
Abstract
Interest in promoting regeneration of the injured nervous system has recently turned toward the use of endogenous stem cells. Elucidating cues involved in driving these precursor cells out of quiescence following injury, and the signals that drive them toward neuronal and glial lineages, will help to harness these cells for repair. Using a biomechanically validated in vitro organotypic stretch injury model, cortico-hippocampal slices from postnatal mice were cultured and a stretch injury equivalent to a severe traumatic brain injury (TBI) applied. In uninjured cortex, proliferative potential under in vitro conditions is virtually absent in older slices (equivalent postnatal day 15 compared to 8). However, following a severe stretch injury, this potential is restored in injured outer cortex. Using slices from mice expressing a fluorescent reporter on the human glial fibrillary acidic protein (GFAP) promoter, we show that GFAP+ cells account for the majority of proliferating neurospheres formed, and that these cells are likely to arise from the cortical parenchyma and not from the subventricular zone. Moreover, we provide evidence for a correlation between upregulation of sonic hedgehog signaling, a pathway known to regulate stem cell proliferation, and this restoration of regenerative potential following TBI. Our results indicate that a source of quiescent endogenous stem cells residing in the cortex and subcortical tissue proliferate in vitro following TBI. Moreover, these proliferating cells are multipotent and are derived mostly from GFAP-expressing cells. This raises the possibility of using this endogenous source of stem cells for repair following TBI.
Introduction
Endogenous stem cells can perform a surprising level of anatomical repair under controlled experimental paradigms, in which apoptosis of neurons is induced, in the company of a minimal inflammatory response. For example, in the hippocampus, after predominantly apoptotic cell death in the CA1 region, anatomical and functional repair is observed with (Nakatomi et al., 2002) and without (Bendel et al., 2005) growth factor augmentation. Similarly, following chromophore-targeted apoptosis, neuronal restoration is observed in the rodent cortex of both corticothalamic neurons (Magavi et al., 2000) and corticospinal motor neurons (Chen et al., 2004). Following a stab injury, which induces a more robust inflammatory response, endogenous stem cells, identified through the expression of glial fibrillary acidic protein (GFAP), proliferate and contribute to gliogenesis in the mouse cortex in vivo (Buffo et al., 2008). Strikingly, in vitro, where the local environmental inflammatory cues have been removed, these cells have the capacity to differentiate, giving rise to neurons, but only following injury (Buffo et al., 2008). These studies suggest that whereas injury is necessary to activate quiescent endogenous neural stem cells, local non-permissive cues (Seidenfaden et al., 2006) as well as the inflammatory response, prevent the generation of new neurons in vivo.
Understanding the signaling by which injury activates quiescent cortical neural stem cells is, therefore, pivotal for harnessing them to effect repair. In view of this, molecular cues that regulate stem cell proliferation are becoming increasingly well defined. Sonic hedgehog (Shh), a member of the hedgehog family of secreted signaling proteins, was originally characterized in the developing nervous system (Marti and Bovolenta, 2002; Ruiz i Altaba et al., 2002). The importance of Shh in regulating proliferation in the developing nervous system is well characterized (Dahmane and Ruiz i Altaba, 1999; Rowitch et al., 1999; Wechsler-Reya and Scott, 1999), but more recently, the role in adult stem cell proliferation has been reported. Genetic fate mapping in adult mice reveals that Shh signaling is active in cells that proliferate to give rise to neurons in both the subventricular zone and the hippocampus (Ahn and Joyner, 2005). Abrogating Shh or its downstream effector, Smoothened, reduces cellular proliferation in both these regions in the postnatal mouse (Han et al., 2008; Lai et al., 2003; Machold et al., 2003). Significantly, Shh signaling is upregulated in reactive astrocytes following a focal freeze injury to the cerebral cortex (Amankulor et al., 2009). Moreover, this expression contributes to an increase in the proliferation of a subset of progenitor cells (Amankulor et al., 2009) raising the hypothesis that Shh signaling in the injured cortex directly influences precursor cells.
In order to potentially harness and redirect these processes to effect repair after TBI, we must first ask whether endogenous stem cells are activated in the cortex after TBI. Using an organotypic stretch injury model for TBI (Morrison et al., 2006), we have developed a slice-neurosphere assay using cultured cortico-hippocampal slices from postnatal mice. After inducing a stretch injury equivalent to a severe TBI, a subset of endogenous cells acquire stem cell properties in the injured cortex, and a greater proportion of these cells express GFAP. We provide evidence of a transient upregulation of Shh-signaling pathway components in the injured cortex and suggest that this may contribute to the acquisition of stem cell properties of a subset of injured cells in the cortex.
Methods
Organotypic slice cultures
Postnatal day 8 C57BL/6 or GFAP-GFP (Nolte et al., 2001) mice were killed by swift decapitation. The brain was removed and dissected into hemispheres, and the mid/hindbrain was removed. The cortico-hippocampal hemisphere was transferred to a McIlwain Tissue Chopper (Mickle Laboratory Engineering, Surrey, U.K.) and hemispheres were sectioned transversely into 400 μm slices. Slices were swiftly transferred to Gey's solution (supplemented with 0.45% glucose), separated, and trimmed. Four of these slices were plated onto a silicone membrane, as centrally as possible (for detailed method of membrane preparation see Morrison and associates [2006]). Slices were incubated at 37°C with 100% humidity and a 5% CO2, 20% O2, 75% N2 atmosphere overnight on a rocker (Denley) at one revolution/50 sec. Media contained 50% minimal essential medium (MEM), 25% Hank's buffered salt solution (HBSS), 25% horse serum, 1 mM glutamine, and 0.45% glucose. Media were replaced with fresh media the following day (Day 1) and with neurobasal media (Neurobasal-A, 2% B27, 1 mM glutamine, and 0.45% glucose) on Day 3. On Day 4 a 50% stretch injury by Lagrangian strain (equivalent to a severe TBI) was applied (see Morrison and associates [2006]), and media were replaced on Day 5. On Day 7 (equivalent to postnatal day 15), tissue was taken for neurosphere culture or for further analysis. In experiments to determine the origin of proliferating cells, following sectioning with the McIlwain chopper, half the slices were microdissected to remove the inner third of the cortex, including the subventricular zone, prior to plating onto the silicone membrane (refer to results in Fig. 3d). Slices were then injured, dissociated, and plated as described.
Neurosphere culture methods
P8 or P15 mouse whole brains were quickly dissected and the mid/hindbrain was discarded. The hemisphere was transferred to the McIlwain chopper and slices at 400 μM thickness were cut and transferred to ice cold Gey's solution (with 0.45% glucose). In separate experiments, equivalent Day 15 organotypic slice culture tissue was microdissected and placed in ice cold Gey's solution. In the Gey's solution, a sharpened tungsten wire was used to dissect out the hippocampus and outer cortex (ensuring the inner subcortical and subventricular zones were not included). The tissue was washed once in prewarmed Neurobasal-A media and incubated in sterile papain solution (2mg/mL in Neurobasal-A with 2% B27) with gentle agitation for 40 min at 37°C. The tissue was washed in neurosphere media (Neurobasal-A, 2% B27, 2mM glutamine, 1% antibiotic, 20ng/mL epidermal growth factor [EGF], 20ng/mL fibroblast growth factor [FGF]-2, heparin 2μg/mL) and gently triturated with a fire-polished glass pipette into suspension. The solution was passed through a 40 μm cell strainer (Beckton Dickinson) and pipetted onto an OptiPrep™ gradient (10% above a 20% solution) and centrifuged at 1900 rpm for 15 min. The middle cellular layer was removed, resuspended in 2mL media, and centrifuged at 1100 rpm for 2 min. The supernatant was discarded and the pellet resuspended in culture media, assessed for viable cells using the Trypan Blue assay (Sigma) and plated at 50,000 cells/mL in neurosphere media (500μL for the neurosphere assay) in an incubator at 37°C, 5% CO2. Care was taken to ensure all samples (both cortical and hippocampal) were plated at an end concentration of 50,000 live cells/mL. Media were half changed on Day 4 and twice weekly thereafter and for the neurosphere assay, the number of neurospheres per well (>100 μm diameter) was determined. For the average neurosphere diameter measurement, all neurospheres per well > 40 μm diameter were included in the count and the average diameter determined.
Generation of clonal or secondary spheres
Individual primary spheres were taken at 10–14 days in vitro (DIV) and mechanically dissociated into a single cell suspension by tritutrating 20–30 times. The suspension was diluted into a total of 300 μL neurosphere media, and after ensuring single cell dissociation, secondary spheres were generated at clonal density after 10–14 DIV. Half the media was changed with fresh neurosphere media twice weekly. Individual spheres were taken and plated on glass cover-slips, previously coated with poly-
To determine the average expansion ratio (Svendsen et al., 1998), dissociated cells were plated at 100,000 cells/mL in neurosphere media, which was half changed twice weekly. After 2 weeks, the neurospheres were collected and incubated in Accutase (Sigma) for 40 min and then triturated into a single cell suspension. The total number of cells was determined using the Trypan Blue assay, and the expansion ratio calculated. Cells were replated at 100,000 cells/mL in neurosphere media for subsequent generations.
Immunohistochemistry
Differentiated spheres were fixed with prewarmed 4% PFA for 15–30 min. After three washes in phosphate-buffered saline (PBS), spheres were incubated in PBST (PBS, 0.1% triton [BDH], 5% donkey serum [Sigma]) for 30 min and then incubated in primary antibody (mouse anti-βIII tubulin [Covance], 1:500; rabbit anti-GFAP [Dako], 1:500; mouse anti-O4 [Chemicon], 1:100) in PBST overnight at 4°C. After three PBST washes, the spheres were incubated with fluorescent secondary antibodies (1:1000 in PBST; molecular probes) for 2 h followed by two PBS washes. DAPI (1:50, Sigma) was applied for 6 min before three further PBS washes and mounting in Mowiol (Harco).
Free-floating slices were fixed in 4% PFA for 4 h, washed in PBS, quenched for 30 min in distilled water with 3% H2O2 and 10% methanol, washed in PBS, and then incubated in blocking serum (PBS, 0.3% Triton, 5% donkey serum). After two washes in PBS, slices were incubated overnight at 4°C in primary antibody (1:200 mouse anti-GFP [Invitrogen], 1:500 rabbit anti-GFAP [Dako]) in blocking serum, incubated in secondary antibody as described previously, before mounting in Mowiol. All images were taken with a Leica SP5 confocal microscope or Leica DM IRBE inverted microscope. To determine the percentage of GFAP cells co-localising with GFP, cells were counted at random from sections (n=3) of either hippocampus or cortex until a minimum of 150 GFAP cells were counted.
Flow cytometry
Single cells suspensions were prepared as described previously. Cells were passed through a 40 μm cell strainer (Beckton Dickinson) before sorting by flow cytometry. Cells were interrogated through a forward/side scatter gate and a GFP-positive/negative gate at a flow rate of 1–2 (Beckton Dickinson FACS Aria II). Cells passed directly in neurosphere media to give an end concentration of 50,000 cells/mL. Subsequent neurospheres were quantified after 8 DIV using the neurosphere assay.
Western blotting and polymerase chain reaction (PCR)
Western blotting was performed as previously described (Zaben et al., 2009). Briefly, for detection of Shh-signalling pathway components, anti-Shh antibody (Santa Cruz H-19; 1:200) and anti-Patched1 antibody (Santa Cruz; 1:50) were applied overnight. Anti-GAPDH antibody (Sigma; 1:50,000) was used as a loading control. HRP-labelled secondary antibodies (1:5000, Vector Labs) were applied for 1 h before detection with an ECL detection system (Amersham Biosciences) onto photographic film (Kodak). Densometry was performed using ImageJ 64 software.
Using flow cytometry, cells were sorted cells directly into TRIzol (Invitrogen). For mRNA expression in injured tissue, samples of microdissected outer cortex or hippocampus was transferred directly into TRIzol at 2, 6, and 24 h post-injury. For all samples, total RNA was extracted and directly reverse-transcribed to complementary DNA (cDNA) using Precision qScript RT Kit (Primer Design). The cDNA was amplified using a one-step PCR kit (mouse custom real-time PCR assay for use with SYBRgreen chemistry; Primer Design) in a real-time thermocycler (Corbett Robotics Rotor-Gene 6000). Custom-made primers used were directed against GFP, mouse Shh and mouse Patched1 (Ptc1) (Primer Design). Fluorescent data were collected at least once during each cycle of amplification, which allowed for real-time monitoring of the amplification. Data were automatically normalized and a threshold was set at the level where the rate of amplification is the greatest during the exponential phase. Ct-values were collected, raw data were processed and analysed using the Comparative Ct method (2[-Delta Delta C(t)] method (2[-ΔΔCt])) (Schmittgen and Livak, 2008) where the comparative expression level equals to 2-ΔΔCt. The expression of mRNA was normalized to a housekeeping gene (SDHA or GAPDH) and expressed as a ratio to control samples. Prism Software (Graphpad) was used for data analysis and graph generation.
GFAP-GFP intensity
Organotypic cortico-hippocampal slices from P8 GFAP-GFP mice were cultured as described. On Days 1, 4, and 6, green fluorescent images were taken for each individual slice using a Leica DM IRBE inverted microscope, with a lens at 5x magnification, Hammamatsu camera and Volocity image capturing software (Perkin Elmer; settings maintained daily at 300 ms exposure and 0 contrast/brightness). On Day 4, half the slices were injured using a 50% stretch. Volocity software was used to determine the percentage of the area of interest above the threshold intensity (threshold set at 500), from a minimum of three slices. The average percentage above threshold intensity was determined at the three time points. Student t tests were performed to determine significance.
In-situ hybridization of cultured cortical slices
Cultured cortical slices were fixed on their supporting silicone membranes in 4% PFA/PBS for several days at 4°C. In-situ hybridization was performed as described by Chapman and associates (2002). Briefly, specimens were dehydrated in methanol, bleached using 6% H2O2/methanol to inactivate endogenous alkaline phosphatases, rehydrated, permeabilized in detergent mix (0.5% deoxycholate, 1% IGEPAL®, 1% SDS, 1 mM EDTA, 150 mM NaCl, 50 mM Tris-HCl, pH 8.0) and fixed in PFA for 20 min. They were hybridized overnight at 70°C using a digoxigenin-labelled antisense probe against mouse Ptc1 at 1 μg/mL (kind gift from Dr Albert Basson, King's College London). After several wash steps, they were incubated overnight at 4°C with an alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche) at 1:2000 and washed extensively, and the color reaction was performed using nitro-blue tetrazolium chloride/5-bromo-4-chloro-3’-indolylphosphate p-toluidine salt as a substrate. The reaction was stopped in PFA, and embryos were equilibrated against 80% glycerol/PBS and mounted for photography. Pictures were taken on a dissection stereomicroscope using a CCD camera and the QCapture-Pro software.
Results
The cortical neurogenic watershed is recapitulated in vitro between postnatal days 8 and 15
Although there is continued neurogenesis throughout life in the hippocampus, in the outer cortex there is a switch at approximately postnatal day 10, after which its neurogenic potential is greatly attenuated (Laywell et al., 2000). We sought to investigate this further by taking P8 dissociated mouse brain and plating cells taken from both the hippocampus and the outer cortex (taking care to dissect out only the outer half so as to not include the inner neurogenic SVZ). After 5 days in vitro, neurospheres were seen in both samples from hippocampus and cortex (Fig. 1a and b). The average diameter of the neurospheres at 5 DIV (hippocampus 55.0 μm +/− 2.6 vs. cortex 47.0 +/− 1.4), 8 DIV (137 μm +/− 8 vs. 105 +/− 7), and 11 DIV (157 μm +/− 9 vs. 151 +/− 11) was equivalent between hippocampus and cortex derived neurospheres (Fig. 1c).
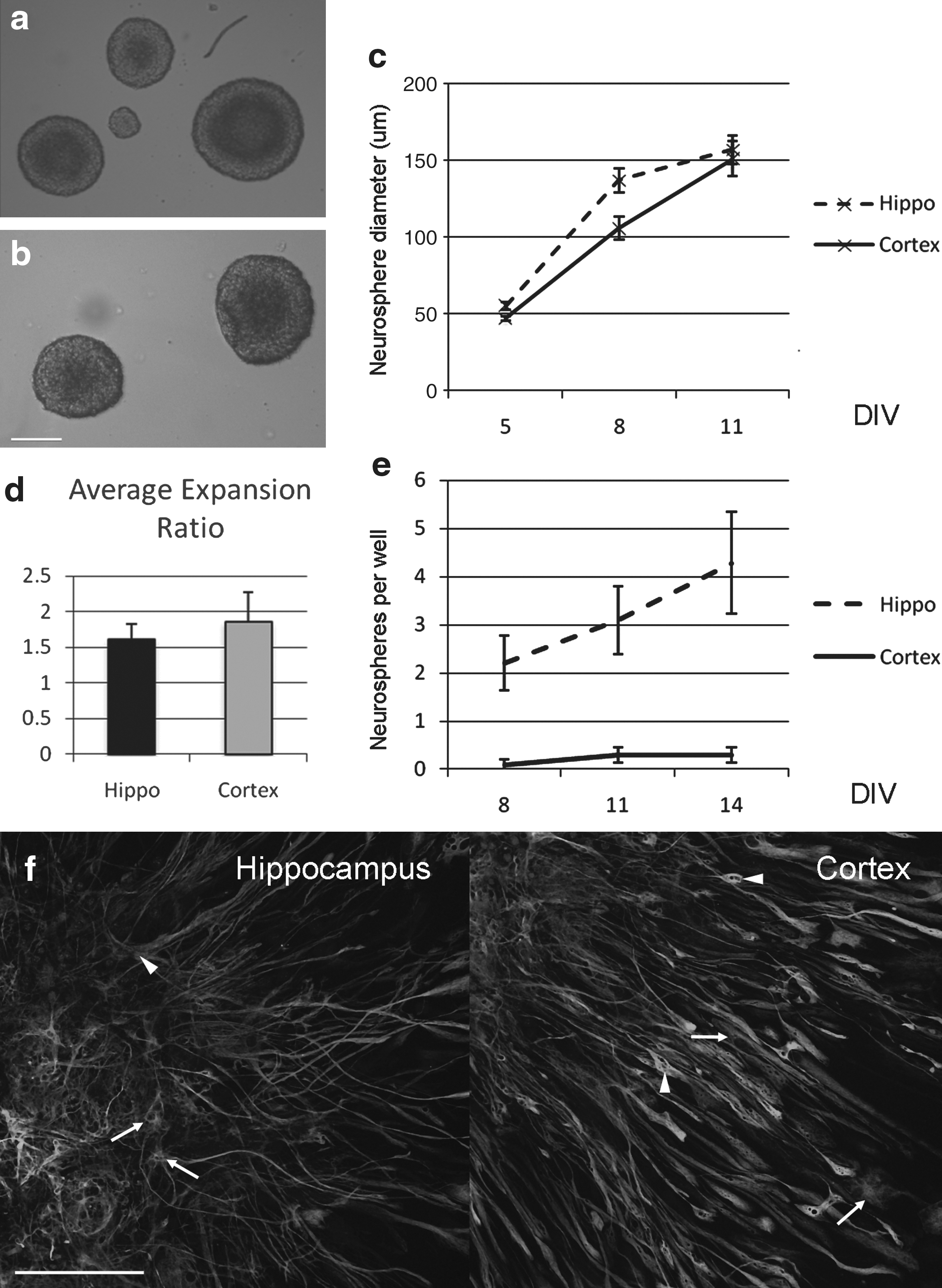
Postnatal proliferative capacity in the cortex is lost by P15.
A key characteristic for any stem cell is the ability to self-renew (Gage et al., 1995), and using the expansion ratio (Svendsen et al., 1998) as a guide, we find that after nine passages, the expansion ratio is 1.6 +/− 0.2 for the P8 hippocampal neurospheres and 1.8 +/− 0.4 for the P8 cortical neurospheres (Fig. 1d).
When tissue from older mice (P15) was cultured, the ability of the cortical tissue to derive neurospheres was greatly reduced. Taking dissociated tissue, in the hippocampus the number of neurospheres after 8 DIV was 2.2 +/− 0.6, at 11 DIV 3.1 +/− 0.7 and at 14 DIV 4.2 +/− 1.0. In the cortex this was attenuated to 0.1 +/− 0.1 at 8 DIV, 0.3 +/− 0.2 at 11 DIV, and 0.3 +/− 0.2 at 14 DIV (Fig. 1e), indicating that by postnatal day 15, the observed postnatal proliferative potential of these cells in the cortex is lost.
Neurospheres derived from P8 mice were subsequently dissociated mechanically and plated at clonal density. Newly generated secondary spheres were plated and differentiated by withdrawing FGF-2 and EGF and adding 2% fetal bovine serum. These clonal spheres from both cortical and hippocampal neurospheres express both neuronal markers (βIII-tubulin) and glial markers (GFAP; Fig. 1f, scale bar 150 μm). Taken together, these data suggest that there are neural stem/progenitor cells (NSPCs) in the cortex at postnatal day 8, but that these lose their proliferative capacity by postnatal day 15.
Cells expressing GFAP account for the majority of neurosphere-forming cells in the postnatal cortex and hippocampus
There is increasing evidence that cells expressing GFAP contribute to constitutive adult neurogenesis (Doetsch et al., 1999; Garcia et al., 2004; Seri et al., 2001). To investigate whether these cells were the source of the proliferating spheres after injury in our model, we used a transgenic mouse in which the enhanced GFP protein was under the control of the human promoter of the astrocytic protein GFAP (Nolte et al., 2001). Both the cortex and hippocampus express the GFP protein at P8 (Fig. 2a). Either dissociated P8 GFP-GFAP cortical or hippocampal cells were sorted by flow cytometry into GFP-positive and negative populations. After 8 DIV, individual cells from both cortex and hippocampus proliferate to give rise to neurospheres from both GFP-positive and GFP-negative cells (Fig. 2b). PCR analysis of flow cytometry sorted GFP cells showed that GFP-negative cells from the hippocampus expressed 1.7% of the mRNA compared to GFP-positive cells (p<0.005, Student t test). Similarly, in cells from the cortex, this was 4.9% compared to GFP-positive cells (p<0.001, Student t test; Fig. 2c), supporting the conclusion that flow cytometry was effective in separating GFP, and therefore GFAP-expressing cells. Under fluorescent microscopy, neurospheres derived from GFP-positive cells did not always exhibit obvious GFP expression. To determine whether this was caused by the loss of GFP and therefore GFAP, or to a loss of fluorescence despite GFP expression, GFP mRNA expression from non-fluorescing neurospheres derived from GFP-positive cells were analyzed by PCR. GFP mRNA, as reflected by the uniqueness of the melt curves for the amplicon synthesized using GFP primers, was expressed by neurospheres generated from the cortex as well as the hippocampus, although there was no visible GFP fluorescence in these neurospheres (data not shown). Triple labeling for GFP (green), anti-GFP (Blue) and anti-GFAP (Red) showed GFP protein co-localizing in cells with anti-GFP antibody staining in both the hippocampus and cortex demonstrating that fluorescence is indicative of protein expression (Fig. 2d and 2e; scale bar 100 μm). Moreover, GFAP staining (red) is more restricted within the cell, suggestive of filamentous staining, unlike the uniform cytosolic staining of the GFP protein (green or blue). This co-localization of GFAP with GFP was not consistent for all GFAP-expressing cells (Fig. 2d and e) indicating that a proportion of GFAP- positive cells do not express GFP. Indeed, the penetrance of the GFP transgene in this strain of mice has been reported to be variable (Nolte et al., 2001), and in our study we found that the number of double positive GFP/GFAP cells in postnatal sections of the hippocampus is 29% and of the cortex is 24% of GFAP cells. We did not find a single cell that was GFP positive but GFAP negative in the cortex or hippocampus. These results show that cellular GFP expression is highly specific but not sensitive, similar to what was found by Nolte and associates (2001). However, as the expression of GFP mRNA in the flow cytometry sorted GFP-negative population <5% of the positive population (Fig. 2c), most likely because there were more GFAP-negative than GFAP-positive cells in the GFP- negative population, we can conclude that the GFP-positive population contains a significant enrichment of GFAP cells.
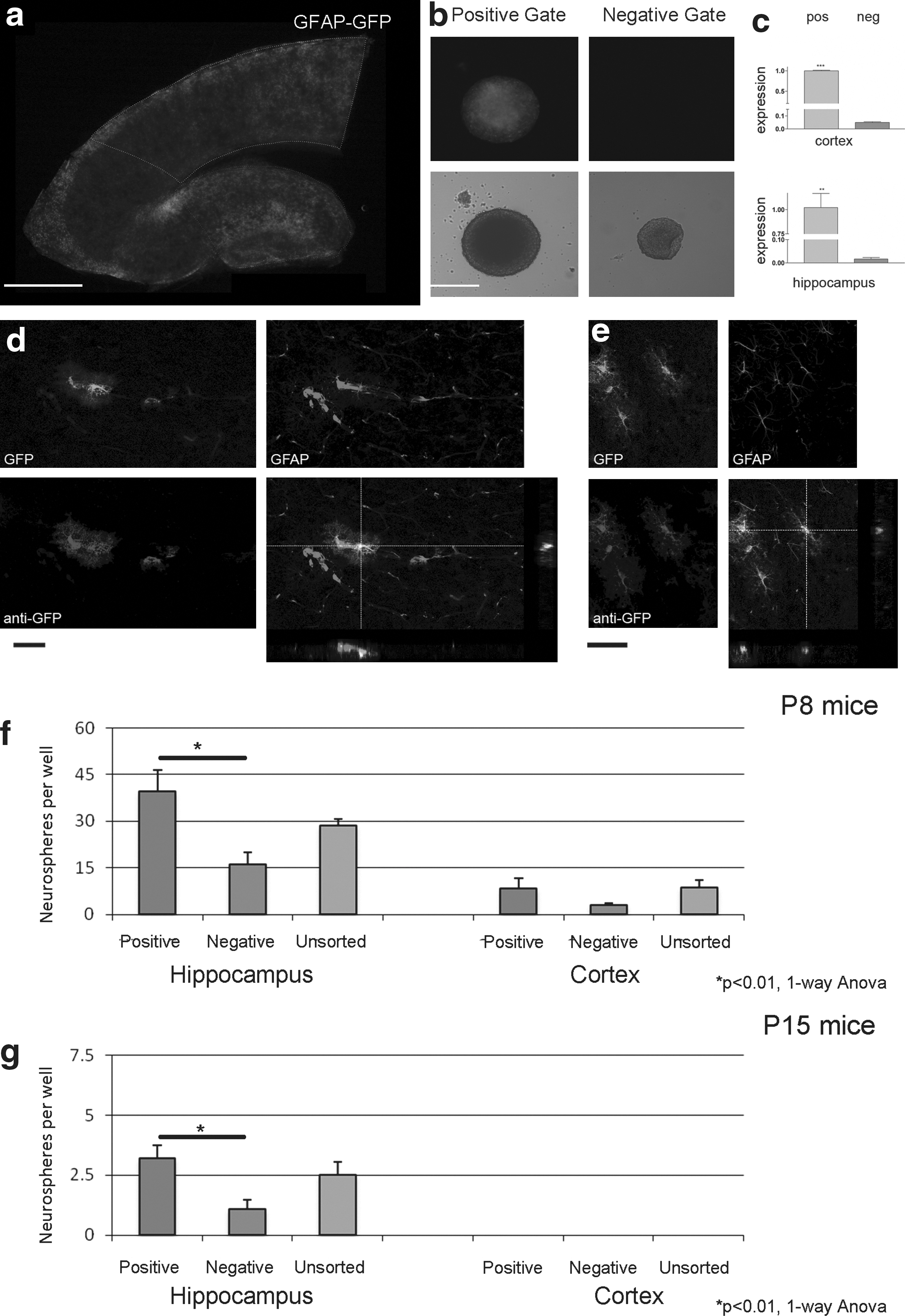
GFAP-GFP expressing cells proliferate to form neurospheres, but do not account for all the formed spheres.
Using flow cytometry, the neurosphere-forming capacity of dissociated outer cortex and hippocampal tissue from P8 and P15 GFAP-GFP mice was determined. Cells were sorted for positive and negative GFP expression, and a third unsorted population. For P8 mice, the numbers of neurospheres per well were 39.3 +/− 7.0 derived from hippocampal GFP-positive cells, 16.1 +/− 3.8 derived from GFP-negative cells and 28.5 +/− 2.3 from the unsorted group . In the cortex, the number of spheres from GFP-positive cells was 8.3 +/− 3.3, with 2.9 +/− 0.7 from GFP-negative cells and 8.7 +/− 2.3 from the unsorted cells (Fig. 2f). From P15 GFAP-GFP mice, the number of spheres per well from the hippocampus was 3.2 +/− 0.5 from GFP-positive cells, 1.1 +/− 0.4 from GFP-negative cells, and 2.5 +/− 0.57 from the unsorted cells. There were no spheres derived from the P15 cortex of these GFAP-GFP mice (Fig. 2g). In the hippocampus, at both P8 and P15, the GFP-positive cells derived a significantly greater number of neurospheres compared to the GFP-negative population (Fig. 2f and g, p<0.01, one-way ANOVA with Tukey post-hoc test). These data suggest that the greater propensity of spheres derived arises from cells that express the GFAP protein.
Injury to the cerebral cortex leads to acquisition of stem cell properties in vitro
We have developed a novel assay to determine the neurogenic potential of the injured brain using an organotypic culture method. The stretch injury protocol uses organotypic cortico-hippocampal slices from P8 mice, which are grown on a silicone membrane for 7 days (P8 [+7] equivalent to P15). After 3 days in full media, in which survival is maximal after the initial culture (data not shown), the slices are switched to a medium without serum, as the presence of serum attenuates endogenous neurogenesis in organotypic slice cultures (Chechneva et al., 2005; Raineteau et al., 2004). After a total of 7 DIV (equivalent postnatal day 15), the outer cortex and the hippocampus are dissected out separately, dissociated, and plated. In our assay, the average number of neurospheres per well was counted as a surrogate for stem cell potential (Coles-Takabe et al., 2008; Reynolds and Weiss, 1992) (Fig. 3a).

A severe stretch injury activates cells in the cortex to proliferate in vitro. These proliferating cells are multipotent.
In control uninjured cortico-hippocampal slices, the number of neurospheres per well after 8 DIV in the hippocampus and cortex was equivalent to that of fresh P15 tissue (1.7 +/− 0.2 increasing to 3.0 +/− 0.3 at 11 DIV for the hippocampus and 0.3 +/− 0.2 at 8 DIV and 0.5 +/− 0.1 at 11 DIV for the cortex; Fig. 3b). However, following a 50% stretch injury, equivalent to a severe TBI (Morrison et al., 2006), there was a significant increase in the number of neurospheres generated, indicative of stem cell activation in the injured hippocampus (3.4 +/− 0.3 at 8 DIV and 6.0 +/− 0.5 at 11 DIV) and more interestingly in the injured cortex (2.5 +/− 0.2 at 8 DIV and 7.1 +/− 1.3 at 11 DIV; p<0.01 two-way ANOVA with Bonferroni post-hoc test; Fig. 3b). These results indicate injury-enhanced neurosphere formation from cells in both the hippocampus and the non-permissive cortex.
Injury-induced NSPCs are multipotent
We next sought to identify whether these proliferating sphere-forming cells were stem/progenitor cells that were capable of differentiating into the three neural stem cell progeny. Following injury, tissue was dissociated and spheres derived from injured cortex or hippocampus were again dissociated and plated at a clonal density. Subsequent secondary clonal spheres were differentiated and stained for neuronal and glial markers. In both hippocampal and cortical clonal spheres derived from the injured brain, double staining showed cells expressing the neuronal marker βIII tubulin and the astrocytic marker GFAP (Fig. 3c). The oligodendrocytic marker O4 was also expressed in cells derived from the clonal spheres. These results indicate that cells in the cortex, which are either native or have migrated from the periventricular zone after injury, are activated after stretch injury to proliferate in vitro and have the capacity to differentiate into all three of the neural stem cell progeny types.
Injury-induced neurosphere-forming cells do not arise from the subventricular zone
To determine whether injury-induced proliferating cells are derived from endogenous cells within the cortical parenchyma (Buffo et al., 2008), or whether they migrate in from the SVZ (Salman et. al., 2004), we microdissected out the SVZ and inner third of the cortex from P8 cortico-hippocampal slices. We compared the neurosphere-forming ability of SVZ-deficient slices compared to control slices (in which the SVZ was microdissected after injury and prior to cell dissociation; Fig. 3d and e) following a 50% stretch injury. The number of neurospheres was comparable (5.8 +/− 0.6 in SVZ deficient slices vs. 6.4 +/− 0.8 in intact slices; p=0.54, Student t test) indicating that the proliferating cells did not arise from the SVZ following a stretch injury.
The majority of neurosphere-forming cells following injury are derived from GFAP-expressing cells
Parenchymal astrocytes from the uninjured healthy adult mouse cortex do not exhibit stem cell like properties when cultured in vitro (Buffo et al., 2008). However, following a stab wound injury to the cerebral cortex, these astrocytes begin to proliferate in vitro to form neurospheres and differentiate in neurons, astrocytes, and oligodendrocytes, key characteristics of stem cells (Buffo et al., 2008). To investigate the source of stem cells in the mouse cortex following TBI, we utilized our TBI model using cortico-hippocampal slices from P8 GFAP-GFP mice. The relative change in GFP expression, and hence GFAP, was determined by quantifying relative change in intensity of fluorescence from the area of interest. In both cortex and hippocampus of freshly sliced tissue, the expression of GFP was low (Fig. 4a). Prior to any injury, in the first 3 days, an increase in GFP expression was a likely consequence of gliosis (data not shown). After 3 days, following a 50% stretch injury, both in the cortex and the hippocampus, the relative intensity of GFP was greater in injured cultures than in controls over time. Quantification showed the proportion of the cortex above the threshold intensity to be significantly increased in the injured (52.1% +/− 4.3%) compared to the control-uninjured cortex (13.5% +/− 4.3%; p<0.01, Student t test) at 6 h and at 72 h post-injury (57.0% +/− 8.3% vs. 14.3% +/− 5.3%; p<0.01, Student t test; Fig. 4b). The results were similarly significant in the hippocampus at 6 h post-injury (61.5% +/− 2.1% vs. 27.6% +/− 5.0%; p<0.01, Student t test) and at 72 h post-injury (76.6% +/− 4.1% vs. 17.1% +/− 8.7%; p<0.01, Student t-test). These results indicate that injury leads to a significant upregulation of GFP, and therefore GFAP, expression.
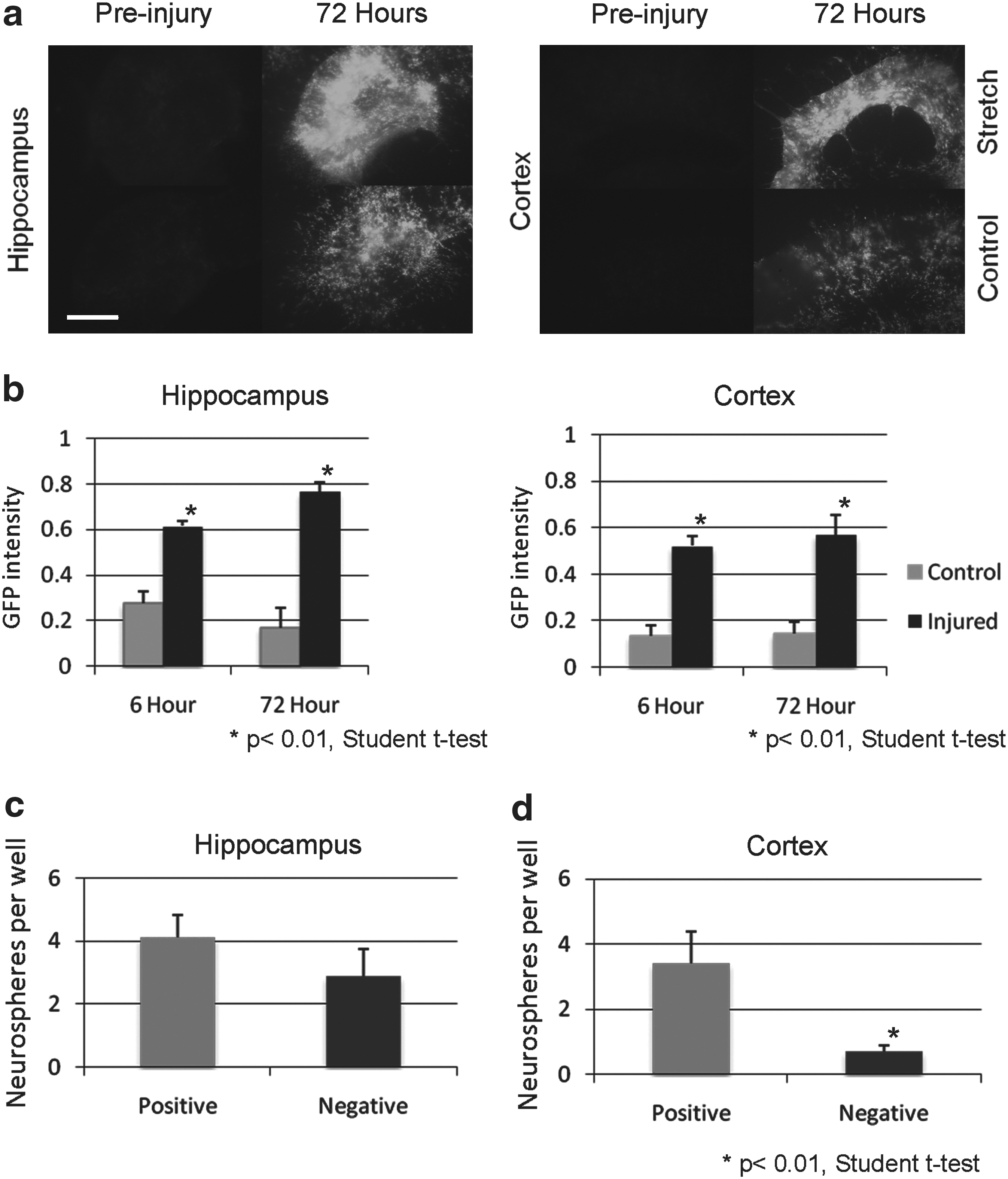
The majority of neurospheres derived from the injured cortex are from GFP-expressing cells.
Following injury to cortico-hippocampal slices derived from P8 GFP-GFAP mice, the upregulation of GFP may have been in part caused by hypertrophy of the astrocyte with an increased expression of GFAP (Sun et al., 2010), because of an increased proliferation of the astrocytes (Buffo et al., 2008, Miyake et al., 1988) or both. To test whether there was an increase in the number of cells expressing GFP, specifically those that continue to proliferate following injury, injured GFP-GFAP cortico-hippocampal slices (at equivalent day P15) were microdissected and dissociated into pools of outer cortex and hippocampus and sorted by flow cytometry into GFP-positive and negative populations. Following injury, in the hippocampus, the neurospheres derived from GFP-positive and negative gates were equivalent (4.1 +/− 0.7 GFP-positive vs. 2.9 +/− 0.8 GFP- negative; p=0.29 Student t test; Fig. 4c). However, a significantly greater proportion of neurospheres derived from the cortex were from the GFP-positive cells (3.4 +/− 1.0) compared to GFP-negative cells (0.7 +/− 0.2; p<0.001 Student t test; Fig. 4d). This implies that following a stretch injury to the cortex, the majority of NSPCs are GFAP expressing.
Components of the Shh-signaling pathway are transiently upregulated in the injured cortex
Shh-signaling regulates the proliferation of stem cells in the postnatal hippocampus and subventricular zone (Ahn and Joyner, 2005; Han et al., 2008; Lai et al., 2003; Machold et al., 2003). More recently, it has been demonstrated that a cortical freeze injury upregulates Shh in astrocytes in the region surrounding the lesion, leading to an increased proliferation of a subset of glial cells (Amankulor et al., 2009). Given these results, we hypothesized that Shh signaling may be upregulated in the cortex following a stretch injury. Using our experimental paradigm, injured or control outer cortex was microdissected and analyzed. We used a combination of quantitative PCR and Western analysis for Shh and the Shh target gene, Ptc1, to assess the level of Shh pathway activation. Ptc1 is a known direct target of Shh signalling, hence, its expression reflects Shh pathway activation (Garcia et al., 2010; Goodrich and Scott, 1998). Shh mRNA was transiently upregulated at 6 h compared to control (1.6-fold increase in Shh mRNA; p<0.05, two-way ANOVA with Bonferroni's post-hoc analysis, Fig. 5a). However, there was no obvious increase in Shh protein expression (Fig. 5b). Similar to Shh mRNA expression, the receptor for Shh in the nervous system, Ptc1, was transiently upregulated at 6 h (a 3.1- fold increase in expression; p<0.0001, two-way ANOVA with Bonferroni post-hoc test; Fig. 5c). This greater expression of Ptc1 mRNA was translated to a 2.7-fold increase in Ptc1 protein at 6 h as measured using a normalized relative band density (Fig. 5B).
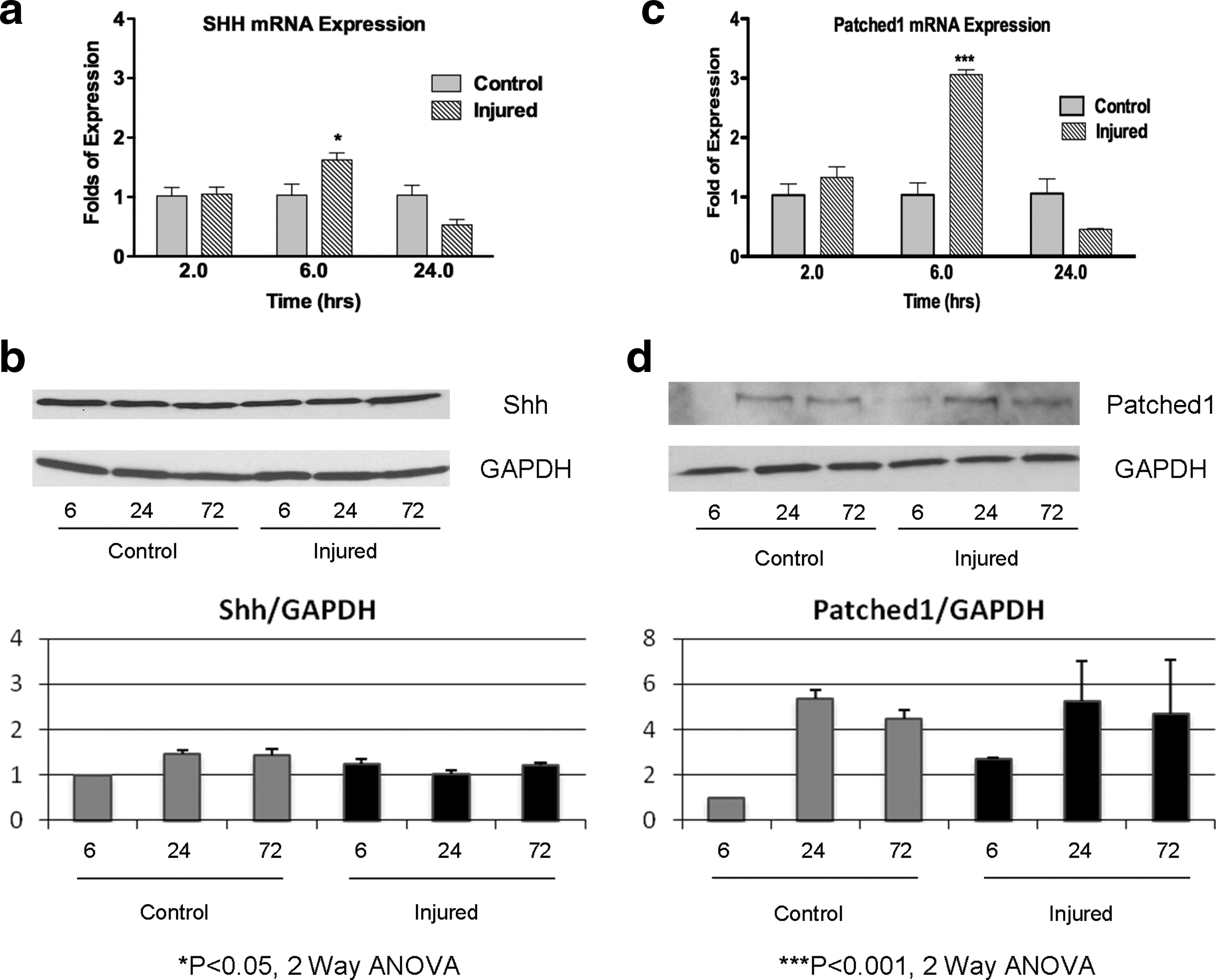
Sonic hedgehog (Shh) signaling components are upregulated in the cortex 6 h after injury.
We also compared the expression of Ptc1 in control and injured cortex by in situ hybridization. Compared to control cortical explants (Fig. 6a), a slight upregulation of Ptc1 expression was already visible in injured cortical explants after 2 h of culture (Fig. 6a’). This relative upregulation became more pronounced after 6 h of incubation (compare Fig. 6b, b’). After 24 h in culture, injured explants either show an even stronger relative upregulation of Ptc1 expression (Fig, 6c,c’; co: n=7, inj.: n=7) or their expression levels fall below those of control explants as seen in our qPCR experiment (not shown; co: n=7, inj.: n=4). This variability could reflect a progressive decline of injured explants in some experimental batches. Taken together, the in situ hybridization experiments support our qPCR analysis and suggest that the Shh signaling pathway becomes activated – at least transiently – in response to a stretch injury. These results raise the intriguing possibility that Shh signaling may regulate proliferative effects as demonstrated in a freeze injury (Amankulor et al., 2009).
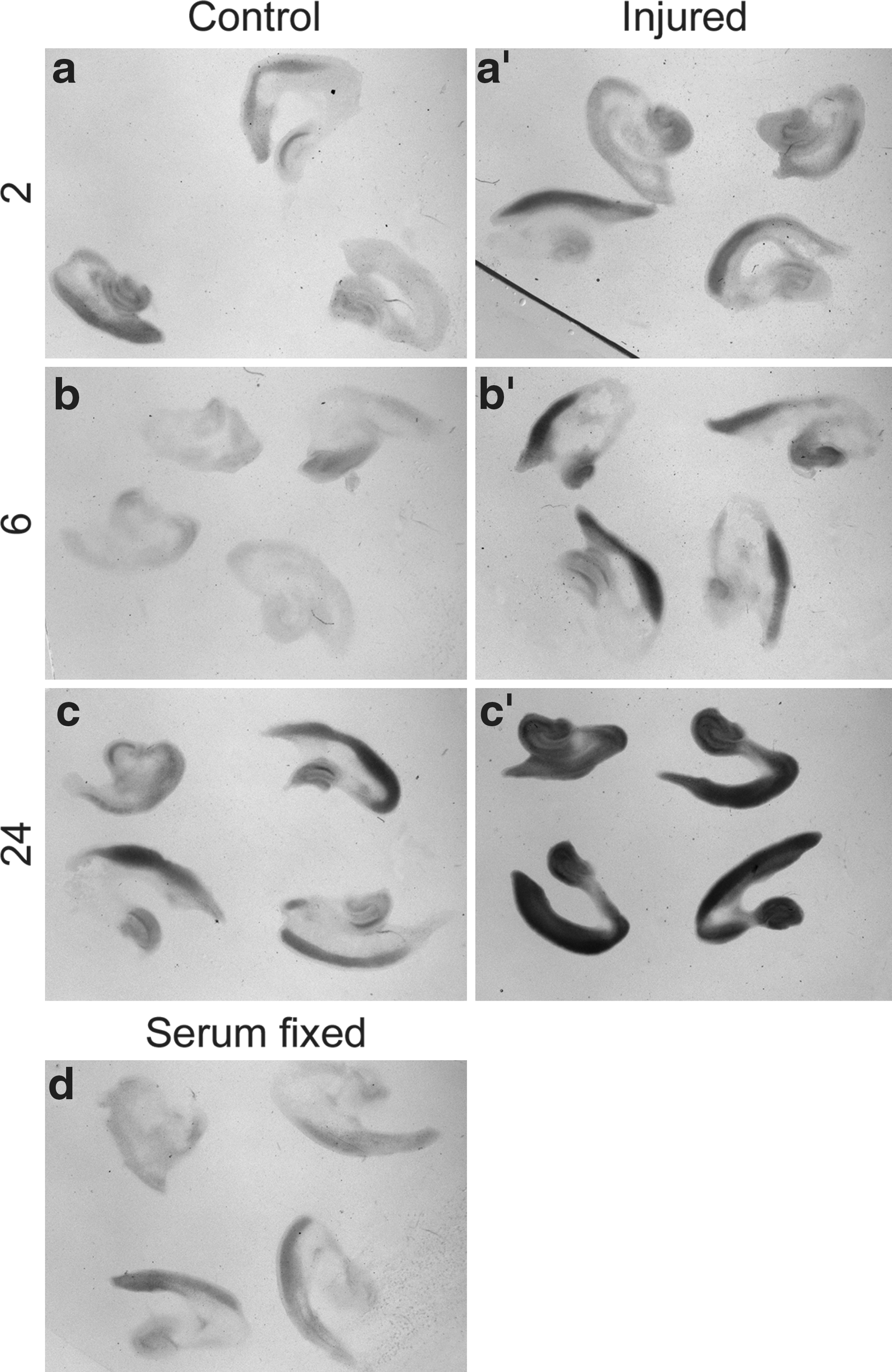
In situ hybridization of Patched1 mRNA indicates an upregulation of Patched1 in the injured outer cortex. 2, 6, and 24 are h post-injury. Patched1 expression in control (
Discussion
A novel model to investigate the effects of TBI on neurogenesis
We describe a novel ex-vivo injury/neurosphere assay that maintains the organotypic architecture while allowing easy manipulation of the culture conditions. This is similar to the recently described slice/neurosphere assay (Jhaveri et al., 2010) that was used to elucidate the effect of antidepressant influence on stem cell/neurogenic precursor proliferation through norepinephrine uptake. Our model uses a biomechanically validated TBI model (an equi-biaxial deformation stimulus [Morrison et al., 2006] that replicates the shear forces equivalent to a 50 km/h impact). This organotypic model thus recapitulates the postnatal quiescence of cortical progenitors and allows quantification of the proliferative recruitment of precursor cells after stretch injury. Moreover, this assay uses cortico-hippocampal slices in which the continuously proliferating hippocampus serves as an internal control. The assay also balances the requirement of serum to minimize initial cell death, especially in cortex (unpublished data), with the inhibitory effects of serum on neurogenesis in organotypic slices (Chechnev et al., 2005; Raineteau et al., 2004).
Our data support the findings of Laywell and colleagues signifying a watershed in cortical neurogenesis in postnatal mice (Laywell et al., 2000). Furthermore, a stretch injury applied to the non-permissive cortex leads to a significant increase in endogenous neurosphere-forming cells when later cultured in vitro. Importantly, secondary clonal spheres were derived following a severe injury and critically, these secondary spheres differentiated to give rise to all three neural stem cell progeny types confirming multipotency of these activated cells in both the injured hippocampus and cortex. In summary, our injury/neurosphere model, in which the niche environment was altered as a result of injury, indicates that injury alters the neurogenic potential of cells residing in the cortex of the rodent brain giving them stem cell-like properties in vitro.
Neurogenesis in niches after injury
Following injury, there is good evidence of regenerative ability in both the SVZ and the subgranular zone of the dentate gyrus, the two established niches of constitutive neurogenesis in the adult rodent (Chirumamilla et al., 2002; Sun et al., 2005). Controversy surrounds whether a similar capacity for neuronal regeneration exists in the cortex, with the traditional view that this region does not support neurogenesis (Caviness and Sidman, 1973; Koketsu et. al., 2003; Kornack and Rakic, 2001). However, a growing body of evidence suggests that under differing paradigms of injury, there is an increase in proliferative cells with neurogenic potential in the injured cortex. Whether these cells migrate in from the SVZ or whether they arise from endogenous cortical cells is debatable. In cortical contusion models for TBI, proliferative cells arise from the SVZ (Ramaswamy et al., 2005) and the dentate gyrus (Dash et al., 2001), and these include migrating neuroblasts toward the injury site (Ramaswamy et al., 2005; Sundholm-Peters et al., 2005). Similarly, stem cells in the SVZ contribute to brain remodeling in the proximal region of a cortical lesion (Salman et al., 2004). Following ischemic injury by prolonged middle cerebral artery occlusion, subventricular neuroblasts migrate to the injured cortex (Jin et al., 2003). The alternative view that quiescent stem cells already residing in the cortex are activated following injury is gaining credence. Following a stab wound, mature protoplasmic reactive astrocytes (identified by genetic labelling) were isolated from the injured cerebral cortex and gave rise to stem cells capable of differentiating into neurons in vitro but not in vivo (Buffo et al., 2008). This process, termed post-injury “dedifferentiation”, suggests that mature cells of one lineage within the cerebral cortex are able to acquire stem/progenitor properties and then follow a different lineage pathway. Under controlled experimental paradigms in which targeted apoptosis of neurons is induced, repair is observed in cortical neurons projecting to the thalamus, (Magavi et al., 2000) or corticospinal motor neurons (Chen et al., 2004). In a recent study, slowly proliferating precursor cells in layer 1 of the cortex were observed, which were activated after ischemia to give rise to GABAergic interneurons (Ohira et al., 2010).
In our study, the injury model induced a stretch equivalent to a severe TBI (Morrison et al., 2006), leading to the activation of possible endogenous proliferative cells both in the hippocampus, and more significantly, in the outer cortex. Multipotency of these cells from both regions was demonstrated in vitro, suggesting that these endogenous cells have the capacity, at least, to form neurons. Moreover, the low level of proliferation we observed in cortex derived from postnatal day 15 mice may represent the recently reported slowly proliferating progenitor cells in the adult cortex (Ohira et al., 2010). Indeed, there are quiescent precursors in non-permissive parenchyma in rodents (Buffo et al., 2008; Palmer et al., 1995) and humans (Arsenijevic et al., 2001). We sought to determine whether the proliferating cells in our study were endogenous within the cortical parenchyma (Buffo et al., 2008), or whether they migrated in from the SVZ (Salman et al., 2004). The proliferative capacity between slices in which the SVZ was either retained or removed was comparable, indicating that in our study, it was unlikely that proliferating cells arose in and migrated from the SVZ. It is likely that in our experiment, the quiescent precursors in the parenchyma proliferate following injury (Buffo et al., 2008; Palmer et al., 1995). However, we cannot exclude the unlikely possibility of cells migrating from the distant hippocampus in our cortico-hippocampal slices. We were unable to determine this definitely, as cortical slices without the hippocampus did not adhere adequately on the silicone membrane.
Our results indicate that following severe injury, the expression of GFAP is increased in both regions of our organotypic slices. This may represent a hypertrophy of the GFAP-expressing cells (Sun et. al., 2010). Indeed, using genetic fate mapping, one study proposed that cortical astrocytes cease dividing by postnatal day 10, and following a stab injury, these astrocytes increase their GFAP expression by hypertrophy rather than proliferation (Burns et al., 2009). Our data support activation of the endogenous GFAP precursors. Approximately half of the neurosphere-forming cells in the hippocampus are derived from GFAP-expressing cells, whereas > 75% of the cortical spheres are as such. This agrees with previous studies (Doetsch et al., 1999; Garcia et al., 2004; Seri et al., 2001), and with the concept that these cells are the predominant source of neurospheres following TBI. In both the SVZ and subgranular zone (SGZ), GFAP expression represents a population of largely quiescent stem cells (Doetsch et al., 1999; Seri et al., 2001) and constitutive neurogenesis in the adult forebrain is a result, in part, of a population of morphologically distinct GFAP-expressing cells (Garcia et al., 2004). This suggests that GFAP-expressing cells in our experiments may indeed be the source of cells that acquire stem cell-like properties in vitro.
From our data, not all the derived neurospheres arise from GFAP-expressing cells. This may be a consequence of a lack of full penetrance of the reporter gene, or perhaps the loss of GFAP expression as the proliferating cells come out of quiescence (Seri et al., 2004). Alternatively, these cells may represent a different population of activated endogenous stem cells. Cycling oligodendrocyte precursors can give rise to new neurons in the piriform cortex (Rivers et al., 2008) or the thalamus (Dimou et al., 2008), and may represent an alternative source of cells after stretch injury. Fate mapping of oligodendrocyte precursors implicates these cells, rather than GFAP expressing cells, as the primary source of reactive gliosis following a stab injury to the cortex (Burn et al., 2009). As a consequence, further characterization of the fate of oligodendrocyte precursors in our model may help determine whether those cells that proliferate, but do not express GFAP, arise from an alternative precursor type. However, this does not detract from the central finding that a population of cells, which proliferate in vitro following injury, are derived from GFAP-expressing cells.
Timing and mechanism of activation
In our experimental paradigm, injured tissue was microdissected and dissociated into a single cell suspension after 3 days (injury at equivalent postnatal day 12 and dissociation at equivalent postnatal day 15). This is consistent with previous reports in which activated cells are observed after an equivalent time point following injury (Buffo et al., 2008; Itoh et al., 2005). Interestingly, Itoh and colleagues (2005) did not observe proliferation at 24 h or at 7 days after injury, suggesting a temporal window of opportunity before activated cells are committed to a differentiation process.
In addition to the timing following injury, the intrinsic changes to the niche environment may contribute to the fate of these activated cells after injury. Exogenous transplantation of neuronal precursors into the cortex results in differentiation into cells with astrocytic or oligodendrocytic characteristics, implicating that in this region of the central nervous system, progenitor cells are influenced by the environment toward a glial lineage (Seidenfaden et al., 2006). In the injured niche environment, there is growth factor upregulation in the hippocampus (Yoshimura et al., 2003) and cortex (Oyesiku et al., 1999). This is accompanied by a robust inflammatory response and gliosis (Kelley et al., 2007; Lenzlinger et al., 2001; Soares et al., 1995). Moreover, disruption of the inflammatory response, specifically microglia activation, results in recovery of neurogenesis in the hippocampus (Ekdahl et al., 2003). This suggests that following an injury, the inflammatory response may contribute to determining the proliferative fate of these endogenous cells.
The secreted protein Shh acts on its receptor Ptc1, to disinhibit the membrane protein Smoothened, and subsequently activate transcription factors of the GLI-Kruppel family (Gli 1 and 2) (Marti and Bovolenta, 2002). In the cerebral cortex, the inflammatory response, induced by lipopolysaccharide injection, leads to the upregulation of the secreted protein Shh, and the downstream effector Gli (Amankulor et al., 2009). Furthermore, the expression of Shh following a freeze injury to the cortex is upregulated in GFAP-expressing cells (Amankulor et al., 2009). In a second study, the proliferative potential of cells from the uninjured adult cortex co-cultured with postnatal astrocytes led to an increase in neurosphere forming ability (Jiao and Chen, 2008). Similar to our observations, the majority of these multipotent sphere-forming cells expressed GFAP (Jiao and Chen, 2008). Interestingly, addition of exogenous Shh to cultures of adult cortical cells resulted in an increase in the number of neurospheres (Jiao and Chen, 2008). Because Shh signalling is crucial in maintaining stem cell self-renewal in the SVZ and the subgranular zone of the hippocampus (Ahn and Joyner, 2005; Han et al., 2008; Wang et al., 2007), we sought to determine whether an increase in Shh signalling correlated with a stretch injury to our organotypic slices. There was a significant increase in Shh mRNA at 6 h, but this did not correlate with an appreciable increase in protein expression. This may be in part because of detection of the full-length uncleaved protein rather than the active secreted form (Bumcrot et al., 1995). However, expression of the receptor for Shh, Ptc1, was increased at 6 h both in mRNA expression and protein expression. The mRNA increase was transient, but resulted in an earlier expression of Ptc1 protein. Indeed, Ptc1 expression is positively regulated by Shh (Goodrich and Scott, 1998) and therefore can be used as a readout for Shh activity in the cortex (Garcia et al., 2010). The region of expression in the cortex did not include the SVZ, as our experimental paradigm ensured that only the outer two-thirds of the cortex was analyzed. This correlation of a transient upregulation raises the possibility that Shh signalling may contribute to our observed increase in cortical cell proliferation following injury. Moreover, the timing of upregulation of Shh signalling (within hours) precedes our observed increase in proliferation at 3 days. Indeed, astrocytes in the adult murine cortex express all the machinery to respond to Shh signalling, including Ptc1, Gli1, Gli2, and Gli3 (Garcia et al., 2010). This would implicate a possible contribution of Shh signalling in astrocytes toward potential stem cell proliferation in the injured cortex.
Conclusion
Using a novel cortical slice‐neurosphere assay, we have demonstrated a population of endogenous GFAP-expressing cells that acquire stem cell properties, but only following a stretch injury that mimics TBI. Our future work is directed toward dissecting the signaling mechanisms to help answer chiefly two questions. What drives these cells out of quiescence and what pushes these cells toward a glial fate in vivo and away from a neuronal fate. This opens up an avenue toward the possibility of manipulating endogenous stem cells in the injured cortex to effect repair.
Footnotes
Acknowledgments
This study was supported by the Wessex Medical Research (A.I.A), the Wessex Neurological Centre Trust (A.I.A), the Royal College of Surgeons of England (A.I.A), and the Medical Research Council (strategic grant G0300356 to W.P.G). We thank Professor Frank Kirchhoff (Göttingen) and Alexei Verkhratsky (Manchester) for supply of the GFAP-GFP transgenic mice, and David Johnston (Biomedical Imaging Unit, University of Southampton) for help with image analysis.
Author Disclosure Statement
No competing financial interests exist.