide which is non-toxic, non-inflammatory, and capable of cellular transduction (Vives et al., 1997). Through TAT-mediated delivery of an appropriate therapeutic cargo, it may be possible to limit GAG production and iNOS activity in activated astrocytes, thereby limiting detrimental effects.
Regulation of the MAPKs p38, ERK, and JNK may play a central role in astrocyte activation (Guan et al., 2006b; Hsiao et al., 2007; Otani et al., 2002). These MAPKs are activated by a variety of stimuli, such as mechanical trauma (Mandell et al., 2001), spinal cord injury (Stirling et al., 2008), ischemia and reperfusion (Guan et al., 2006a; Namura et al., 2001), and cytokine (Hsiao et al., 2007; Waetzig et al., 2005) and LPS stimulation (Bhat et al., 1998; Nakajima et al., 2004). Inhibition of MAPK cascades by the small-molecule inhibitors of p38 (Piao et al., 2003; Yoo et al., 2008), JNK (Guan et al., 2006a; Hua et al., 2002), and ERK (Mandell et al., 2001) decreased the stimulated upregulation of iNOS in vitro. In the present study, small-molecule inhibitors of p38 MAPK (SB203580), JNK (SP600125), and ERK (U0126) verified the essential role of the MAPK cascade in each of the different activation paradigms employed. Taken together, these results suggest that therapeutic modulation of astrocyte activation may be possible via the MAPK pathways.
However, a significant limitation of existing small-molecule inhibitors is that they cannot be targeted to a specific cell population, in this case activated astrocytes. In our study we chose to focus on JNK as a possible therapeutic target because, while the roles of p38 and ERK in astrogliosis are still controversial (Cole-Edwards et al., 2006), the role of JNK in neurodegeneration after CNS insults has been well-established (Cole-Edwards et al., 2006; Guan et al., 2006a). Peptide inhibitors for JNK are commercially available (Axxora), and the TAT-JNK inhibitor has been well-characterized (Bonny et al., 2001; Borsello et al., 2003). In contrast, the peptide inhibitor of p38 has only recently been reported in literature (Fu et al., 2008).
An alternative strategy to enhance repair is to target the glial scar directly. Degradation of the glial scar with chondroitinase ABC, or attenuating its formation through the use of a GAG synthesis inhibitor have been shown to promote axonal regrowth (Bradbury et al., 2002; Moon et al., 2001; Silver and Miller, 2004). Unfortunately, the use of chondroitinase ABC as a therapeutic is not ideal, as cleaving the GAG side chains leaves behind debris that remains inhibitory to neurite outgrowth (Silver and Miller, 2004; Ughrin et al., 2003). Similarly, general inhibition of GAG production may have unwanted side effects, as certain GAGs, such as heparan sulfate GAGs, are important in guiding axonal growth, promoting neurite outgrowth, and facilitating synapse formation (Properzi et al., 2008).
Successful glial-specific therapies will likely need to strike a fine balance between inhibiting negative consequences of astrocyte activation while maintaining their homeostatic functions. Inhibiting their supporting function could exacerbate the pathology given their roles in the normal function of the brain. For example, reactive astrocytes were selectively ablated after spinal cord injury in mice expressing a GFAP-herpes simplex virus-thymidine kinase transgene with ganciclovir (Faulkner et al., 2004). In that study, the ablation of all reactive astrocytes after injury resulted in the failure of blood–brain barrier repair, leukocyte infiltration, local tissue disruption, severe demyelination, neuronal and oligodendrocyte death, and pronounced motor deficits. Therefore a more subtle modulation of activation may be required for improved outcome. In our study, we aimed to preserve homeostatic astrocytic processes by downregulating activated astrocytes rather than eliminating them altogether. This strategy may prove more beneficial for ameliorating the detrimental effects of activated astrocytes by maintaining their presence in the injury site. However, further in vivo studies are necessary to clarify the relationship between modification of astrocyte activation and wound healing. Astrocytes grown in serum-containing medium have been used as models of reactive gliosis due to their proliferative nature (Audouy et al., 1999). However, in vitro studies involving astrocyte cultures lack critical input regarding systemic effects and environmental cues which could potentially impact the results reported here, highlighting the need for additional in vivo work.
Reactive astrocytes play a key role in the formation of the glial scar that is inhibitory to axon growth (Wanner et al., 2008) due to injury-induced upregulation of GAG production (McKeon et al., 1995). We hypothesized that the increase in GAG content of activated astrocytes could be exploited to deliver a therapeutic molecule, since intracellular delivery of GFP by TAT was increased after activation induced by different stimulation paradigms (Figs. 1C and 2C). In confirmation of our hypothesis, a TAT-JNK peptide inhibitor significantly prevented the functional consequences of astrocyte activation after stretch-induced injury, cytokine, or LPS stimulation, as measured by reduced nitrite and GAG production (Fig. 5). Our study is the first to report direct modulation of astrocyte activation with a peptide JNK inhibitor. By exploiting this characteristic of activated astrocytes, our delivery strategy may be highly specific for activated astrocytes.
Previous studies have attempted to reduce astrogliosis after injury (Ghirnikar et al., 1994; Spigolon et al., 2010; Yu et al., 1991, 1993; Zhu et al., 2007). GFAP synthesis was inhibited in primary astrocytes after scratch injury by antisense RNA (Yu et al., 1991). However, the inhibitory effect lasted only 3–5 days after injury due to the limited stability of the antisense RNA, resulting in only a delay in reactive astrogliosis. Utilization of the CDK inhibitor olomoucine arrested the developmental astrocytic cell cycle and reduced astrocyte activation after hypoxia, scratch-wound, and ischemia (Zhu et al., 2007). Newly proliferating astrocytes have been implicated in the astrogliotic scar (Kernie et al., 2001); however, previously existing astrocytes may contribute significantly to the inhibitory effects of the glial scar as well. Systemic injections of kainic acid into the adult rat hippocampus induced marked increases of GFAP (Spigolon et al., 2010). Administration of D-JNKI-1, the protease-resistant all-D-retroinverso form of the peptide inhibitor used in our study, significantly attenuated neuronal cell death and partially prevented increased GFAP expression, although not significantly. Although suggestive, a direct effect of the peptide inhibitor on astrocytes could not be distinguished from an indirect effect on astrocyte activation due to the significantly reduced neuronal death. The results of our study demonstrate that the TAT-JNK peptide inhibitor exerts a direct effect on astrocyte activation in response to multiple stimuli.
IL-1β- and LPS-mediated inflammation and neurotoxicity have been used to model a broad array of neurodegenerative diseases such as bacterial meningitis (Quagliarello et al., 1991), Alzheimer's disease (Lee et al., 2008) and Parkinson's disease (Yang et al., 2008). Utilizing a peptide construct to deliver a JNK inhibitor to a specific population of cells to reduce the effects of IL-1β- and LPS-mediated activation and subsequent neurotoxicity could potentially be a targeted therapeutic for these and other neurodegenerative disorders. The ability to deliver a therapeutic cargo via TAT transduction to a specific cell population could be of practical use considering the wide range of cell types involved in these complex neuropathologies.
Footnotes
Author Disclosure Statement
No competing financial interests exist.