
Abstract
Although traumatic brain injury (TBI) is recognized as one of the leading causes of death from trauma to the central nervous system (CNS), no known treatment effectively mitigates its effects. Lithium, a primary drug for the treatment of bipolar disorder, has been known to have neuroprotective effects in various neurodegenerative conditions such as stroke. Until this study, however, it has not been investigated as a post-insult treatment for TBI. To evaluate whether lithium could have beneficial effects following TBI, lithium at a dose of 1.5 mEq/kg was administered after injury. Assessed at 3 days and 3 weeks post-injury using hematoxylin and eosin staining, lithium treatment was found to reduce lesion volume. Lithium at doses of 2.0 and 3.0 mEq/kg also significantly reduced lesion volume at 3 days after injury, and the therapeutic window was at least 3 h post-injury. TBI-induced neuronal death, microglial activation, and cyclooxygenase-2 induction were all attenuated by lithium at 3 days after injury. In addition, lithium treatment reduced TBI-induced matrix metalloproteinase-9 expression and preserved the integrity of the blood–brain barrier. As for behavioral outcomes, lithium treatment reduced anxiety-like behavior in an open-field test, and improved short- and long-term motor coordination in rotarod and beam-walk tests. Lithium robustly increased serine phosphorylation of glycogen synthase kinase-3β (GSK-3β), suggesting that the underlying mechanisms responsible for lithium's protective effects are triggered by increasing phosphorylation of this kinase and thereby inhibiting its activity. Our results support the notion that lithium has heretofore unrecognized capacity to mitigate the neurodegenerative effects and improve functional outcomes in TBI.
Introduction
I
The complex pathology of TBI suggests that treating it will require an agent capable of interfering with multiple pathways of cell survival or death (Margulies and Hicks, 2009). One such agent is lithium. For more than half a century, lithium has been the primary medication used to treat bipolar disorder. Prolonged use of this drug is even reported to reverse bipolar patients' loss of gray matter volume (Chuang and Manji, 2007; Sassi et al., 2002). In addition, our and other laboratories have found robust neuroprotective effects of lithium in animal models of a variety of neurological and neurodegenerative diseases, including cerebral ischemia, spinal cord injury, Huntington's disease, Alzheimer's disease, and amyotrophic lateral sclerosis (Chiu and Chuang, 2010; Quiroz et al., 2010).
Lithium, a direct inhibitor of glycogen synthase kinase-3β (GSK-3β; Klein and Melton, 1996), can also indirectly inhibit this kinase by enhancing serine phosphorylation through activation of the serine/threonine kinase Akt (Beaulieu et al., 2004; Chalecka-Franaszek and Chuang, 1999), the protein kinase A (Jope, 1999), and the protein kinase C (Kirshenboim et al., 2004). Inhibition of GSK-3 results in enhanced expression of several neuroprotective and neurotrophic proteins, such as heat-shock protein 70 (HSP70; Ren et al., 2003), brain-derived neurotrophic factor (BDNF; Yasuda et al., 2009), and Bcl-2 (Liang and Chuang, 2006; Senatorov et al., 2004). By reducing microglial migration, lithium also modulates inflammation and attenuates inflammation-induced neurotoxicity (Beurel et al., 2010; Yuskaitis and Jope, 2009). Despite its well-known and multifaceted neuroprotective/neurotrophic effects, however, lithium has not been investigated as a post-insult treatment for TBI. The effectiveness of post-insult lithium treatment might be more clinically relevant, since it could realistically be administered to human victims.
In this study, we determined the severity of brain injury produced by a controlled cortical impact (CCI) device with different combinations of parameters. The potential therapeutic effects of lithium were then tested in two different CCI injury conditions. Our results indicate that post-insult administration of lithium reduced lesion volume, neuronal degeneration, microglial activation, and upregulation of cyclooxygenase-2 (COX-2). Lithium treatment also attenuated TBI-induced disruption of the blood–brain barrier (BBB) and upregulation of matrix metalloproteinase-9 (MMP-9). Further, lithium administered after TBI improved functional outcomes on multiple behavioral tests. The beneficial effects of lithium in the TBI model are likely due to its ability to inhibit GSK-3β activity.
Methods
TBI induction and lithium treatment
All animals were treated in accordance with guidelines of the Uniformed Services University of the Health Sciences and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. A CCI device (Leica, Wetzlar, Germany) was used to induce TBI in 8-week-old male C57BL/6 mice. Briefly, prior to TBI mice were anesthetized with 2.0% isoflurane in O2 and mounted on a stereotaxic frame (Kopf Instruments, Tujunga, CA). An approximately 4-mm-diameter craniotomy was performed over the left parietal cortex between the bregma and the lambda sutures. The skullcap was carefully removed with no disruption of the dura. The point of impact was identified midway between the lambda and the bregma sutures, as well as midway between the central suture and the left temporalis muscle. The CCI injury was performed using a 3-mm-diameter convex tip set to compress the brain with combinations of velocity at 5.0 m/sec and different levels of deformation of 1.0–2.5 mm (1.0, 1.5, 2.0, and 2.5 mm). The craniotomy was then closed by repositioning the bone flap. A group of sham-injured mice underwent identical craniotomy procedures without CCI injury. Body temperature was maintained at 37±0.5°C using a heating pad coupled to a rectal probe.
Lithium administration and plasma lithium level measurement
Lithium chloride (1.5 mEq/kg dissolved in normal saline; Sigma-Aldrich, St. Louis, MO) or an equal volume of saline was injected intraperitoneal (IP) 15 min after TBI, followed by injections once daily for up to 2 weeks. For the dose-response study, lithium was injected at different doses (1.0, 1.5, 2.0, 3.0, or 5.0 mEq/kg) 15 min after injury, followed by daily injections for 3 days. For the time-course study, lithium (1.5 mEq/kg) was injected 3 or 6 h after TBI, followed by injections once daily for 3 days. Whole blood samples were collected 12 h after the last of the 2-week lithium injections, and plasma lithium levels were measured using inductively coupled plasma/mass spectrometry by Medtox Scientific Inc. (St. Paul, MN; test code 60063).
Evaluation of lesion volume
Lesion volume was measured at 3 days and 3 weeks after CCI injury. Animals were deeply anesthetized and then transcardially perfused with heparin saline followed by 4% formaldehyde. Brains were collected and fixed in the same fixative for 24–48 h and then in 30% sucrose for 24–48 h. Sections 30 μm thick were cut using a cryostat (Leica) starting at 600 μm anterior to the bregma. Serial sections at 540-μm intervals were stained with hematoxylin and eosin (H&E) and scanned with an Epson scanner. Each section was measured for the area of normal H&E staining with ImageJ software from the National Institutes of Health. Cavitation, hemorrhage, or loss of normal H&E staining were considered as lesions. The lesion area was calculated as the area of the contralateral side minus that of the ipsilateral side. The lesion volume of a given animal was calculated according to a previous report with slight modification (Dash et al., 2010), where A stands for the lesion area (mm2) for each slice, and X stands for the distance (mm) between two sequential slices: {0.5A1+0.5(A1+A2)+…+0.5(An−1+An)+0.5An} X.
Fluoro-Jade B staining
Fluoro-Jade B (FJB) is a fluorescent dye that can be used to label degenerating neurons specifically. This makes it a valuable tool for evaluating neuronal degeneration after TBI (Hall et al., 2008). To perform FJB staining, serial sections at 540-μm intervals were first immersed in a solution containing 1% sodium hydroxide in 80% ethanol for 5 min followed by 2 min in 70% ethanol. Brain sections were then transferred to a solution of 0.06% potassium permanganate and gently shaken for 10 min. After immersion in a 0.0004% FJB solution, the sections were air-dried and cleared in xylene for at least 1 min before mounting with DPX (Sigma-Aldrich). At 20× magnification the FJB-positive cells were counted in the hippocampal dentate gyrus area of all sections from a given animal.
Immunohistochemical analysis
To assess the inflammatory response after injury, sections were first blocked with 5% normal donkey serum, then incubated overnight at 4°C with a monoclonal rat anti-mouse F4/80 antibody (1:50; Abcam, Cambridge, MA), or a polyclonal rabbit anti-COX-2 antibody (1:100; Cayman Chemical, Ann Arbor, MI). The sections were then reacted for 1 h at room temperature with a FITC-conjugated donkey anti-rat antibody (1:100; Jackson ImmunoResearch Laboratories, West Grove, PA), or a Cy 3-conjugated donkey anti-rabbit secondary antibody (1:100; Jackson ImmunoResearch Laboratories). Finally, the sections were stained with 4,6-diamino-2-phenylindole (DAPI; Sigma-Aldrich), and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Sections were visualized via fluorescence microscopy (Olympus, Center Valley, PA). Cell density was measured according to a method previously reported (Clausen et al., 2009; Shein et al., 2009) with slight modifications. Briefly, immunostaining of three sections approximately 50 μm apart at bregma −2.0 mm level from a given animal was performed. For each section, three high-power (40×) photomicrographs were taken in the area adjacent to the injury site. For COX-2 immunostaining, photomicrographs were taken in layer II of the cortex adjacent to the injury site. Positive cells were identified and counted using Adobe Photoshop (Mountain View, CA) software and averaged for a given animal. Cell counting was performed by an observer blinded to treatment status.
Double-fluorescent staining
To study cell-type-specific expression of COX-2, we double-fluorescence stained COX-2 with neuronal nuclear antigen (NeuN for neurons), glial fibrillary acidic protein (GFAP for astrocytes), and F4/80 (for microglia/macrophages). Briefly, the sections were incubated overnight at 4°C with a polyclonal rabbit anti-COX-2 antibody (1:100; Cayman Chemical), together with a biotin conjugated anti-NeuN antibody (1:100; Millipore, Billerica, MA), an Alexa Fluor 488-conjugated mouse GFAP antibody (1:100; Cell Signaling, Danvers, MA), or a monoclonal rat anti-mouse F4/80 antibody (1:50; Abcam). The sections were then incubated with the respective secondary antibodies at room temperature for 1 h. Finally, the sections were stained with DAPI (Sigma-Aldrich) and mounted with Vectashield mounting medium. Sections were visualized via fluorescence microscopy (Olympus) for staining. A confocal laser microscope (Zeiss LSM 510; Carl Zeiss, Oberkochen, Germany) was used to verify the co-localization of NeuN and COX-2 at a thinner slice layer.
Western blotting analysis
Western blotting was performed as previously described (Kim et al., 2007). Brain tissue was collected from the cortex posterior to the bregma on the injury side and sonicated for 30 sec in lysis buffer (T-PER tissue protein extraction reagent; Thermo Scientific, Rockford, IL) with phosphatase inhibitors I and II (Sigma-Aldrich), and protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). The lysates were centrifuged at 12,000 rpm for 20 min. Protein concentrations were determined using the BCA method. An aliquot containing 30 μg of protein was loaded to each lane, and proteins were separated by electrophoresis on SDS-polyacrylamide gels of 4–12% (Invitrogen, Carlsbad, CA), followed by transferring to a polyvinylidene difluoride membrane. The membranes were incubated overnight at 4°C with monoclonal rabbit anti-phospho-GSK-3β (1:500; Cell Signaling), monoclonal rabbit anti-total GSK-3β (1:2,000; Cell Signaling), monoclonal rabbit anti-MMP-9 (1:1000; Origene, Rockville, MD), polyclonal rabbit anti-phospho-Akt (1:1000; Cell Signaling), polyclonal rabbit anti-Akt (1:2,000; Cell Signaling), or monoclonal mouse anti-β-actin (1:5000; Sigma-Aldrich) antibodies. The membranes were then incubated with goat anti-rabbit IRDye 800CW-conjugated or goat anti-mouse IRDye 700CW-conjugated secondary antibodies and scanned with an Odyssey machine (LI-COR Biosciences, Lincoln, NE).
Evans blue extravasation
With slight modifications, we used Evans blue extravasation to evaluate the integrity of the BBB (Wang et al., 2011). At 68 h after TBI, 2% Evans blue (Sigma-Aldrich) in saline at 5 mL/kg of body weight was administered through the jugular vein. Four hours later, mice were transcardially perfused with heparin saline to remove the intravascular Evans blue. Both ipsilateral and contralateral hemispheres of the brain were weighed and then homogenized individually in 0.5 mL of 50% trichloroacetic acid. The lysates were further centrifuged at 4000g for 40 min. The supernatant was measured by spectrophotometry at a wavelength of 620 nm. The results were calculated using its standard curve and expressed as μg/g of tissue.
Beam-walk test
To evaluate fine motor coordination, a beam-walk test was performed as previously described (Fox et al., 1998; Loane et al., 2009). The beam-walk device contains a narrow wooden beam 6 mm in width and 120 cm in length suspended about 30 cm above a table. Before surgery, each mouse was trained to walk from one end of the beam to the other. The number of foot faults for the right hindlimb was counted over 50 steps. Animals with a basal level of < 10 faults per 50 steps were selected for the experiment. The beam-walk test was performed on days 1, 3, 7, 14, and 21 post-injury, and the number of foot faults per 50 steps was recorded.
Rotarod test
A rotarod test was used to evaluate motor coordination. For four consecutive days before surgery, the mice received training on a rotarod device (Ugo Basile, Collegeville, PA) with an accelerating protocol. Briefly, the rotarod was accelerated from 4 to 40 rpm in 4 min and maintained at 40 rpm for 1 min. Latency to fall from the device or to cling and rotate for two full rotations was recorded. After surgery, rotarod performance was evaluated on days 1, 3, and 7 post-injury with four trials each day; the animals were given at least 5 min to rest between trials. The best performance of the four trials on each day was recorded as previously reported (Walker et al., 2010).
Open-field test
To evaluate gross motor function, the animals underwent an open-field test 10 days post-TBI. Each mouse was gently placed in the center of an acrylic arena (40×40 cm with black walls) and allowed to explore freely for 10 min. The inner square of 20×20 cm was defined as the center zone, while the remainder of the structure was defined as the peripheral zone. The total distance animals traveled and the time spent in the center zone were video-recorded and analyzed using ANY-Maze software (Stoelting Co., Wood Dale, IL). The open field test was performed on the same group of animals subjected to the beam-walk test under the 2.0 mm condition. All other behavioral tests were performed on different cohorts of animals.
Statistical analysis
All data analyses were carried out using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). Two-way repeated measures analysis of variance (ANOVA) with post-hoc Bonferroni comparison was used to analyze results of the beam-walk and rotarod tests. One-way ANOVA was used to compare data among multiple groups, and Student's t-test was used to compare between two groups. Results were quantified and expressed as mean±standard error of the mean (SEM). Statistical significance was defined as p<0.05.
Results
Characterization of combinations of velocity and different levels of deformation on the severity of TBI
To characterize the injury severity caused by different levels of impact, we tested four different CCI conditions with velocity at 5 m/sec and various levels of deformation: 1.0, 1.5, 2.0, and 2.5 mm (Fig. 1A and C). In the 2.5-mm injury condition, a lesion was noted in the cortex and the entire hippocampus on the ipsilateral side, as detected by H&E staining. In the 1.5-mm and 2.0-mm injury conditions, a lesion was observed in the cortex and part of the ipsilateral hippocampus. By contrast, in the 1.0-mm condition, a lesion occurred only in the cortex but not in the hippocampus. We chose the 1.5-mm and 2.0-mm injury conditions for our study. The 1.5-mm condition was mainly used for short-term (3 days) experiments, as better tissue preservation was observed compared with the 2.0-mm condition. For long-term (3 weeks) studies and most behavioral tests, we selected the 2.0 mm condition as the optimal injury, because it consistently induced hyper-locomotor activity and anxiety-like behavior in the open-field test.
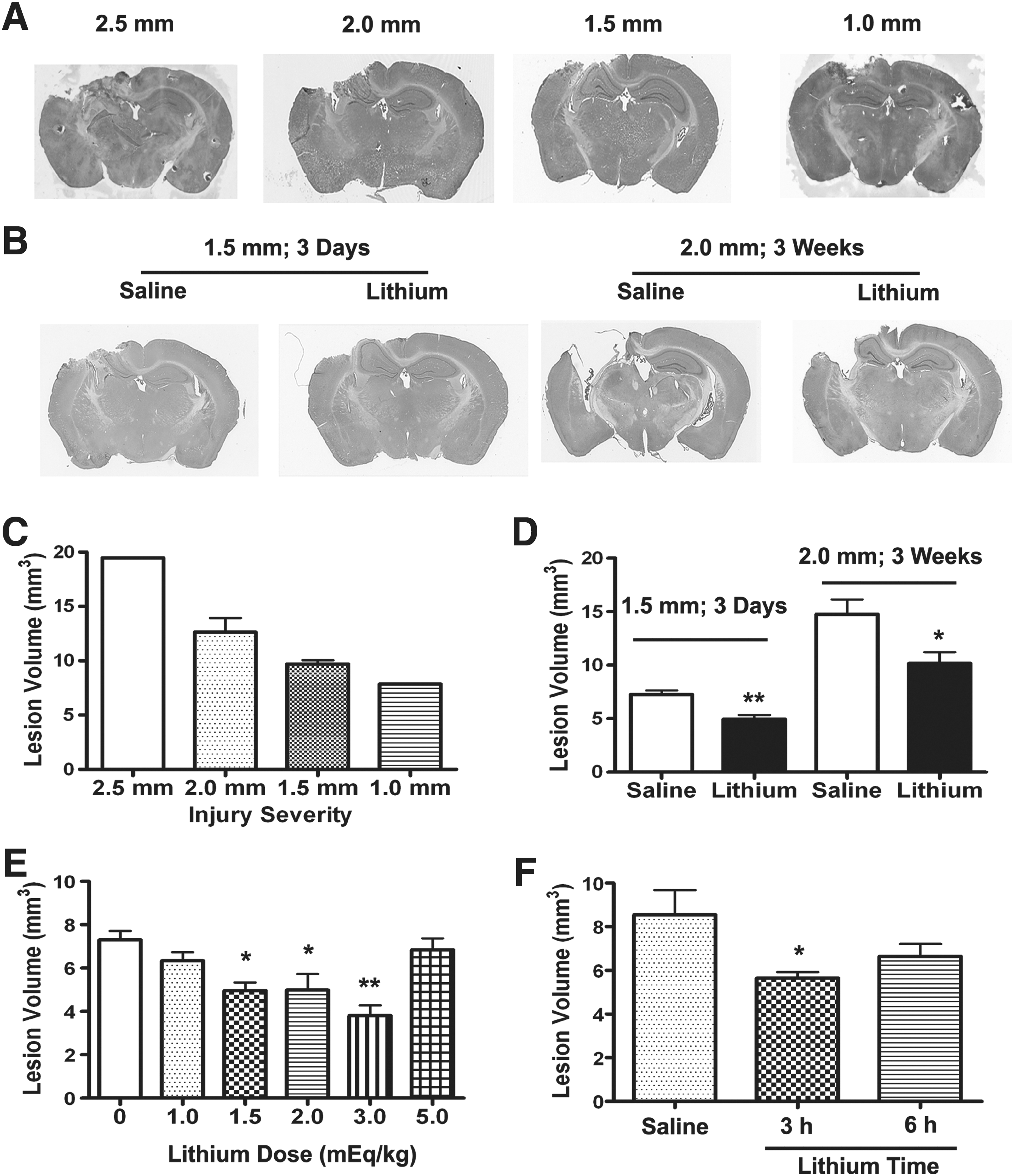
Post-insult lithium treatment reduces controlled cortical impact (CCI)-induced brain lesion in traumatic brain injury (TBI) mice. Hematoxylin and eosin
Post-insult lithium treatment reduces TBI-induced brain lesions in mice
To test whether post-insult lithium treatment reduces TBI-induced brain lesions, we evaluated lesion volume with H&E staining at 3 days post-injury in the 1.5 mm condition, and 3 weeks after injury in the 2.0 mm condition. In the first test (3 days post-injury in the 1.5 mm condition), daily injection with 1.5 mEq/kg lithium chloride decreased lesion volume from 7.26±0.38 mm3 in the saline-treated group to 4.96±0.38 mm3 in the lithium-treated group (Fig. 1B and D ; p<0.01). Similarly, at 3 weeks after injury in the 2.0 mm condition, post-insult lithium treatment for 2 weeks reduced lesion volume from 14.76±1.39 mm3 in the saline-treated group to 10.18±1.03 mm3 in the lithium-treated group (Fig. 1B and D ; p<0.05). In both conditions, no lesion was found in the contralateral cerebral hemisphere. The dose-response study showed that lithium at doses of 1.5, 2.0, and 3.0 mEq/kg, all significantly reduced lesion volume at 3 days after injury, while lithium did not reduce lesion volume at doses of 1.0 or 5.0 mEq/kg (Fig. 1E). The time-course study showed that lithium still significantly reduced lesion volume when administered 3 but not 6 h post-injury (Fig. 1F). Therefore, the lowest effective dose (1.5 mEq/kg) was chosen for this study. The plasma lithium level was 0.14±0.01 mEq/mL 12 h after the last of 2-week lithium injections (n=4).
Lithium treatment decreases the number of degenerating neurons in the dentate gyrus of TBI mice
To evaluate neurodegeneration in the hippocampus, we used FJB staining, a valuable tool for measuring neuronal degeneration after TBI (Hall et al., 2008). The hippocampus is a vulnerable area to central nervous system (CNS) injuries, and selective death of newborn neurons in the hippocampal dentate gyrus has been reported after TBI (Gao et al., 2008). Three days after TBI, the number of FJB-positive cells in the dentate gyrus was significantly reduced in the lithium-treated group, compared with the saline-treated group. The average number of FJB-positive cells in the dentate gyrus was 232±34 in the saline-treated group, and 98±4 in the lithium-treated group (Fig. 2; p<0.05). No FJB-positive cells were found in the dentate gyrus of the sham-injured animals.
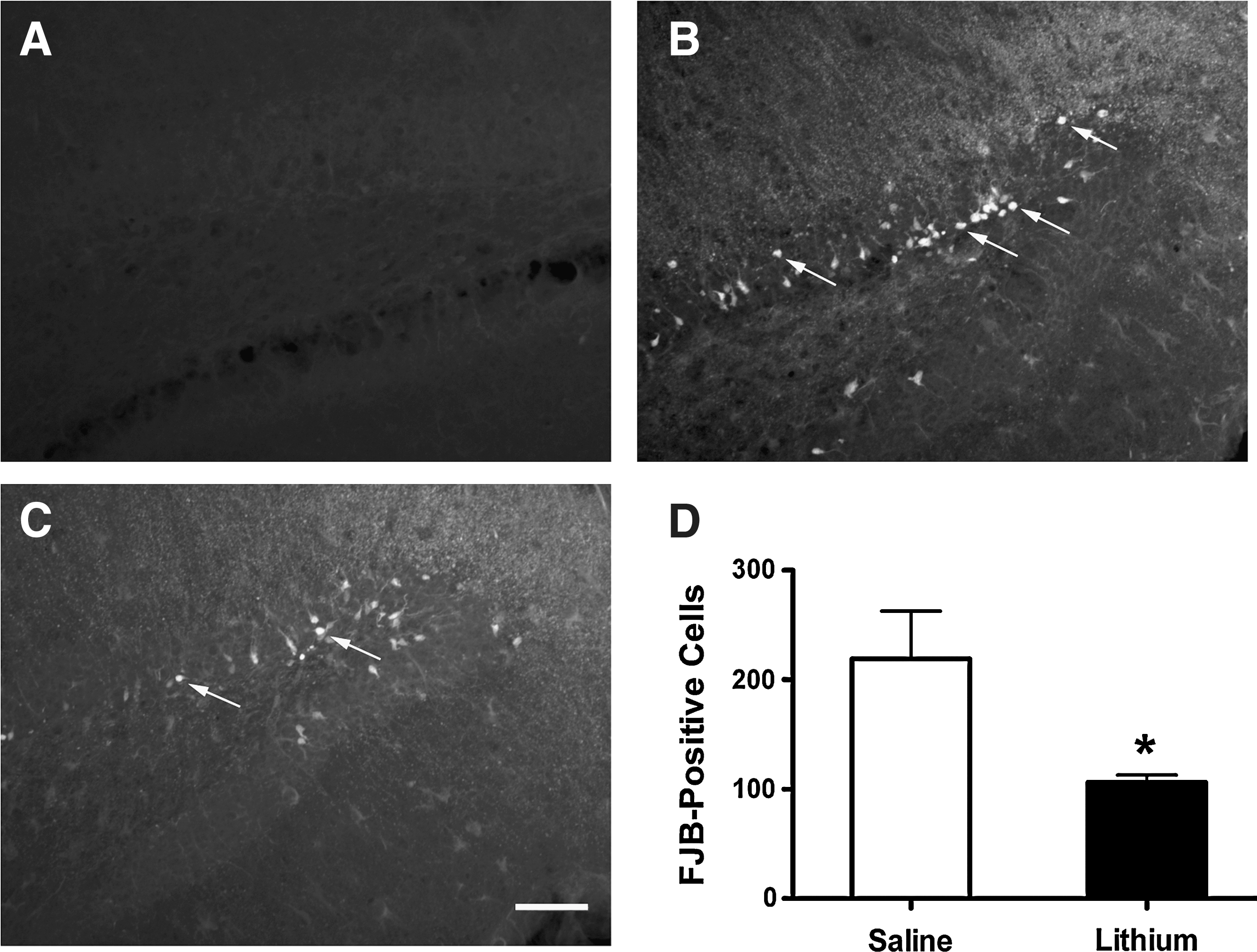
Post-insult lithium treatment decreases the number of degenerating neurons in the dentate gyrus of traumatic brain injury (TBI) mice. Fluoro-Jade B (FJB) staining was performed to evaluate neuronal degeneration. No FJB-positive cells were observed in the dentate gyrus of the sham-injured group (
Lithium suppresses TBI-induced neuroinflammation
Recognizing that neuroinflammation plays an important role in the pathogenesis of TBI, we used F4/80 immunostaining to study lithium's effect on microglial activation. In sham-injured animals, the ipsilateral cortex contained no positive cells for F4/80 (Fig. 3B). By contrast, in animals measured 3 days post-TBI, the levels of F4/80-labelled microglia/macrophages around the injury site in the ipsilateral cortex increased markedly. In the saline-treated group, average microglia/macrophage cell density rose to 466±32 cells/mm2 (Fig. 3C and 3I). In animals treated with lithium, however, levels of microglia/macrophages declined to 228±26 cells/mm2, or less than half of the levels in the untreated group (Fig. 3D and I; p<0.01).
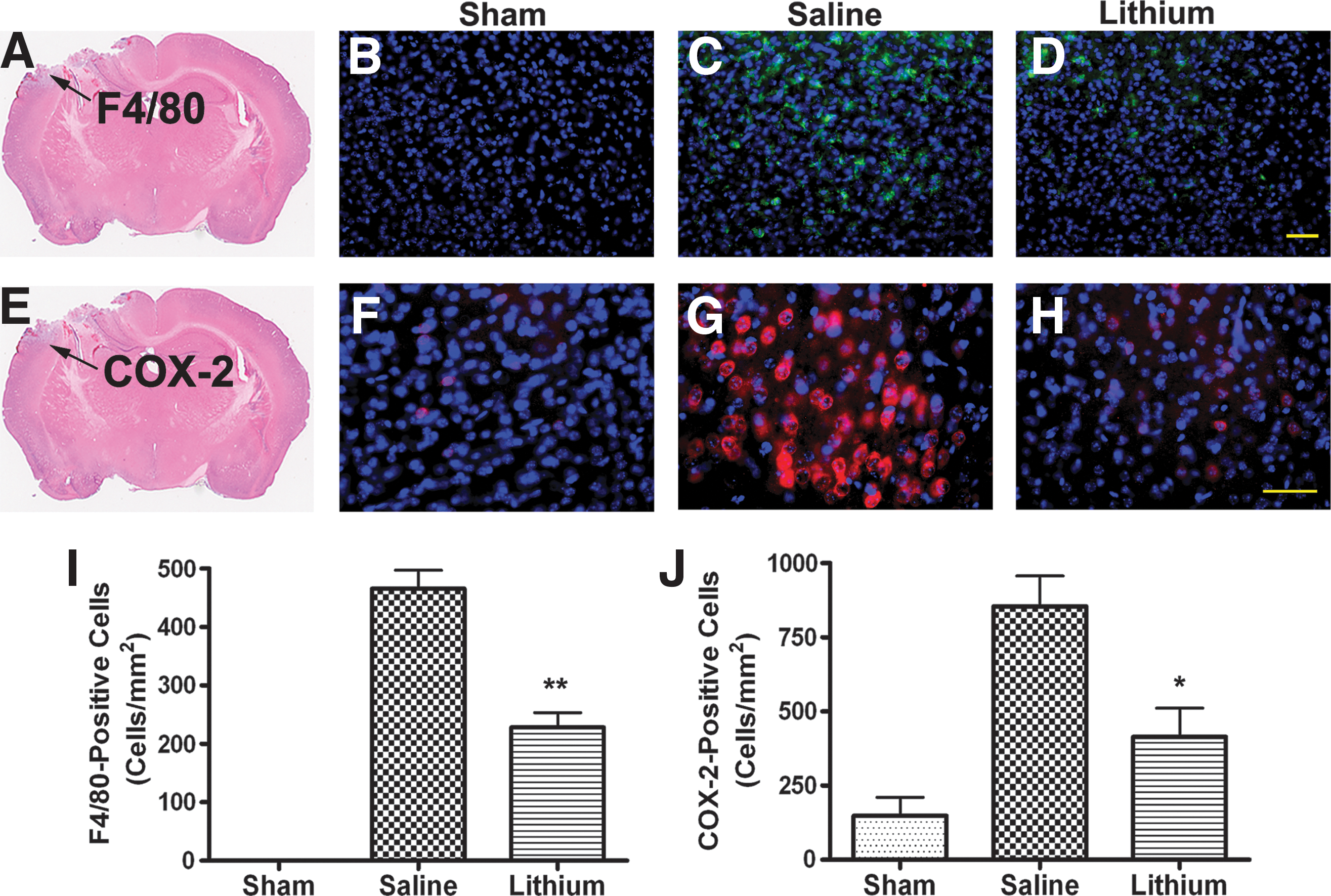
Lithium suppresses microglial activation and cyclooxygenase-2 (COX-2) expression in the cortex of traumatic brain injury (TBI) mice. Representative photomicrographs of F4/80 staining for microglia activation (
COX-2 is the rate-limiting enzyme that catalyzes the metabolism of arachidonic acid into inflammatory prostaglandins (Andreasson, 2010). When compared with the sham-injured group (Fig. 3F), TBI robustly increased the expression of COX-2 in the cortex adjacent to the injury site (Fig. 3G), while in the lithium-treated group, the expression of COX-2 was suppressed (Fig. 3H). In the saline-treated group, the average density of COX-2-positive cells in the cortex area was 855±103 cells/mm2, while in the lithium-treated group, it decreased to 414±97 cells/mm2 (Fig. 3J; p<0.05).
To further explore cell-type-specific expression of COX-2, we performed double-fluorescence staining using COX-2 and selective markers from different cell types. We found that COX-2 was mainly expressed in neurons located in the area adjacent to the injury site (Fig. 4 upper panel), and there was no co-localization of COX-2 with markers for astrocytes (Fig. 4 lower panel and Supplementary Fig. 1; see online supplementary material at

A majority of cyclooxygenase-2 (COX-2)-positive cells are neuronal nuclear antigen (NeuN)-positive neurons. Representative photomicrographs of double immunofluorescence staining of COX-2 with NeuN or glial fibrillary acidic protein (GFAP) are shown. Upper panels: Double staining of COX-2 (red;
Lithium attenuates BBB breakdown and MMP-9 expression after TBI
To investigate BBB integrity, we detected Evans blue extravasation at 3 days after TBI. The extravasation of Evans blue in ipsilateral brain tissue was markedly increased in the saline-treated group (4.94±0.23 μg/g tissue), and this was significantly lower in the lithium-treated group (3.29±0.36 μg/g tissue; Fig. 5A; p<0.01). In the contralateral side of the brain there was only a small amount of Evans blue, with no significant difference between the saline-treated (1.49±0.19 μg/g tissue) and lithium-treated (1.26±0.10 μg/g tissue) groups. We further examined protein levels of MMP-9, an enzyme largely responsible for the degradation of the extracellular matrix proteins that maintain BBB integrity (Rosell et al., 2008). In the ipsilateral cortex 3 days after TBI, levels of MMP-9 in saline-treated animals were enhanced by more than twofold compared with sham-injured animals (Fig. 5B; p<0.05). By contrast, lithium treatment completely blocked this increase (Fig. 5B; p<0.05).
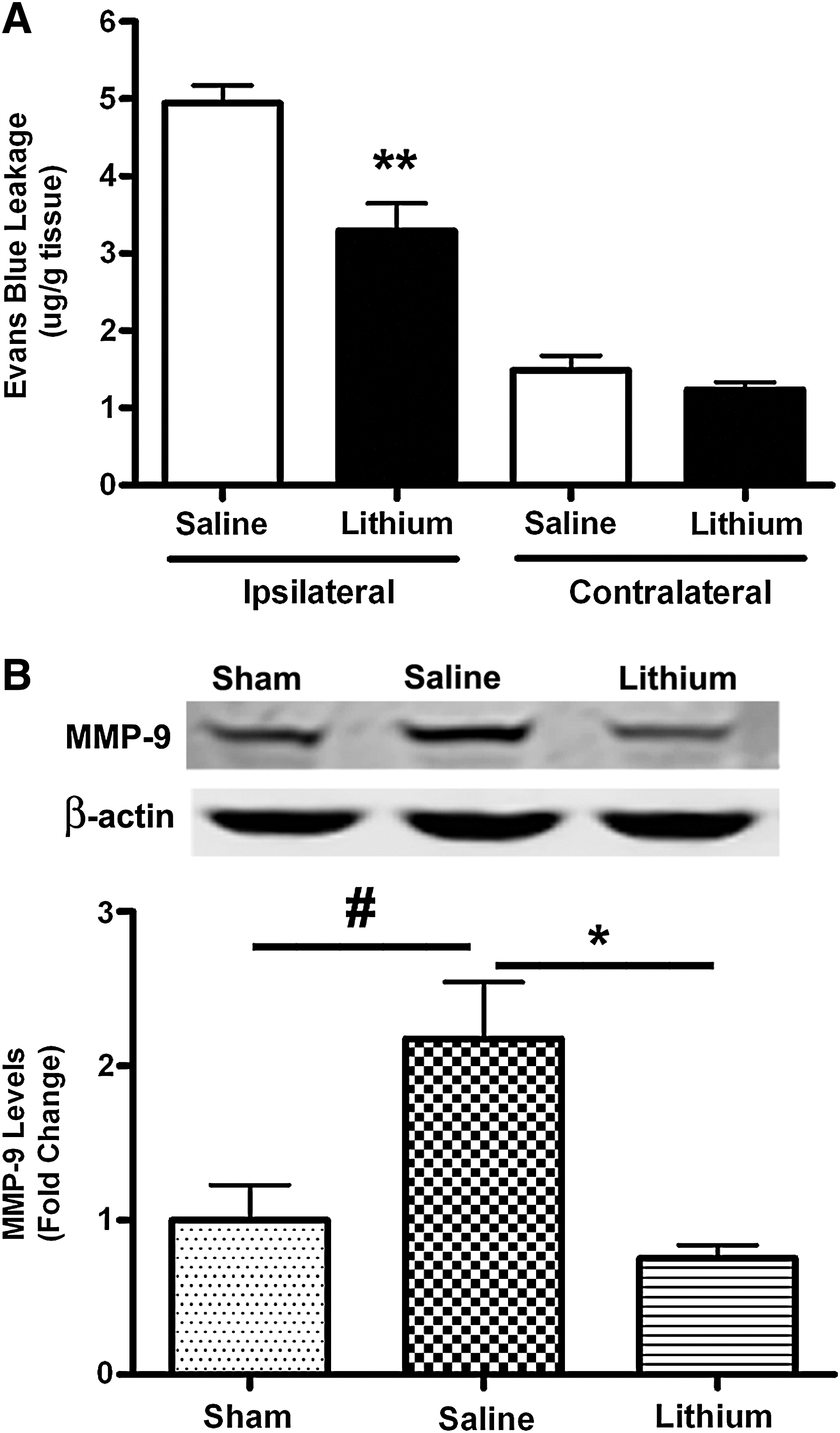
Lithium reduces blood–brain barrier (BBB) disruption and matrix metalloproteinase-9 (MMP-9) expression after traumatic brain injury (TBI). BBB integrity was evaluated by Evans blue extravasation (
Lithium improves motor coordination after TBI
To evaluate fine motor movement, the beam-walk test was performed for the two selected injury conditions. In both cases, TBI induced a significant increase in the number of foot faults, with a trend toward recovery in both the lithium- and saline-treated groups. At 7 days after injury in the 1.5-mm condition, the number of foot faults was significantly reduced, from 46±3 in the saline-treated group to 31±3 in the lithium-treated group (Fig. 6A; p<0.01). At 14 days after injury, the number of foot faults was markedly reduced, from 34±3 in the saline-treated group to 17±2 in the lithium-treated group (Fig. 6A; p<0.01). At 21 days after injury, the number of foot faults was significantly decreased, from 35±3 in the saline-treated group to 13±3 in the lithium-treated group (Fig. 6A; p<0.01). Similar results were also found in TBI induced under the 2.0-mm CCI condition (Fig. 6B). The foot faults were 44±4 (saline) and 30±4 (lithium) at 14 days post-TBI, and were 44±2 (saline) and 29±4 (lithium) at 21 days post-TBI (Fig. 6B; p<0.01). Therefore treatment with lithium demonstrably improves and speeds recovery of fine motor coordination.
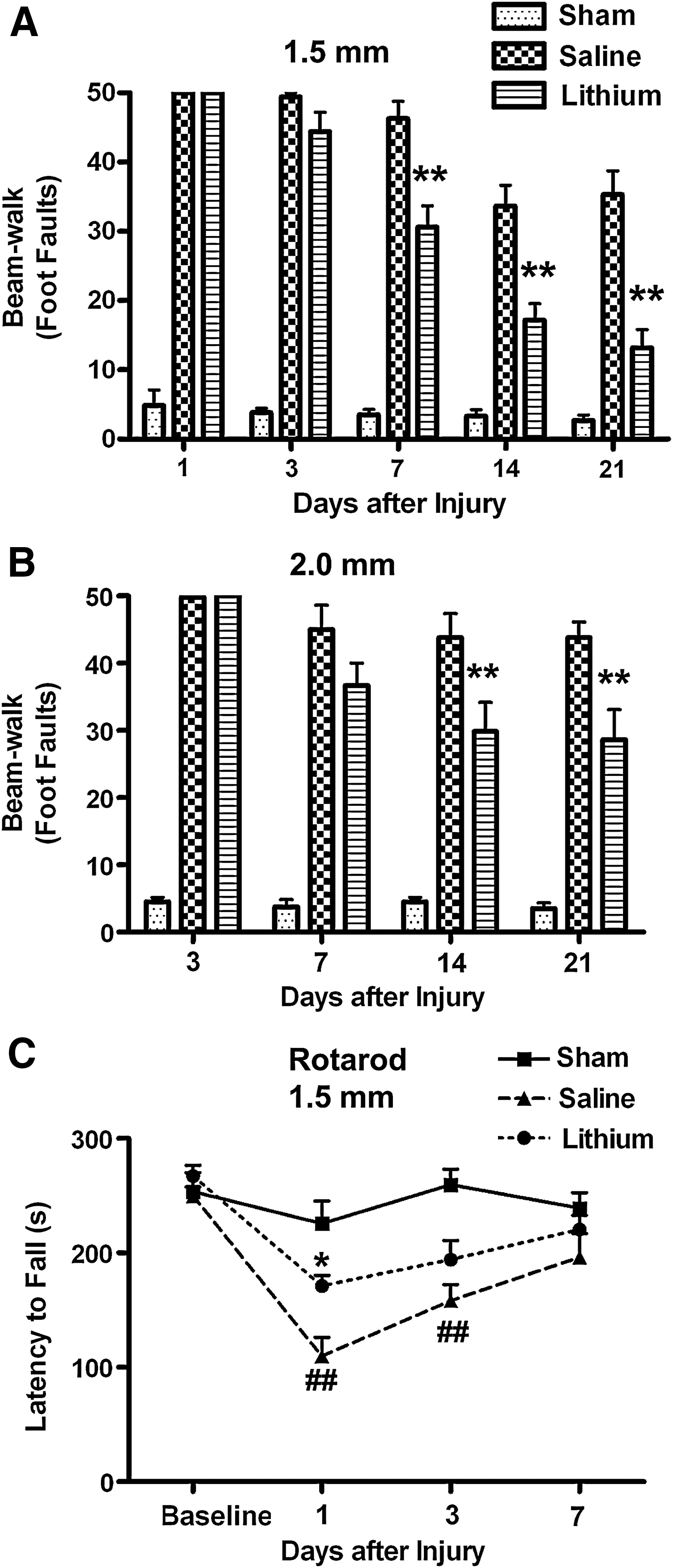
Lithium markedly improves motor coordination in traumatic brain injury (TBI) mice. TBI increased the number of foot faults in both the saline- and lithium-treated groups under the condition of 1.5 mm (
The rotarod test was used to evaluate motor coordination in the 1.5 mm CCI paradigms of TBI. At 1 day after injury, lithium treatment significantly improved motor coordination by increasing average latency to fall from 109.7±16.41 sec in the saline-treated group to 171.0±9.47 sec in the lithium treated group (Fig. 6C; p<0.05). Note that CCI-induced deficits in rotarod performance at 1 and 3 days post-injury were largely recovered at 7 days after injury (Fig. 6C; p<0.01).
Lithium attenuates TBI-induced hyper-locomotor activity and anxiety-like behaviors
The open-field test was used to evaluate gross motor function and anxiety-like behaviors in the 2.0 mm injury condition 10 days after injury. We found that after TBI, mice traveled a significantly longer distance (34.89±2.26 m) than those in the sham-injured group (26.47±2.25 m; Fig. 7B; p<0.05), but lithium treatment robustly reduced the distance traveled of the injured mice to nearly the same level as that of the sham-injured group (27.76±1.70 m; Fig. 7B; p<0.05). To measure the anxiety levels in TBI mice, we tracked the amount of time they spent in the center zone, since spending less time in the center zone has been shown to indicate an anxiety-like behavior in mice (Prut and Belzung, 2003). We found that TBI mice spent less time (21.45±3.29 sec) in the center zone than did sham-injured mice (41.37±4.50 sec; Fig. 7C; p<0.01). Lithium treatment significantly increased the time TBI mice spent in the center zone, to 32.33±3.32 sec (Fig. 7C; p<0.05). These results support the finding that lithium attenuates hyper-locomotor activity and anxiety-like behaviors after TBI.
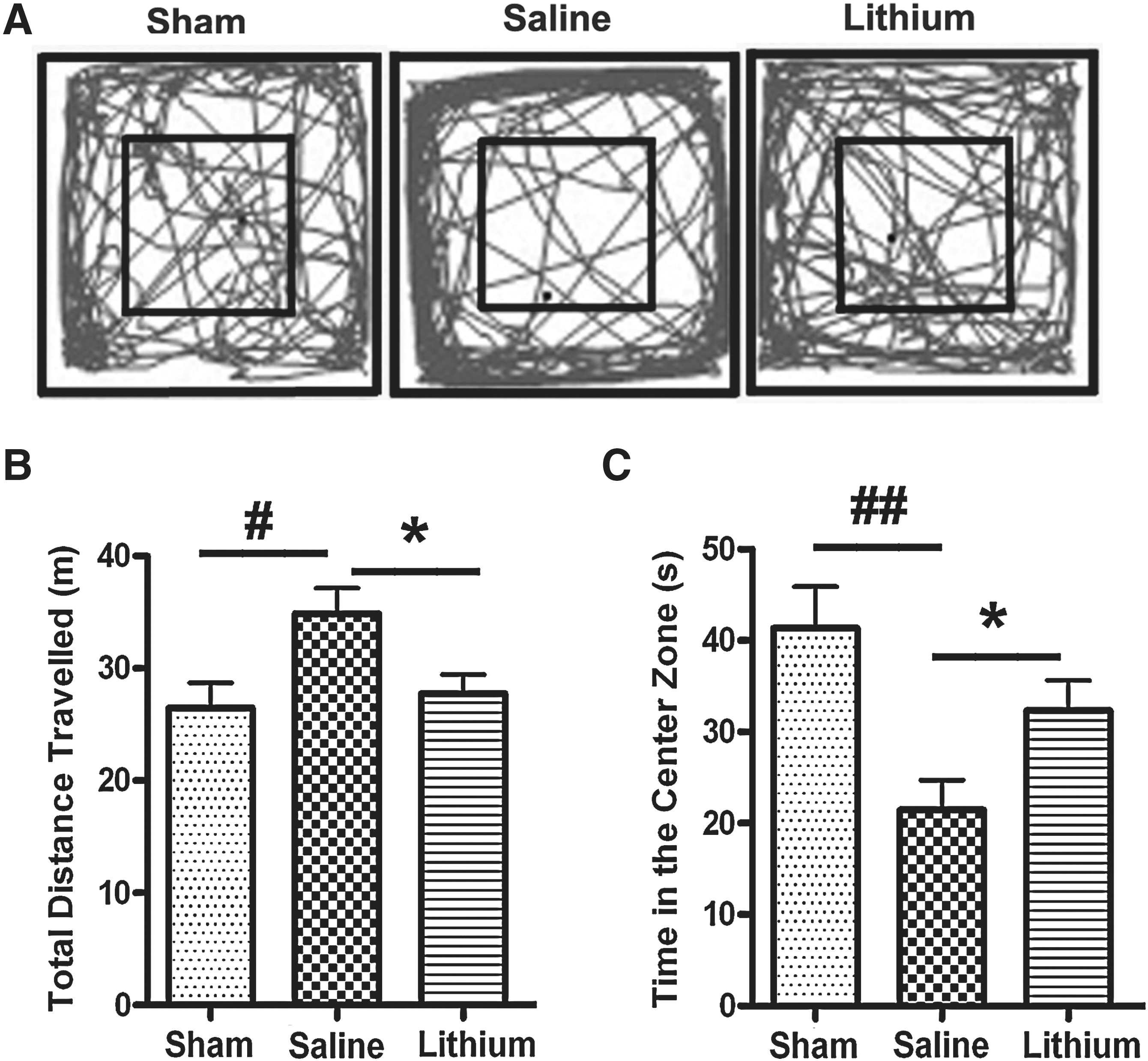
Lithium attenuates hyper-locomotor activity and anxiety-like behaviors. Representative movement tracks in the open-field test 10 days post-injury in the 2.0 mm group are shown (
Lithium increases GSK-3β phosphorylation after TBI
Mounting evidence supports the observation that lithium inhibits GSK-3β, both directly and indirectly, and that GSK-3 inhibition underlies much of the actions and effects of this drug (Chiu and Chuang, 2010; Li and Jope, 2010). To study the effects of lithium and the role of GSK-3β inhibition, we used Western blotting to assess the phosphorylation of GSK-3β (p-GSK-3β) at 3 days after TBI. Although there was no significant difference in the levels of p-GSK-3β at Ser 9 between the TBI and sham-injured groups, lithium treatment induced a more than threefold increase in the serine phosphorylation of GSK-3β (Fig. 8A; p<0.05), while protein levels of total GSK-3β (t-GSK-3β) were unchanged (Fig. 8A). By examining the phosphorylation of Akt (p-Akt), one of the upstream serine/threonine kinases that phosphorylate and inhibit GSK-3, we found a significant increase in p-Akt at Ser 473 in the lithium-treated group compared with the sham-injured group (Fig. 8B; p<0.05), but no significant difference between the sham-injured and the saline-treated groups was observed. In all groups, the protein levels of total Akt (t-Akt) were also unaltered (Fig. 8B). These results suggest that the phosphorylation and subsequent inhibition of GSK-3β likely contribute to lithium's beneficial effects against TBI.
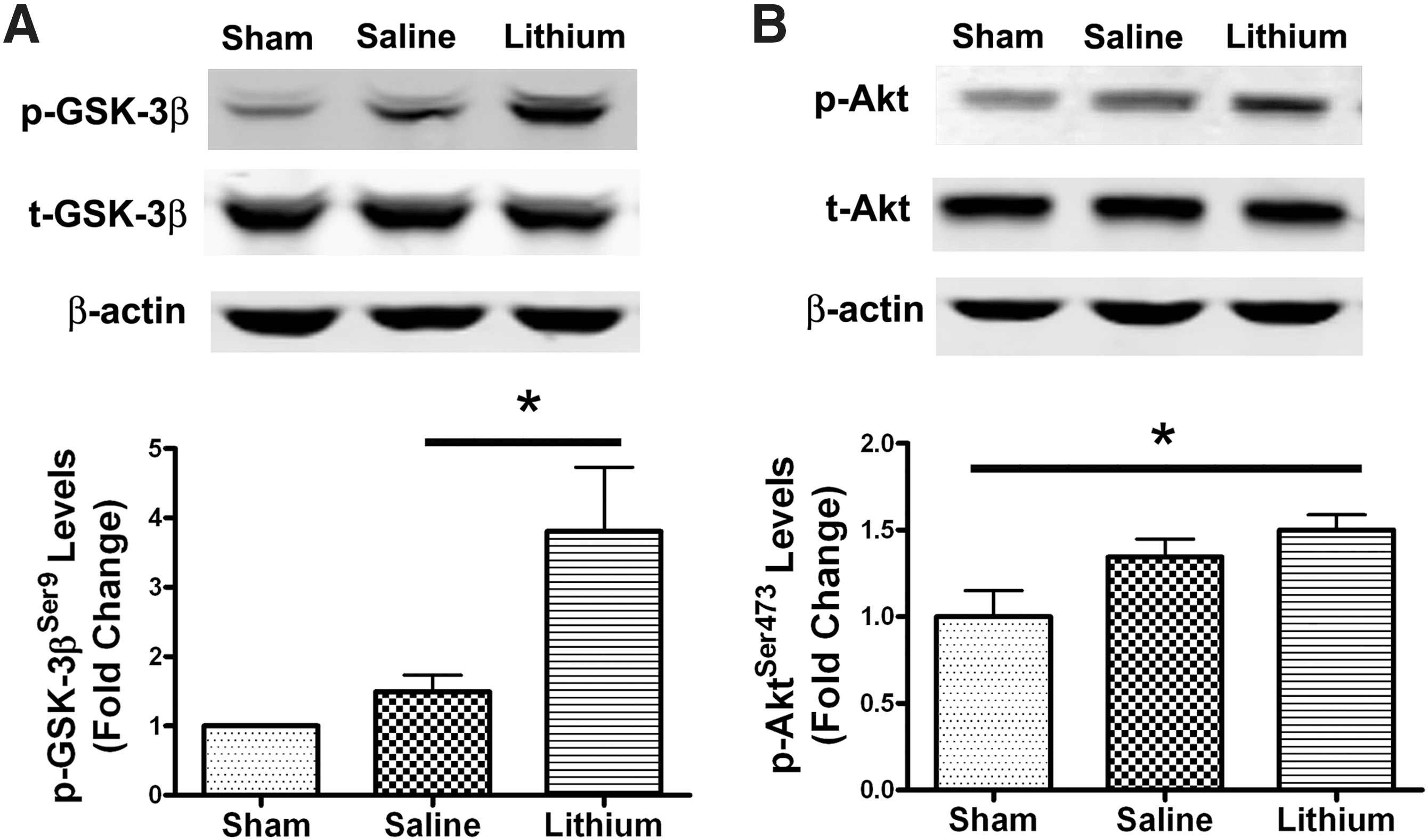
Lithium increases glycogen synthase kinase-3β (GSK-3β) phosphorylation after traumatic brain injury (TBI). Representative Western blots showed that lithium significantly increased GSK-3β Ser 9 phosphorylation levels 3 days after TBI (
Discussion
To our knowledge, this is the first study to demonstrate that post-insult treatment with lithium has beneficial effects in a mouse model of TBI. Specifically, we found that IP delivery of lithium (starting at 15 min after injury and followed by daily injection for 3 days or 2 weeks) significantly reduced lesion volume at 3 days and 3 weeks, and attenuated neuronal degeneration in the hippocampal dentate gyrus at 3 days after CCI-induced injury. Lithium treatment also suppressed activation of microglia, induction of COX-2, and disruption of the BBB at 3 days after TBI, giving further proof of the ability of lithium to suppress neuroinflammation after TBI. Finally, the improved performance on the beam-walk, rotarod, and open-field tests indicates that lithium treatment also improved behavioral outcomes following TBI.
Chronic pre-insult treatment followed by post-insult treatment with lithium has also been shown to reduce lesion volume, ameliorate hippocampal neurodegeneration, attenuate levels of proinflammatory interleukin-1β (IL-1β), and improve Morris water maze performance in a mouse model of TBI (Zhu et al., 2010). However, without long-term pre-insult treatment, post-insult treatment with lithium at a dose of 1.0 mEq/kg failed to show beneficial effects in TBI (Zhu et al., 2010). In contrast, our dose-response study showed that post-insult treatment with lithium at doses of 1.5, 2.0, and 3.0 mEq/kg significantly reduced TBI-induced lesion volume. In addition, treatment with lithium at a dose of 5.0 mEq/kg produced no benefit in reducing lesion volume. These results are consistent with the preclinical and clinical observation that lithium has a relatively narrow dose window, and therefore the lack of neuroprotection at supratherapeutic doses, such as 5 mEq/kg, could be related to its toxic effects (Chiu and Chuang, 2010).
It is well known that neuroinflammation contributes to the pathogenesis of TBI (Loane and Faden, 2010). To evaluate lithium's effect on neuroinflammation, we focused on microglial activation and COX-2 expression. Microglia, which play an important role in the inflammatory response to injury or infection in the brain (Yuskaitis and Jope, 2009), have been found in areas surrounding the injury site from 3 days to 8 weeks after TBI (Yu et al., 2010). Attenuation of microglia activation via inhibition of interleukin-1β (IL-1β) and histone deacetylase inhibition contributes to the neuroprotective effect against TBI (Clausen et al., 2009; Shein et al., 2009). COX is the rate-limiting enzyme responsible for the generation of prostaglandins, which are important mediators of inflammation (Andreasson, 2010). COX-2 is an inducible form of COX expressed in glutamatergic neurons in normal conditions (Niwa et al., 2000). COX-2 is markedly upregulated in animal models of TBI (Ahmad et al., 2008; Kunz et al., 2002, 2006). However, the effects of COX-2 inhibition after TBI are inconsistent. For example, pharmacological inhibition of COX-2 with nimesulide was reported to have beneficial effects against TBI (Cernak et al., 2002), whereas inhibition of this enzyme with another selective inhibitor rofecoxib showed no significant neuroprotection, despite a reduction in the number of TUNEL-positive cells (Kunz et al., 2006). In addition, genetic knockdown of COX-2 had only minor effects in sparing cortical tissue, and had no effect on behavioral outcome after TBI (Kelso et al., 2009). Another report also showed that genetic disruption of COX-2 did not change the lesion volume or alter Morris water maze performance after TBI (Ahmad et al., 2008). The results of the present study suggest that lithium's ability to attenuate microglia activation and COX-2 upregulation may help to reduce neuronal damage following TBI.
BBB disruption, which occurs in hours to days after TBI, is associated with neuroinflammation and cell death in both animal models and patients with head trauma (Shlosberg et al., 2010). Attenuation of BBB disruption in animal models of TBI, spinal cord injury, and cerebral ischemia has been shown to be beneficial (Lotocki et al., 2009; Wang et al., 2011; Yu et al., 2008). The permeability of BBB is increased by MMP-9, a major form of gelatinase that degrades extracellular-matrix and tight-junction proteins (Rosell et al., 2008). The MMP-9 gene promoter contains putative nuclear factor-κB (NF-κB) p65 binding sites, and inhibition of NF-κB following ischemic insult blocks MMP-9 gene expression (Cheng et al., 2006; Van den Steen et al., 2002; Wang et al., 2011). In the same way, lithium administered following TBI might induce suppression of neuroinflammation by inhibiting activation of NF-κB, which in turn reduces the upregulation of MMP-9 and disruption of the BBB.
TBI frequently results in behavioral deficits in motor and cognitive function (Fujimoto et al., 2004). In our study, lithium markedly suppressed both short-term and long-term motor deficits in rotarod and beam-walk performance after TBI. Lithium attenuated anxiety-like behavior, as measured by the time spent in the center zone in an open-field test. Our results in the open-field test further showed that lithium treatment attenuated TBI-induced hyper-locomotor activity, a behavior generally interpreted as a sign of depression in mice (Pandey et al., 2009). Accumulating evidence suggests that the hippocampal dentate gyrus is involved in neurogenesis and regulation of behaviors associated with anxiety and depression in rodents (Adachi et al., 2008; Omata et al., 2011). In the present study, we found that lithium attenuated neuronal degeneration in the hippocampal dentate gyrus after TBI. We therefore suggest that the apparent anxiolytic and antidepressant-like effects of lithium stem from its ability to attenuate neurodegeneration in this brain region.
GSK-3, consisting of α and β isoforms, is a serine/threonine kinase involved in diverse cellular functions (Chiu and Chuang, 2010; Li and Jope, 2010; Rowe et al., 2007). Serine phosphorylation of GSK-3β or GSK-3α negatively regulates GSK-3 activity (Chiu and Chuang, 2010; Jope, 1999, 2003; Li and Jope, 2010). GSK-3β inhibition or gene silencing effectively suppresses glutamate excitotoxicity in neuronal cultures (Liang and Chuang, 2007). In vivo studies found that GSK-3 inhibition reduces infarct volume and improves neurological deficits after cerebral ischemia in rodents (Koh et al., 2008). In a mouse model of TBI, GSK-3 inhibition by lithium is also beneficial in alleviating TBI-induced depressive behavior (Shapira et al., 2007). The effects of GSK-3 inhibition by lithium may be attributable to enhanced expression of important neuroprotective proteins such as heat shock protein 70 (HSP70), BDNF, and Bcl-2 (Liang and Chuang, 2006; Ren et al., 2003; Senatorov et al., 2004; Yasuda et al., 2009). GSK-3 inhibition promotes the activation of cell survival transcription factors, including cyclic adenosine monophosphate response element-binding protein (CREB), AP-1, β-catenin, and heat-shock factor-1 (Bijur and Jope, 2000), and to downregulate the pro-apoptotic protein p53 and Bax (Chiu and Chuang, 2010; Jope, 2003). These effectors further regulate members of the Bcl-2 protein family, which modulate the balance of cell death and survival signaling (Chiu and Chuang, 2010). GSK-3 has also been shown to crucially regulate immune responses in the CNS (Beurel et al., 2010). Other studies have noted that inhibition of GSK-3 via lithium, as well as other GSK-3 inhibitors, reduces microglial migration, cytokine release, and neurotoxicity (Yuskaitis and Jope, 2009). Notably, GSK-3β and Akt phosphorylation levels increased after TBI (Shapira et al., 2007), and the present study found that there was a trend toward an increase in both p-GSK-3β and p-Akt 3 days after TBI. Lithium treatment robustly increased p-GSK-3β and p-Akt levels, compared with the saline-treated and sham-injured groups, respectively. We believe that through GSK-3β inhibition, lithium modulates cell death/survival signaling pathways to reduce cell death. Lithium also suppresses neuroinflammation and reduces neuronal toxicity, which further contributes to neuronal survival. Taken together, lithium reduces lesion volume and attenuates neurodegeneration, leading to the improvement of functional outcome after TBI.
We have reported that long-term pre-insult treatment with lithium is beneficial in an animal model of stroke (Nonaka and Chuang, 1998), suggesting that lithium can be used prophetically in patients with a high risk of stroke such as transient ischemia attack victims. Pre-insult treatment with lithium also has beneficial effects against TBI (Shapira et al., 2007; Zhu et al., 2010). Unlike the case of stroke, the major causes of TBI involve motor vehicle accidents and other unpredicted incidents (Faul et al., 2010). Therefore, it is more clinically relevant to identify the effects of post-insult treatment against TBI. The present study demonstrated multiple beneficial effects of post-insult treatment of lithium, providing a realistic solution to apply this knowledge to the clinical situation.
Based on the results of the present study, we therefore suggest that the neuroprotective, anti-inflammatory, and beneficial behavioral effects of lithium in the TBI mouse model are likely mediated through GSK-3β inhibition and modulation of its downstream effectors. In view of lithium's long history in clinical use, its safety is not in question. Our results that clearly demonstrate its benefits in the mouse model therefore pave the way for early clinical trials as a potential treatment for TBI patients.
Footnotes
Acknowledgments
Support for this work included funding from the Department of Defense in the Center for Neuroscience and Regenerative Medicine, the Blast Lethality Injury and Research Program, and the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (NIMH-NIH). The authors wish to gratefully acknowledge the editorial assistance of Dr. Elizabeth Sherman, Ioline Henter, and Peter Leeds of the NIMH. We are also grateful to Dr. Oz Malkesman, Laura Tucker, and Dr. Amanda Fu from the Center for Neuroscience and Regenerative Medicine for help with behavioral tests and CCI surgery.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.