
Abstract
Previous studies on the therapeutic potential of agents affecting the dopamine system in traumatic brain injury (TBI) suggest that dopamine dysregulation may have a major role in behavioral deficit after TBI. We have previously identified that TBI reduces striatal dopamine synthesis and release at 7 days post-injury. In order to reverse deficits in the activity of tyrosine hydroxylase and dopamine release following TBI, we administered nicotine by intraperitoneal injection into rats for 7 days. Tyrosine hydroxylase activity assay demonstrated recovery of activity with nicotine treatment in injured animals. Microdialysis experiments using potassium stimulation to induce dopamine release showed recovery of dopamine release in injured animals receiving nicotine treatment. There was no effect of nicotine injection on extracellular dopamine metabolite levels, indicating the specificity of nicotine's effect on dopamine synthesis and release. Also, the activation of downstream postsynaptic molecule dopamine and cAMP regulated phosphoprotein 32 (DARPP-32) was assessed by Western blots for DARPP-32 phosphorylated at threonine 34 (pDARPP-32-T34). Injury reduced pDARPP-32-T34 levels, but nicotine treatment of injured animals did not alter pDARPP-32-T34 levels, indicating that postsynaptic dopamine signaling is complex, and the recovery of dopamine release may not be sufficient for the recovery of DARPP-32 activity.
Introduction
Striatal dopamine signaling involves a phosphoprotein present in all of the medium spiny neurons: dopamine and cyclic adenosine 3′,5′-monophosphate-regulated phosphoprotein, 32kDa (DARPP-32). This protein is a major downstream regulator of dopamine signaling pathway, involved in virtually all aspects of striatal dopamine signaling ranging in biochemical, electrophysiological, and transcriptional events (Greengard et al., 1999; Svenningsson et al., 2004). Phosphorylation of DARPP-32 at threonine 34 (pDARPP-32-T34) by cAMP-dependent protein kinase (PKA) converts it into a potent inhibitor of protein phosphatase-1 (PP-1), which regulates the activity of various ion channels and neurotransmitter receptors (Greengard et al., 1999). Data recently generated from our group showed that TBI decreases striatal pDARPP-32-T34 levels and increases the activity of striatal PP-1 in rats (Bales et al., 2011).
Changes in PP-1 activity have been described as affecting synaptic plasticity in hippocampal neurons by induction of long-term depression or long-term potentiation (Brown et al., 2000; Mulkey et al., 1994). Similarly, decreased pDARPP32-T34 and increased PP-1 activity levels in striatum may contribute to the loss of striatal function, leading to motor and memory deficits. Animal studies have shown that rats with striatal lesions have spatial memory deficits (Block et al., 1993; Devan et al., 1999) and motor deficits (Erinoff et al., 1979; Ungerstedt et al., 1971). Such deficits were also characterized in animal models of TBI (Dixon et al., 1999; Hamm, et al. 1996; Scheff et al., 1997; Smith et al., 1991). Therefore, alterations in striatal pDARPP-32-T34 signaling may be a major underlying mechanism for behavioral deficits after TBI. Currently, it is unknown if TBI-induced alterations in pDARPP-32-T34 are associated with decreased evoked dopamine release.
Nicotine, an agent that activates nicotinic acetylcholine receptors on neurons, has been found to improve cognitive functions and behavioral outcomes in both normal physiology and injury settings. It enhances locomotor activity (Smith et al., 2010) and memory (Levin et al., 1990, 1996) in rats. In addition, nicotine treatment has been shown to increase TH activity and levels (Carr et al., 1989; Hiremagalur et al., 1993; Sabban and Gueorguiev, 2002), as well as dopamine release (Marshall et al., 1997; Rahman et al., 2007). Striatal slice experiments have also shown that applications of higher concentrations of nicotine can increase pDARPP-32-T34 levels by activation of dopamine D1 receptors (Hamada et al., 2004). Therefore, to reverse the dopamine synthesis and release deficits as well as decreased pDARPP-32-T34, we administered nicotine into rats by intraperitoneal injection for 7 days after TBI. We report here the effects of 7-day nicotine treatment on potassium stimulus evoked dopamine release and TH activity, as well as on activity of DARPP-32, by monitoring phosphorylation at threonine 34 (pDARPP-32-T34) and threonine 75 (pDARPP-32-T75). Also, phosphorylation level of downstream signaling molecule extracellular-regulated kinase (ERK) was monitored to correlate with changes in DARPP-32 phosphorylation.
Methods
Animals
Seventy-two Sprague-Dawley rats (Harlan Laboratories) weighing 280–350 g were used in this study under the guidelines set by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Animals were housed in 12-h light/dark cycle with food and water given ad libitum. Animals were grouped into four conditions: Sham + Saline, Injured + Saline, Sham + Nicotine, Injured + Nicotine. Each group had n=5–7 for microdialysis, TH activity assay, and Western blots.
Surgery and drug administration
Animals assigned to injury groups received TBI using the controlled cortical impact (CCI) device as previously described (Dixon et al., 1991). Briefly, animals were first anesthetized using 5% isoflurane. Intubation was performed, and animals were mounted on a stereotaxic frame. Anesthesia was maintained using 2% isoflurane in 2:1 N2O/O2 and a parasagittal craniectomy (center of craniectomy at AP: +4.0 mm, L: +2.8 mm from lambda) ∼ 8 mm in diameter was performed to expose the brain to allow access for the impactor tip of the CCI device. TBI was induced at 4 m/s (50 ms dwell time) and 2.6 mm deformation from the cortical surface. In our hands, these impact parameters produce overt cortical and hippocampal histopathology (Singleton et al., 2010). After injury, the surgical area was closed by silk sutures and animal recovery was monitored by watching for tail pinch and righting reflexes. Sham animals underwent craniectomy only and no CCI. After the surgery, animals were returned to the housing facility.
Rats were intraperitoneally injected with either 0.9% saline or nicotine (2mg/kg, suspended in 0.9% saline) starting 24 h after surgery. The injection schedule continued twice daily for 7 days until euthanasia on day 7.
TH activity assay
The activity levels of TH was determined using the method previously described (Shin et al., 2011). Briefly, dopa decarboxylase inhibitor 3-hydroxybenzylhydrazine (NSD-1015) (Sigma, St. Louis, MO) was intraperitoneally injected into rats 30 min before euthanasia (100 mg/kg, suspended in 0.9% saline) at 7 days post-injury. The striata were dissected out, frozen in liquid nitrogen, and stored in −70°C until analysis. For analysis, the tissues were sonicated in 0.2M HClO2 at 5mg/μL concentration. Sonicated tissues were centrifuged at 13,000xg for 30 min, and supernatants were loaded into high-performance liquid chromatography (HPLC) for detection of
Microdialysis and high-performance liquid chromatography
Six days following surgery, microdialysis probes were implanted as previously described (Dixon et al., 1997; Shin et al., 2010). A Microdialysis probe (SciPro, Sanborn, NY) with 3 mm membrane length, 0.6 mm diameter, and 35 kDa permeability cutoff was stereotaxically implanted into the striatum (AP: +0.0 mm, L: +2.8 mm, DV:-4.0 mm from bregma) 1 day before the experiment. Artificial cerebrospinal fluid (ACSF) with 126.5mM NaCl, 2.4mM KCl, 1.1mM CaCl2, 0.83mM MgCl2, 27.5mM NaHCO3, and 0.5mM KH2PO4 was infused at 0.2μM/min overnight. On day 7 post-injury, 1 h before sample collection began, flow rate was increased to 2μM/min. Sample collection was performed every 20 min in a tube containing 5μL of 0.3M HClO2 and immediately analyzed using HPLC. At the 60th min after sample collection began, dopamine release was stimulated by infusion of ACSF with high potassium: 48.9mM NaCl, 80mM KCl, 1.1mM CaCl2, 0.83mM MgCl2, 27.5mM NaHCO3, and 0.5mM KH2PO4 for 40 min. At the end of the stimulus, high-potassium ACSF was switched back to original ACSF. Samples were collected for 160 min.
Dopamine and dopamine metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) in the dialysates were also monitored using HPLC. The HPLC system consisted of four-channel analytical cell CoulArray Detector (ESA, Chelmsford, MA), eight coulometric electrodes using −120 to +300 mV potentials in 60 mV increments, and C18 column.
Western blot
Rats were anesthetized by pentobarbital at 100 mg/kg before dissection of the striata. After dissection, tissues were immediately frozen in liquid nitrogen and stored at −70°C until analysis. On the day of the analysis, tissues were sonicated in lysis buffer (0.1M NaCl, 0.01M Tris-HCl (pH 7.6), 0.001M EDTA (pH 8.0), 1μg/mL phenylmethylsulfonyl fluoride, Phosphatase Inhibitor Cocktails 1 and 2 (1:100, Sigma, St. Louis, MO), Protease Inhibitor Cocktail (Complete Mini, Roche Applied Science, Mannheim, Germany). The sonicated tissues were centrifuged at 13,000xg for 30 min. Protein concentration was determined using BCA protein assay kit (Pierce, Rockford, IL), and each sample was loaded for Western blot equally at 40 μg. Samples were electrophoresed using 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to polyvinylidene fluoride membranes. Membranes were blocked for 1 h in 5% bovine serum albumin (BSA) (Sigma, St. Louis, MO) suspended in 0.05M tris-buffered saline with 0.05% Tween-20 (TBST). Then, the membranes were incubated with primary antibody for pDARPP-32-T34, pDARPP-32-T75, or phosphorylated ERK (pERK) (1:1000, Cell Signalling, Boston, MA) overnight. The membranes were washed using TBST and were incubated for 1 h with anti-mouse or anti-rabbit immunoglobulin G-conjugated to peroxidase (1:20,000, Pierce). After washing the membranes again with TBST, they were exposed to chemiluminescence solution (Western Lighting, Perkins Elmer, Boston, MA). Membranes were exposed to autoradiographic X-ray film to visualize the proteins. The Western blots were quantified by measuring optical densities using Scion Image PC software (Frederick, MD). Afterwards, the membranes were stripped by incubation in 100mM glycine (pH 2.3) at 55°C for 1 h. β-actin levels in each sample were analyzed by using anti-β-actin monoclonal antibody (1:10,000, Sigma-Aldrich) and anti-mouse immunoglobulin G as described.
Statistical analysis
All data for Western blot, TH activity assay, and area under the curve for dopamine release were analyzed using one-way ANOVA. For DOPAC and HVA levels, repeated-measures ANOVA was used. If statistical significance was found, one-tailed Dunnett's post-hoc test was used to compare each group to Injured + Saline group. All calculations were performed using PASW Statistics 19 (SPSS Inc., Chicago, IL) software.
Results
The TH activity assay was first used to assess the effects of nicotine on
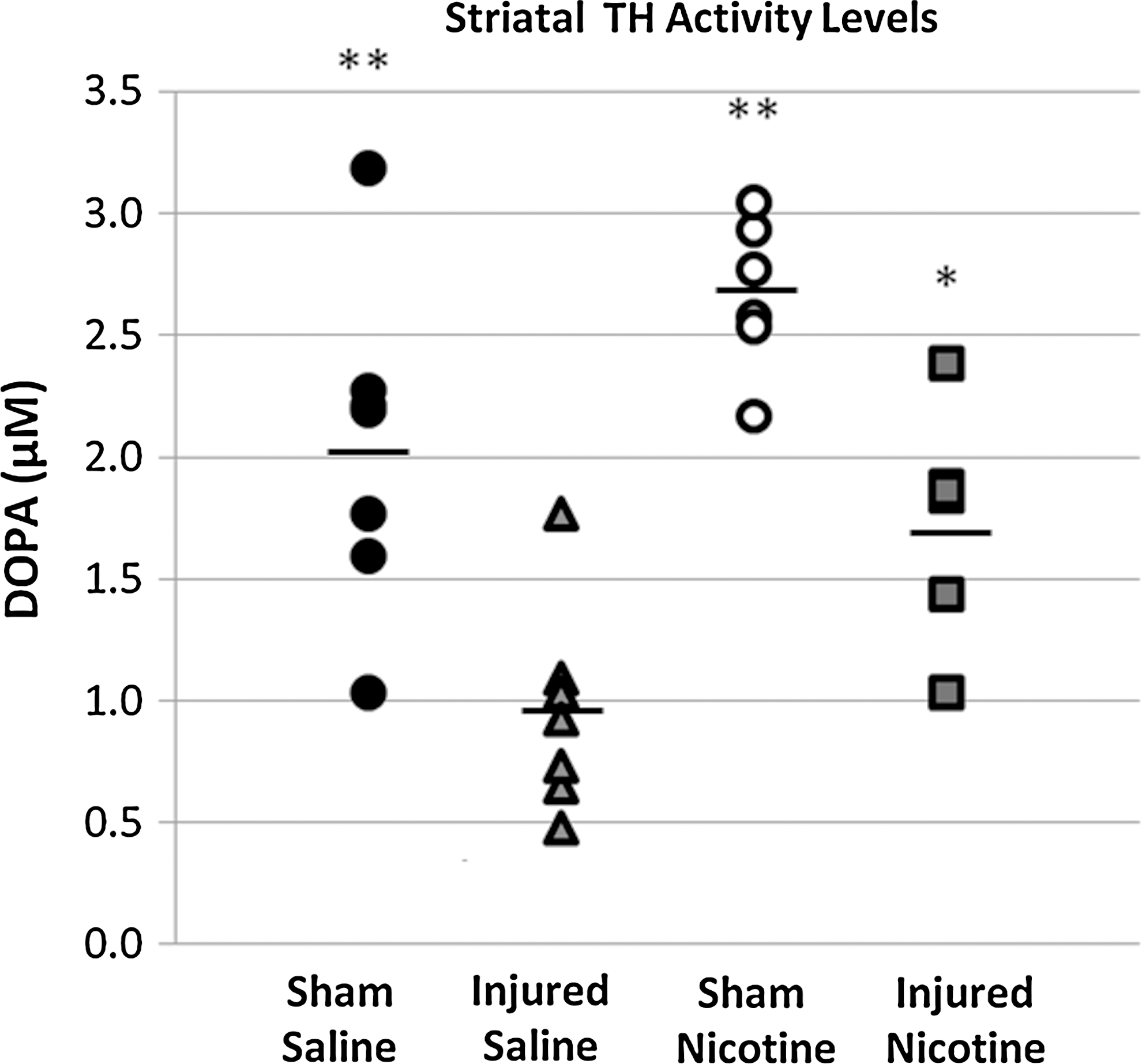
TH activity assay. Dopa accumulation in striatal tissue was monitored using HPLC after injection of NSD-1015, a dopa decarboxylase inhibitor. Vertical scatter plot shows TH activity of each sample, and mean TH activity level is indicated by a line. **p≤0.001, *p≤0.01 compared to the Injured + Saline group.
Next, dopamine release was assessed by using microdialysis and HPLC. Dopamine levels in the microdialysates were analyzed by two different methods: by peak dopamine levels and dopamine area under the curve (Fig. 2). Peak dopamine levels showed significant between-group differences (F 3,17=4.372; p≤0.05), with the Injured + Saline group showing significant decrease compared to the Sham + Saline and Sham + Nicotine groups (p≤0.05). The Injured + Nicotine group showed significantly increased peak dopamine levels compared to the Injured + Saline group (p≤0.05), demonstrating that nicotine treatment induces recovery from dopamine release deficits after TBI.
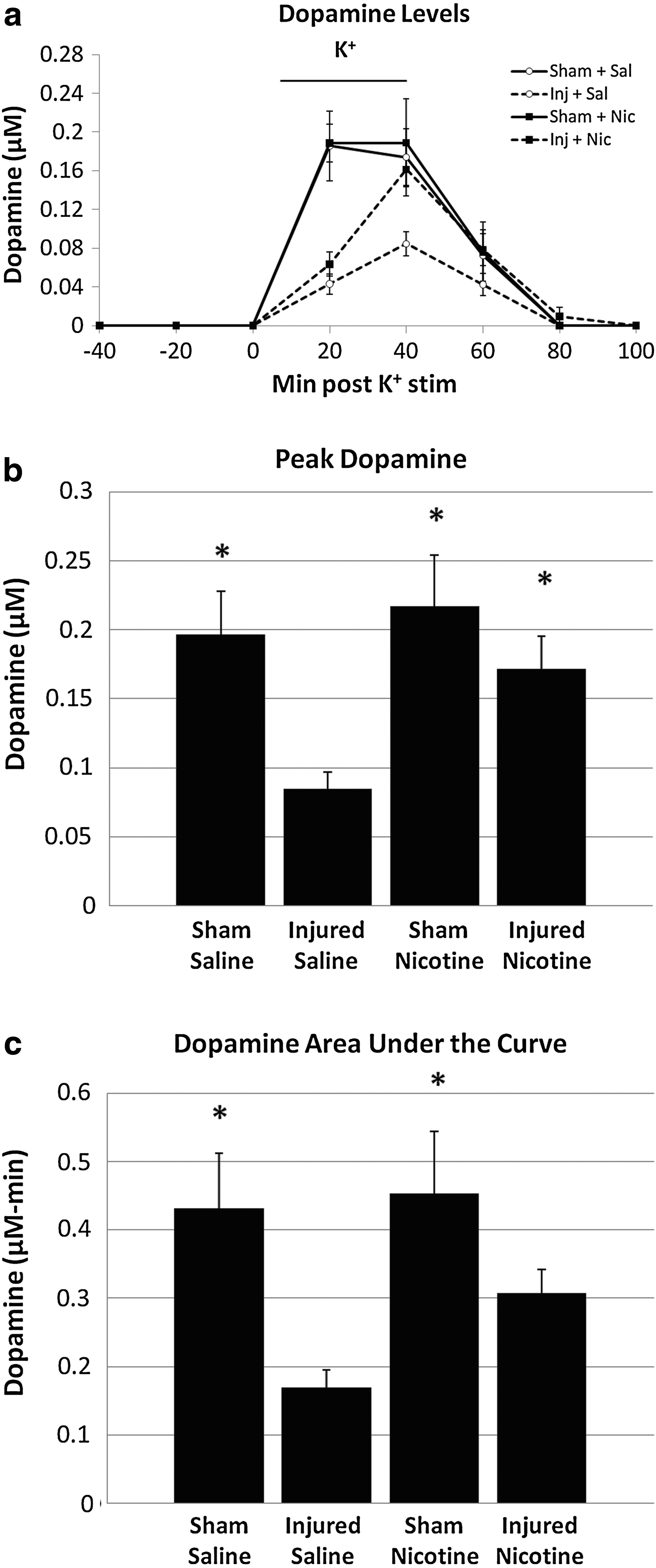
Microdialysis for dopamine. When high potassium ACSF was infused, the Injured + Saline group had decreased dopamine release compared to the Sham + Saline and Sham + Nicotine groups
Dopamine area under the curve was analyzed for each of the four groups to assess for differences in dopamine levels over the entire duration of the potassium stimulus. There was significant between-groups difference (F 3,17=4.241; p≤0.05). Both the Sham + Saline and Sham + Nicotine groups had significantly higher levels compared to the Injured + Saline group (p≤0.05). However, the Injured + Nicotine group showed no significant difference from Injured + Saline group, unlike for the peak dopamine levels.
Dopamine metabolites were also analyzed using microdialysis and HPLC (Fig. 3). There was no significant difference among the four groups (DOPAC: F 3,17=0.824, p>0.05; HVA: F 3,16=0.316, p>0.05). Therefore, nicotine reverses the dopamine deficit induced by TBI but induces no effect on extracellular dopamine metabolite levels.
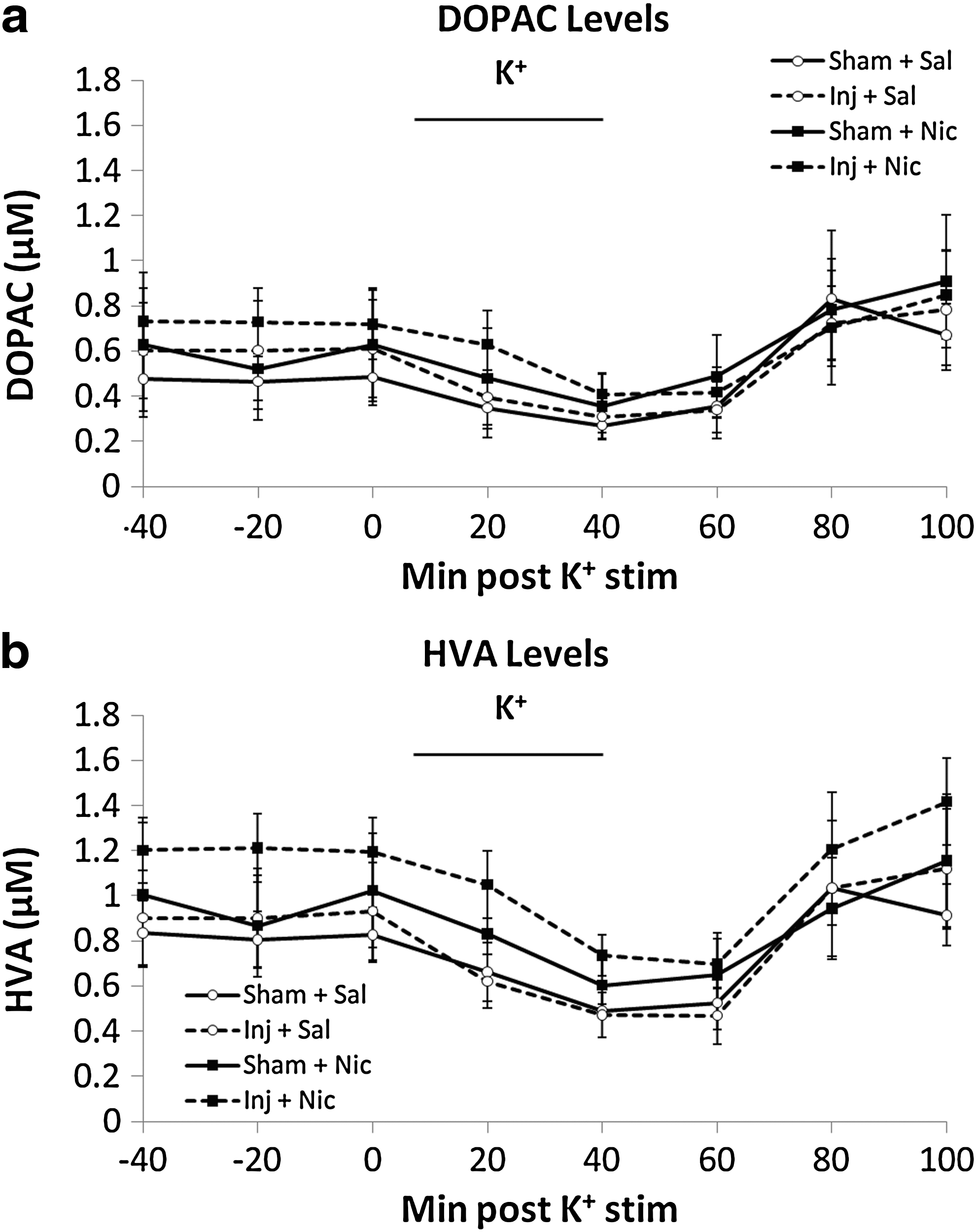
Microdialysis for dopamine metabolites. High potassium ACSF infusion induced temporary decrease in DOPAC
The level of pDARPP-32-T34 showed significant between-group difference (F 3,16=8.816; p≤0.001) (Fig. 4), with the Injured + Saline (44.2±9.4%) and Injured + Nicotine (31.9±7.3%) groups having significantly lower levels compared to the Sham + Saline group (p≤0.05). When pDARPP-32-T75 levels were monitored, no significant between-group difference was found (F 3,21=2.171; p>0.05; data not shown). No statistically significant differences were found for pERK among the four groups (F 3,20=1.557; p>0.05).
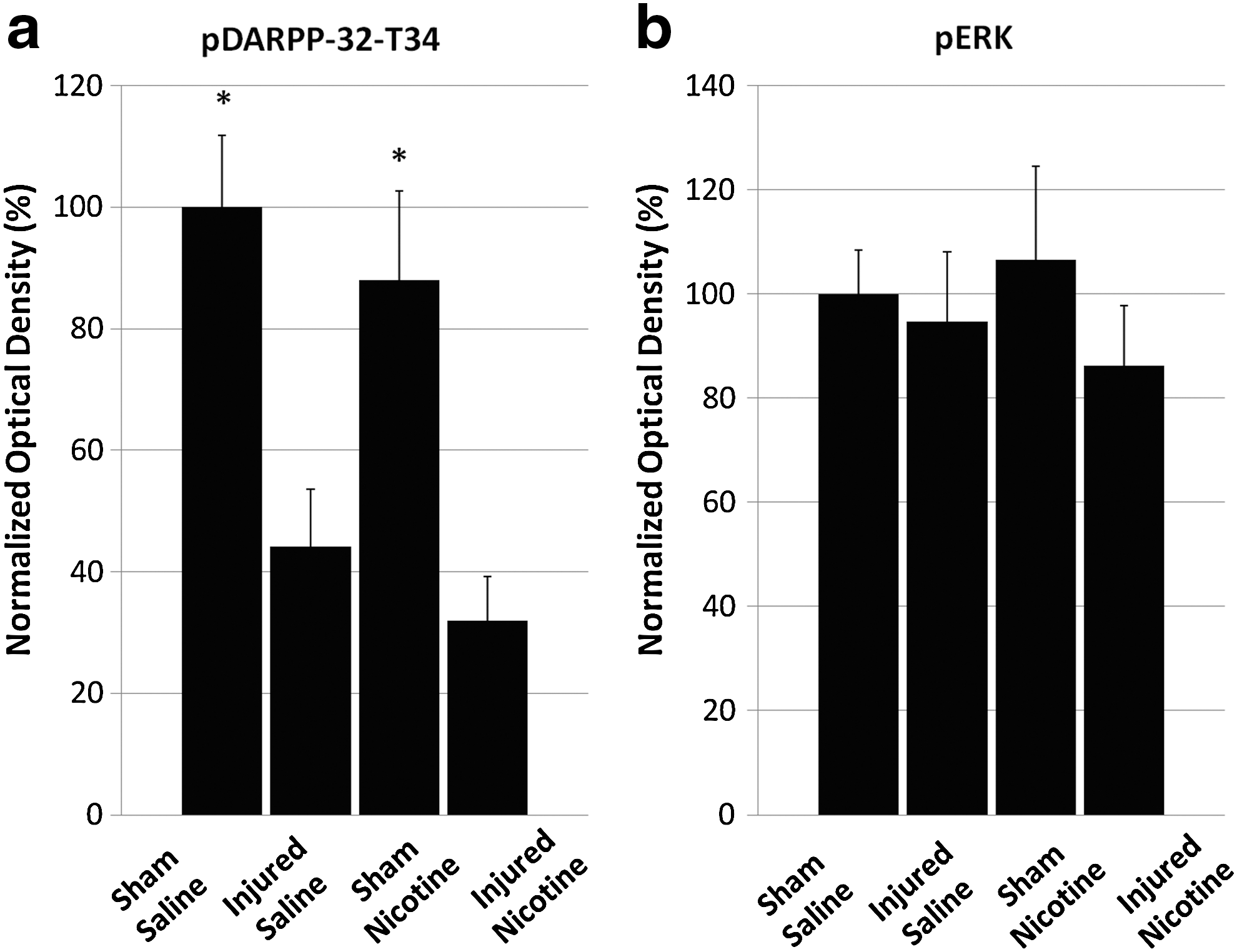
Western blot data of pDARPP-32-T34 and pERK. Injury reduces pDARPP-32-T34 levels
Discussion
In this study, we assessed the effects of nicotine on striatal presynaptic dopamine synthesis and release, as well as postsynaptic signaling by examining DARPP-32 phosphorylation. Nicotine treatment reversed TH activity and dopamine release deficit following CCI. However, nicotine did not reverse deficits in postsynaptic markers of dopamine signaling, suggesting the complexity of postsynaptic deficits in DARPP-32 signaling, which may involve various mechanisms other than decreased presynaptic dopamine signaling.
There are several suggested mechanisms of TH activation by nicotine treatment. Nicotine was previously demonstrated to increase TH transcription (Osterhout et al., 2005) and phosphorylation (Haycock et al., 1993). Also, in vitro studies showed nicotinic receptor activation increasing TH phosphorylation, activity, and protein levels (Bobrovskaya et al., 2007; Craviso et al., 1992; Sabban and Gueorguiev, 2002). Nicotine induces a rapid rise in the levels of intracellular calcium (Sabban and Gueorguiev, 2002), and the regulation of TH activity levels by intracellular Ca2+ has been previously reported (Bustos et al., 1980). Because Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a major kinase for TH at serine 19 (pser19TH) (Dunkley et al., 2004), increased intracellular Ca2+ can lead to CaMKII activation, which will subsequently lead to pser19TH increase. Increased pser19TH induces conformational change of TH, allowing phosphorylation at serine 40 (pser40TH) at an increased rate (Dunkley et al., 2004). In addition, Ca2+ activation of CaMKII can lead to activation of specific subtypes of adenylate cyclase (Wong et al., 1999), which can further contribute to pser40TH increase.
Another possibility of nicotine's effect on TH activity is via activation of protein kinase C (PKC). Nicotine has been demonstrated to induce PKC activation (Tang et al., 1997), which can phosphorylate TH and induce activation. Increase in pser40TH was shown to depend upon protein kinase C (PKC) 24 h after nicotine stimulus, whereas other kinases may contribute to pser40TH levels at earlier times (Bobrovskaya et al., 2007). Therefore, PKC and CAMKII activation may be the major contributors to increased TH activity by nicotine treatment.
The peak dopamine levels after potassium stimulus showed that nicotine treatment induces recovery of dopamine release in injured animals, and that nicotine treatment of sham animals does not significantly change dopamine release compared to saline-treated sham animals. In our current report, nicotine-induced increase in dopamine release is in agreement with increase in TH activity. Because potassium-stimulated dopamine release has been shown to depend upon newly synthesized dopamine (Fairbrother et al., 1990), various mechanisms by which nicotine can activate TH activity may translate to increased dopamine release.
When assessing the area under the curve, nicotine-treated injured animals showed no significant difference compared to saline-treated injured animals. These results indicate that in the setting of TBI, nicotine may improve maximum dopamine release, but does not fully recover dopamine release pattern measured over the stimulus duration. In Figure 2, dopamine release curves show the temporal delay in reaching peak dopamine levels for Injured + Nicotine and Injured + Saline groups compared to both sham groups. Because potassium exposure depolarizes neurons and leads to dopamine release, dysregulation of ion channels occurs following TBI (Lima et al., 2008; Lu et al., 2008; Stys, 2004) and may possibly contribute to delays in potassium-stimulated dopamine release. It is likely that nicotine treatment in injured animals may upregulate maximum TH activity as previously explained, but it is unable to overcome injury-induced delay in dopamine release.
As previously shown (Shin et al., 2011), injury at a 1 week time point did not alter the levels of extracellular dopamine metabolites DOPAC and HVA. Nicotine treatment did not change the levels of these metabolites, indicating the specificity of nicotine's enhancement of dopamine release. The levels of these metabolites depend upon the activity of enzymes such as monoamine oxidase, aldehyde dehydrogenase, and catechol-O-methyltransferase. Current findings suggest that nicotine alters TH activity specifically, while not affecting these enzymes.
Dopamine D1 receptor activation has been demonstrated to increase pDARPP-32-T34 levels (Svenningsson et al., 1998), and it is possible that the TBI-induced pDARPP-32-T34 deficit may be in part caused by an upstream dopamine release deficit. In the current study, injury significantly decreased pDARPP-32-T34 levels, and nicotine treatment did not reverse this deficit. The levels of pDARPP-32-T75 were also monitored, as it functions as an inhibitor of PKA, indirectly reducing pDARPP-32-T34 and modulating PP-1 inhibition (Bibb et al., 1999). There were no significant alterations of pDARPP-32-T75 by injury or nicotine treatment in our study.
The activity of DARPP-32 is modulated by both dopamine and glutamate signaling, which oppose each other in the medium spiny neurons (Halpain et al., 1990). Activation of N-methyl-D-aspartate (NMDA) receptors on medium spiny neurons leads to Ca2+influx (Choi, 1987; Martinez-Sanchez et al., 2004), which can activate protein phosphatase 2B (PP-2B) a major phosphatase for pDARPP-32-T34 (Halpain et al., 1990; King et al., 1984). Nicotine administration can contribute to the decrease in pDARPP-32-T34, as it not only induces dopamine but also glutamate release, leading to activation of PP-2B. By a similar mechanism, protein phosphatase 2A (PP-2A) can be activated, which can decrease pDARPP-32-T75 levels. It is also possible that the level of dopamine release induced by current nicotine treatment regimen was not high enough to activate D1 receptors mainly. Low level of dopamine release by nicotine in the striatum may activate mainly D2 receptors (Hamada et al., 2004), and the activation of D2 receptors has been shown to reduce pDARPP-32T34 by either reducing PKA activity or increasing PP-2B activity (Nishi et al., 1997).
To further monitor postsynaptic striatal dopamine signaling, Western blot was performed for pERK a molecule downstream of pDARPP-32T34, because of its important roles in learning and memory (Deng and Karin, 1994; Sgambato et al., 1998). In agreement with pDARPP-32-T34 levels, no changes were noted for pERK with nicotine treatment. There are several possible reasons for the lack of nicotine's effect on ERK phosphorylation level. Glutamate release can activate NMDA receptors of medium spiny neurons, leading to phosphorylation ERK (Shiftlett and Balleine, 2011). However, this activation of ERK is transient, because Ca2+ influx by NMDA receptor leads to PP-2B activation, which dephosphorylates and activates striatal enriched phosphatase (STEP) (Paul et al., 2003). This activation of STEP then leads to dephosphorylation of ERK (Shiftlet and Balleine, 2011). Also, D2 receptor activation can decrease pDARPP-32-T34 levels (Nishi et al., 1997), which will reduce the activation of ERK. Therefore, the lack of nicotine's effect on pERK level may be caused by these opposing effects of upstream signaling pathways.
The differences in nicotine's effects on presynaptic dopamine synthesis and release compared to postsynaptic signaling proteins indicate that the complexity of striatal dopamine signaling probably involves multiple signaling systems. Gaining insights into the mechanisms behind the differential effects of nicotine on pre- and postsynaptic dopamine signaling in the striatum is an important goal for future studies.
The experiments in this report have a few limitations that should be considered for designing future studies on this subject. In this report, a dose–response study was not performed for nicotine. Although only one dose was assessed, we found it to be sufficient to reverse deficits in dopamine synthesis and release. Because deficits in phosphorylation of downstream signaling molecules such as DARPP-32 were not affected by this current dose of nicotine, future efforts can focus on testing multiple doses and a more chronic regimen to enhance DARPP-32 signaling. It is also possible that nicotine treatment may have been initiated too early following the injury. Following TBI, there is an immediate overflow of glutamate inducing excitotoxicity followed by an elevation in glutamate release for up to 2 days (Hinzman et al., 2010). Nicotine induces glutamate release as previously mentioned, and injection of nicotine beginning at 1 day post-injury may possibly exacerbate the injury. This issue can be clarified by testing an injection regimen beginning 2 weeks following injury, when the initial excitotoxic effects would have subsided.
In addition, although the use of microdialysis to assess neurotransmitter levels in specific regions of the brain is common and well established, newer techniques such as fast-scanning cyclic voltametry have been shown to provide advantages in temporal and spatial resolution (Hinzman et al., 2010; Wagner et al., 2005). Future studies using these novel methods to assess neurotransmitter changes following TBI may provide additional insights into subregion-specific effects of pharmacological agents.
Postsynaptic dopamine signaling in the striatum is regulated by a complex interaction of various pathways, and current findings show a mechanistic insight into the complexity of dopamine neurotransmission in the striatum. Although the nicotine treatment regimen in this report was able to induce recovery of TH activity and evoked dopamine release deficits, no measured postsynaptic signaling deficit was reversed, possibly because of the combination of factors mentioned previously. Therefore, in future studies focused on treating neurotransmission deficits, combinational approaches may be needed to separately target pre- and postsynaptic mechanisms. As pharmacological agents such as amantadine, which has both NMDA antagonist and dopamine agonist effects, was successful in reversing TBI-induced pDARPP-32-T34 deficits (Bales et al., 2011), a combination of agents such as nicotine with amantadine may enhance recovery of dopamine signaling following TBI.
Footnotes
Acknowledgments
This work was funded by National Institutes of Health grants 1F30NS067731-01 and 5R01NS060672-02, and Veterans Administration Rehabilitation Research and Development grant B6761R.
Author Disclosure Statement
No competing financial interests exist.