
Abstract
Transplantation of neural stem cells (NSCs) improves functional outcomes following traumatic brain injury (TBI). Previously we demonstrated that human NSCs (hNSCs) via releasing glial cell line-derived neurotrophic factor (GDNF), preserved cognitive function in rats following parasagittal fluid percussion. However, the underlying mechanisms remain elusive. In this study, we report that NSC grafts significantly reduce TBI-induced axonal injury in the fimbria and other brain regions by blocking abnormal accumulation of amyloid precursor protein (APP). A preliminary mass spectrometry proteomics study revealed the opposite effects of TBI and NSCs on many of the cytoskeletal proteins in the CA3 region of the hippocampus, including α-smooth muscle actin (α-SMA), the main stress fiber component. Further, Western blot and immunostaining studies confirmed that TBI significantly increased the expression of α-SMA in hippocampal neurons, whereas NSC grafts counteracted the effect of TBI. In an in vitro model, rapid stretch injury significantly shortened lengths of axons and dendrites, increased the expression of both APP and α-SMA, and induced actin aggregation, effects offset by GDNF treatment. These GDNF protective effects were reversed by a GDNF-neutralizing antibody or a specific calcineurin inhibitor, and were mimicked by a specific Rho inhibitor. In summary, we demonstrate for the first time that hNSC grafts and treatment with GDNF acutely reduce traumatic axonal injury and promote neurite outgrowth. Possible mechanisms underlying GDNF-mediated neurite protection include balancing the activity of calcineurin, whereas GDNF-induced neurite outgrowth may result from the reduction of the abnormal α-SMA expression and actin aggregation via blocking Rho signals. Our study also suggests the necessity of further exploring the roles of α-SMA in the central nervous system (CNS), which may lead to a new avenue to facilitate recovery after TBI and other injuries.
Introduction
T
Recent studies from several groups including ours revealed that stem cells may increase the levels of various neurotrophic factors in injured brains either by directly secreting such factors or stimulating endogenous cells to release them (Gao et al., 2006; Kim et al., 2010; Shindo et al., 2006). These neurotrophic factors include glial cell lined-derived neurotrophic factor (GDNF), nerve growth factor, brain-derived neurotrophic factor (BDNF) and neurotrophin 3. Our group previously reported that TBI reduced GDNF expression in rat hippocampal formations (a region critical for learning and memory), whereas human fetal neural stem cells (hNSCs) secreted significant amounts of GDNF into injured brains after transplantation in vivo (Gao et al., 2006). Furthermore, hNSC transplantation reduced cognitive deficits in rats after parasagittal fluid percussion injury (Gao et al., 2006). However, it is unclear to what extent GDNF contributes to hNSC-mediated prevention of cognitive deficits in TBI, and what molecular mechanisms underlie the neuroprotective effects of GDNF.
Originally discovered in glial cell lines (Lin et al., 1993), GDNF is expressed in both neurons and glia in the central nervous system (CNS) (Batchelor et al., 2002; Miyazaki et al., 2001; Schaar et al., 1993). GDNF serves as a potent survival factor for dopaminergic and motor neurons (Beck et al., 1995; Henderson et al., 1994; Lin et al., 1993; Oppenheim et al., 1995; Sauer et al., 1995; Tomac et al., 1995), protects hippocampal (Bonde et al., 2000; Kim et al., 2001) and cortical neurons (Kitagawa et al., 1999; Minnich et al., 2010; Wang et al., 1997), and induces neurite outgrowth (Akerud et al., 1999; Coulpier et al., 2004). Particularly interesting from the perspective of cognitive function is that adult mice heterozygous for a null mutation in the GDNF gene show an impaired water maze learning performance (Gerlai et al., 2001), the intrahippocampal delivery of an exogenous GDNF gene by lentiviral vector transduction improves spatial learning in aged rats (Pertusa et al., 2008), and grafting of mouse C17.2 transformed progenitor cells, engineered to secrete GDNF, improved cognitive function following TBI (Bakshi et al., 2006). Therefore, it is reasonable to speculate that protection of host neurons by hNSC-produced GDNF is one of the mechanisms by which hNSCs contribute to cognitive improvement.
In a recent study using an unbiased quantitative mass spectrometry and bioinformatic proteomics approach, we observed apparently opposite effects of TBI and hNSC grafting on many of the structural proteins, including neurofilaments and actins in the CA3 region of rat hippocampi (unpublished observation). Furthermore, amyloid β-precursor protein (APP or βAPP), a marker for abnormal axonal transport (Geddes et al., 2000; Gentleman et al., 1993; Marmarou & Povlishock, 2006; Pierce et al., 1996; Stone et al., 2000), was increased by TBI. All these point to the possibility of hNSC grafts modulating axonal damage following TBI.
Traumatic axonal injury (TAI) is a well-recognized pathological phenomenon in all degrees of TBI, causing “diffuse axonal injury” mainly after severe injuries (Adams et al., 1991; Geddes et al., 2000; Hurley et al., 2004; Maxwell et al., 1997; Povlishock, 1992). White matter damage caused by shear and tensile forces is usually present as the result of serial molecular, physiological, and structural changes, including axolemmal disruption, intracellular calcium accumulation, loss of microtubules, neurofilament compaction, mitochondrial damage, calpain-mediated proteolysis, axonal swelling, and secondary axotomy (Maxwell et al., 1997). TAI also causes disruption of neural networks and thus contributes to both mortality and morbidity in TBI patients (Adams et al., 1991; Geddes et al., 2000; Hurley et al., 2004; Maxwell et al., 1997; Povlishock, 1992). Because TAI is a process persisting hours to days after initial injury, amelioration of TAI could result in a significant functional improvement in TBI patients (Buki et al., 1999). To this end, we show in this study how hNSCs and GDNF affect TAI using both in vivo and in vitro traumatic injury models.
Methods
Proliferation and differentiation of hNSCs
Most reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. The K048 line of hNSCs, derived from the forebrain of an 8-week human fetus, was provided by C.N. Svendsen (Svendsen et al., 1998; Wu et al., 2002). Cells were maintained as described in our previous publication with minor modifications (Wu et al., 2002). Briefly, K048 cells (0.3–0.4×106/mL) were cultured in basic medium supplemented with 20 ng/mL EGF (R&D Systems, Inc., Minneapolis, MN), 20 ng/mL bFGF (R&D Systems Inc.), 5 μg/mL heparin (Invitrogen Carlsbad, CA), 10 ng/mL LIF (Chemicon International Inc., Temecula, CA), 100 μg/mL transferrin, 100 μM putrescine, 20 nM progesterone, 25 μg/ml insulin, and 30 nM sodium selenite. The basic medium was composed of Dulbecco's Modified Eagle Medium (DMEM; Mediatech, Inc., Herndon, VA):F12 (Invitrogen) (3:1), 15 mM HEPES, 1.5% glucose, 2 mM
For transplantation, neurospheres (passage number 15–24) were seeded at 8×104 cells/cm2 in a T25 culture flask precoated with 0.01% poly-D-lysine and 0.5 μg/cm2 laminin (Invitrogen). Cells were primed (Wu et al., 2002) with 10 ng/mL bFGF, 2.5 μg/mL heparin and 1 μg/mL laminin (FHL) for 5 days with half of the medium replaced every other day and then switched to DMEM/F12 medium containing B27 (Invitrogen) for 2 days. Prior to transplantation, primed hNSCs were dissociated by incubation with 0.025% trypsin/200 U/mL deoxyribonuclease (DNase) for 10–20 min at 37°C followed by mechanical trituration. Numbers of live cells were determined by a trypan blue exclusion assay. Cells with >85% viability were centrifuged and resuspended in basic medium supplemented with B27, 250 U/mL DNase, 0.6% glucose, and 3 nM FK506 (Alexis Corp., Switzerland) at a density of 0.5×105 cells/μL and then stored on ice until grafting.
For in vitro differentiation, small spheres of hNSCs were seeded at a density of 2.5–6.2 x104/cm2 onto flexible silastic membranes in Flex I® Culture Plates (Flexcell International Co., Hillsborough, NC), which were pre-coated with 0.01% poly-D-lysine and 1 μg/cm2 mouse laminin (Invitrogen). Cells were incubated with ELL media containing 20 ng/mL
Fluid percussion TBI and transplantation of hNSCs
All surgeries were approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch and performed under aseptic conditions in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Twenty-six male Sprague–Dawley (SD) rats (300–350 g, Harlan) were randomly divided into three groups: Sham (n=9), TBI plus vehicle injection (TBI+Veh) (n=9) and TBI plus hNSC grafting (TBI+hNSCs) (n=8). They were further split into two sets, one for morphological analyses and the other for Western blotting. Parasagittal fluid percussion TBI was performed according to our previous description (Gao et al., 2006). Briefly, isoflurane-anesthetized rats were subjected to parasagittal fluid percussion TBI at a pressure of 2.0 atm. Sham-injured rats were connected to the trauma device without injury.
One day after TBI, hNSCs were transplanted into TBI injured SD rats according to our previous description (Gao et al., 2006). Briefly, 100,000 hNSCs in 1 μL were injected into the hippocampal region (Bregma−4.3 mm; ML: −3.5 mm; Dura −3.2 mm) through the edge of the injury site at 0.2 μL/min. To minimize possible immune rejection, all animals were treated with the immunosuppressor NEORAL cyclosporine (Novartis Pharmaceuticals Corp., East Hanover, NJ) at 100 μg/mL in drinking water from 1 to 2 days before grafting throughout the course of the experiment. Three days after grafting, 12 rats (4 per group) were euthanized for brain collection and histological examination, whereas the other 14 (4 to 5 per group) were subjected to protein extraction for Western blot analysis. The time point, 3 days after transplantation or 4 days post-injury, was chosen based on the spreading of hNSCs after implantation, as well as the extent and time course of APP accumulation.
Rapid stretch injury (RSI)
The Flex I® Culture Plates containing ELL-primed and differentiated hNSC-derived neurons were connected to a 94A Cell Injury Controller (Biomedical Engineering Facility, Medical College of Virginia). The silastic membranes were deformed by controlled nitrogen gas pulse to achieve a predetermined degree of stretch for a predetermined duration (Ellis et al., 1995; Wu et al., 2002). Cells were subjected to different levels of stretch injury (0, 10, 20, 30, 40, and 50 psi). Thirty minutes after stretch injury, two thirds of the medium was replaced with fresh differentiation B27 media containing various reagents and cultured for additional 4 days. The reagents included 15 ng/mL GDNF (R&D Systems), 6 μg/mL GDNF neutralizing antibody (R&D Systems), and 40 μM calcineurin autoinhibitory peptide (EMD Chemicals, Gibbstown, NJ). Subsets of hNSC-derived neurons were treated either with 40 μM calcineurin autoinhibitory peptide or 1 ug/mL cell permeable Rho inhibitor (Cytoskeleton Inc., Denver, CO) at 4 days post-injury for 4 h. The specific doses were determined based on the titration studies in hNSCs and/or previously published literature. At the end of incubation, cells were subjected to either fixation or protein extraction.
Calcineurin activity assay
The activity of calcineurin (CAN or PP2B) was assayed in protein extracts using a commercially available colorimetric Calcineurin Cellular Activity Assay Kit (EMD Biosciences, San Diego, CA) by using RII phosphopeptide as a substrate according to the manufacturer's instructions.
Histological analyses
Immunostaining procedures were previously described in detail (Gao et al., 2006; Tarasenko et al., 2004). Briefly, rat brain tissues were fixed with 4% paraformaldehyde 3 days post-transplantation via intracardiac perfusion. Postfixed tissues were embedded in OCT compound (Fisher) and cryostat-sectioned at a thickness of 30 μm. Cultured cells were fixed with ice-cold 4% paraformaldehyde for 20–30 min. Tissue sections and cells were blocked with 5% normal serum plus 0.3% bovine serum albumin (Sigma) and permeablized with 0.25% Triton X-100 (Sigma) in Tris-buffered saline (TBS). Incubation with primary antibodies took place at 4°C overnight. All antibodies were empirically tested for optimal concentrations, including rabbit polyclonal anti-APP (1:100, Invitrogen), mouse monoclonal anti-α-smooth muscle actin (α-SMA, 1:100, Sigma), mouse monoclonal anti-pan-axonal neurofilament marker (SMI312, 1:500), mouse monoclonal anti-neuronal class III β-tubulin (TuJ1, 1:2,500, Covance), and rabbit polyclonal anti-microtubule-associated protein 2 (MAP2, 1:250, Fisher Scientific/Chemicon). Signals were visualized by incubation with species-specific secondary antibodies conjugated with Alexa Fluor® 568 or 488 (Invitrogen/Molecular Probes, 1:400) at room temperature for 2 h in the dark. Nuclei were counterstained with 1 μg/mL DAPI (Sigma).
For F-actin labeling, cultured cells were incubated with 0.25% bovine serum albumin (BSA) at room temperature for 20 min and then with Alexa Fluor® 488 phalloidin (Invitrogen, 1:500) in 0.1% BSA at 37°C for 1 h. Labeled cells and tissue sections were examined with a Nikon Eclipse E1000 epifluorescent microscope and a Nikon TE2000-E microscope connected with the C1si confocal system (Nikon), respectively. Analyses of the images were conducted with the NIS-Element BR imaging software (Nikon Instruments Inc., Lewisville, TX).
Tissue/cell processing and Western blotting analyses
The analyses were performed based on a previously described protocol with modifications (Tarasenko et al., 2004). For tissue collection, anesthetized rats were perfused with 25 mL of phosphate buffered saline containing 1 mM phenylmethylsulfonyl fluoride (PMSF, Enzo Life Sciences, Inc/Alexis Biochemicals, Farmingdale, NY), 1X Protease Inhibitor Cocktail (Sigma), 30 mM sodium fluoride (Sigma) and 1 mM sodium vanadate (Sigma). Immediately following decapitation, brains were removed and the hippocampal formation was dissected out with the alvial surface down. The CA3 region along the smaller curve of the medial side of the hippocampus was separated from the dentate gyrus. Tissues were then homogenized in sodium dodecyl sulfate (SDS) lysis buffer containing 5 mM EDTA, 50 mM Tris, 2% SDS, 1 mM dithiothreitol (DTT), 1 mM PMSF, and 1% Protease Cocktail Inhibitors (Sigma) and centrifuged at 20,000×g for 20 min. Cultured cells were treated with Cell Lysis Buffer (Cell Signaling Technology Inc., Danvers, MA) supplemented with 1 mM PMSF (Sigma). Protein concentrations were determined using the Bio-Rad Protein Assay Dye (Bio-Rad Laboratories, Hercules, CA).
Total protein extracts were electrophoresed through 4-20% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Following blocking, the membranes were probed with primary antibodies at 4°C overnight and then with peroxidase-conjugated secondary antibodies (1:2,000). Primary antibodies included mouse monoclonal anti-APP (1:500, Invitrogen), goat polyclonal anti-calcineurin B1 (PP2B-B1, 1:200, Santa Cruz Biotechnology, Inc.), goat polyclonal anti-calcineurin B2 (PP2B-B2, 1:200, Santa Cruz), goat polyclonal anti-calcineurin Aα (PP2B-Aα, 1:200, Santa Cruz), goat polyclonal anti-calcineurin Aβ (PP2B-Aβ, 1:200, Santa Cruz), and mouse monoclonal anti-αSMA (1:500, Sigma). Blots were reprobed with internal controls including monoclonal anti-GAPDH (1:1,000, Santa Cruz) or monoclonal anti-β-actin (1:20,000, Sigma). Chemiluminescent detection of immunoreactive signals was subjected to densitometry analyses using AlphaEase FC software (Alpha Innotech).
Histological and statistical analyses
For in vivo immunohistochemical analyses, four rats per group were used for quantitative analyses of APP and α-SMA. APP was measured in the fimbria of the hippocampus from three sections per rat (Bregma −4.00 to −4.33 mm). APP+ profiles (>5 μm) were counted in each section and averaged from four rats per group. Quantitative measurements of α-SMA immunoreactive intensity were performed using the NIS Element densitometry software (Nikon).
For immunocytochemical analyses in vitro, the length of each neurite was measured from the edge of soma to the longest traceable terminal. Approximately 200 neurons labeled with TuJ1 from 10 randomly selected fields were included for quantitative assessments.
All other assays were performed in at least three independent experiments. Statistical analyses were expressed as means±SEM after Student's t test or one-way ANOVA with the aid of GraphPad Prism software (GraphPad Software, Inc. CA). Data were considered significant at p<0.05.
Results
Neural stem cell transplantation blocks TAI-associated APP accumulation in the white matter of rat brains after fluid percussion TBI
Parasagittal fluid percussion TBI increased APP expression in the CA3 region compared to the sham control (Fig. 1A, B). On the other hand, transplantation of hNSCs did not significantly decrease the overall expression of APP, albeit with a reducing trend. To further examine the localization of APP, brain sections from a different set of animals (four rats per group) were collected for immunofluorescent staining. As shown in Fig. 1C, APP immunoreactivity was enhanced in parts of the corpus callosum, the subcortical white matter of parietal cortex, and the alveus and fimbria regions of hippocampus at the injury site. This APP accumulation was significantly reduced by hNSCs grafted 24 h after injury. The fimbria, which contains both afferents and efferents connecting hippocampus and septum, showed morphological and quantitative changes of APP immunoreactive profiles (>5 μm in size) 4 days after injury or 3 days after cell transplantation (Fig. 1D,E). Higher magnification imaging revealed both APP single-labeled axonal bulbs, indicating a secondary axotomy, and a few axonal swellings double labeled with APP and an axon-specific marker SMI312 (Fig. 1F). These data indicate that hNSC grafts block APP accumulation in white matter and thus reduce TAI 4 days after TBI.
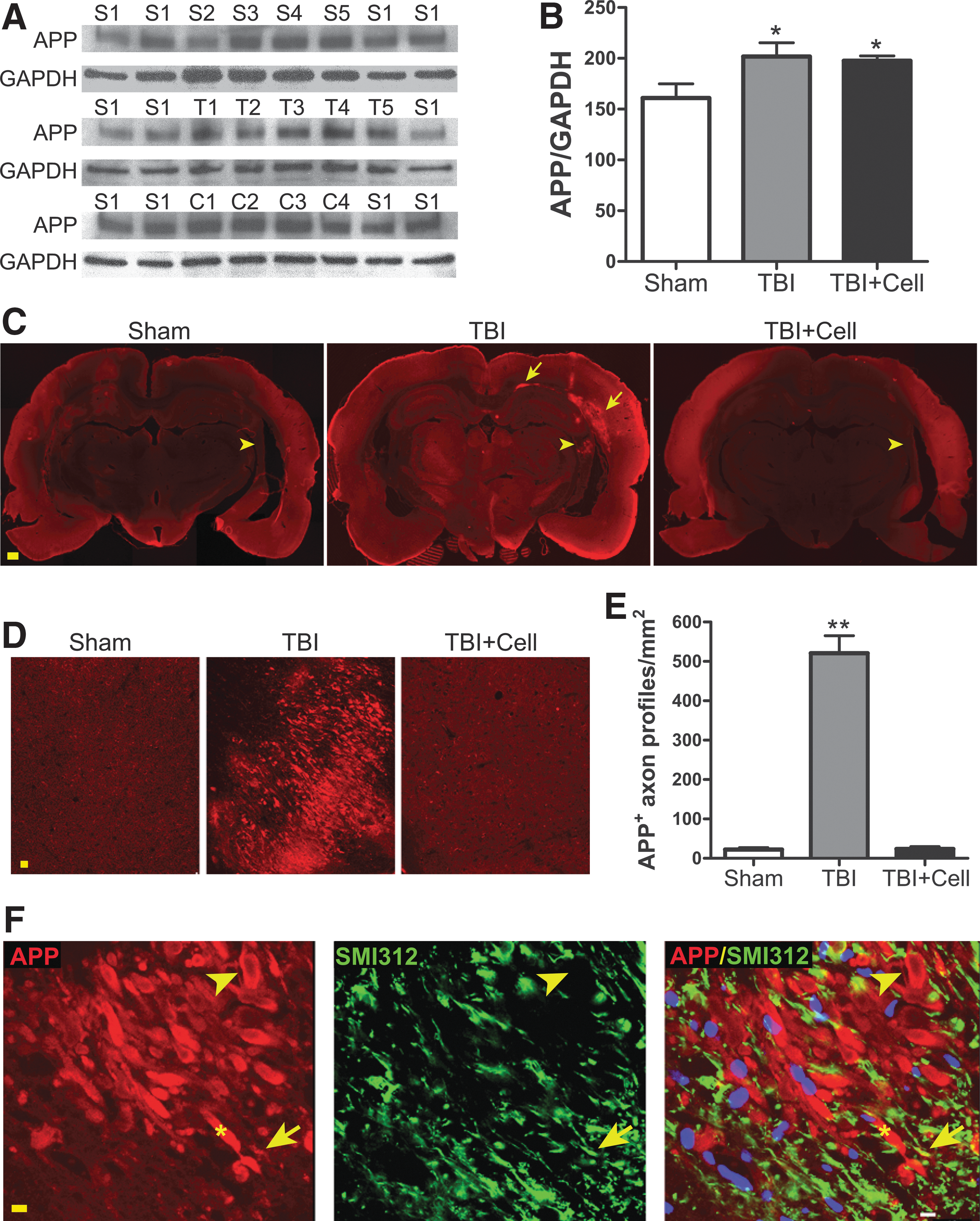
Abnormal amyloid precursor protein (APP) accumulation and traumatic axonal injury in rat brains after parasagittal fluid percussion TBI. (
Neural stem cell transplantation reduces α-SMA expression in rat hippocampi after fluid percussion TBI
To further dissect the cellular and molecular changes after TBI and transplantation, we focused on the proteins that were affected differentially by injury versus cell grafting. One such protein emerging from our preliminary mass spectrometry proteomics study is α-SMA, an actin isoform whose expression has never been reported in neurons. Applying quantitative Western blot and immunofluorescent analyses, we found that normal rat brain expressed only a low level of α-SMA. In contrast, parasagittal fluid percussion TBI increased the overall immunoreactivity of α-SMA on the injury site (data not shown), and particularly increased α-SMA expression in the CA3 region (Fig. 2A–C). The injury-enhanced α-SMA immunoreactivity was more obvious in the cell bodies of the CA3 pyramidal neurons (Fig. 2B). Transplantation of hNSCs 1 day post-injury significantly reduced the injury-induced α-SMA elevation (Fig. 2A–C).
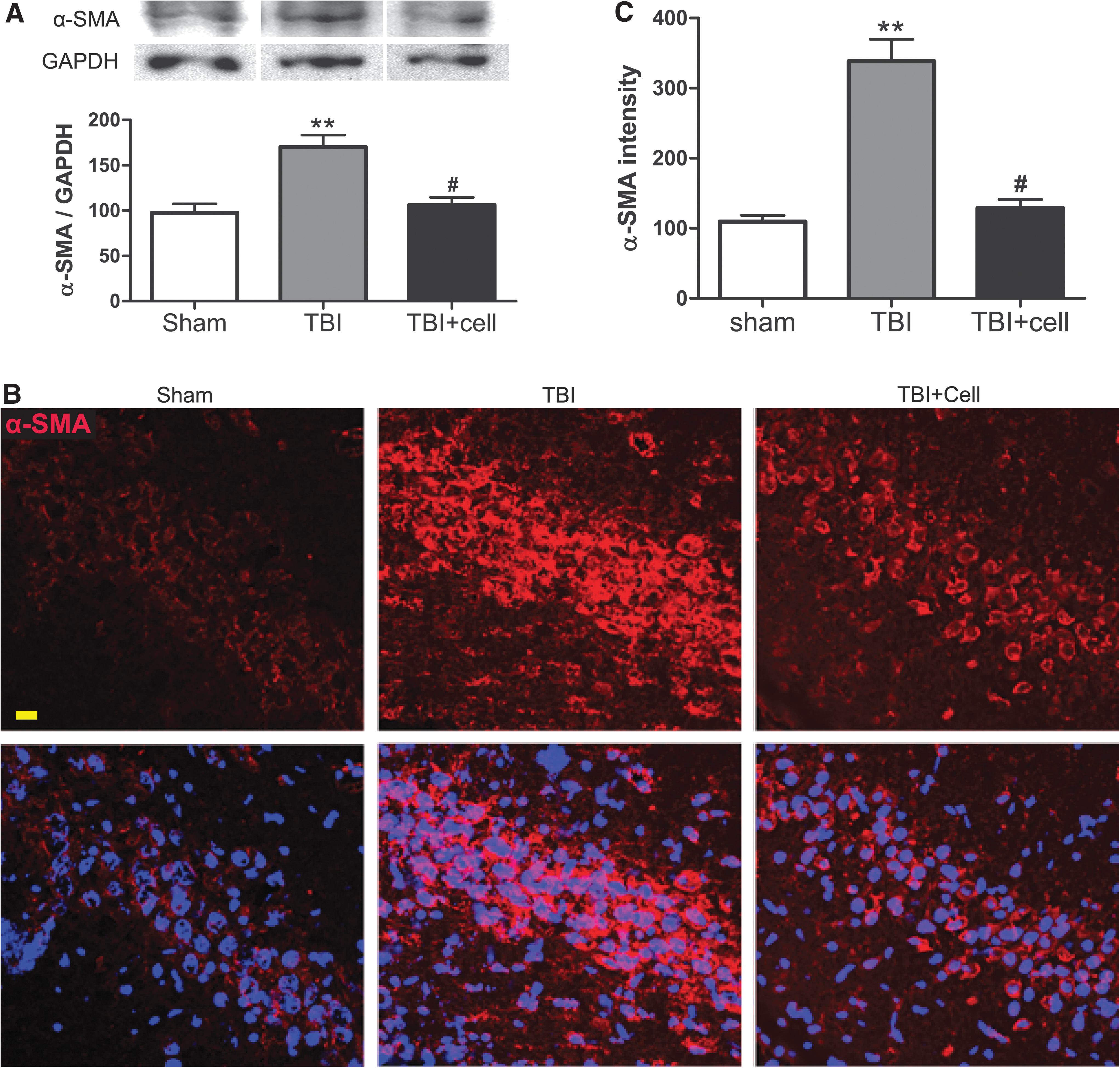
Increased expression of α-smooth muscle actin (SMA) in rat hippocampus after parasagittal fluid percussion TBI.
GDNF protects neurites after rapid stretch injury in vitro
Previously we demonstrated that hNSC transplantation prevents cognitive deficits after acute fluid percussion TBI (Gao et al., 2006), and suggested that this might be attributed to GDNF released from grafted cells. To further elucidate the mechanisms underlying the effect of hNSC-released GDNF on neurons, we used an in vitro rapid stretch injury (RSI) model. Neurons (glutamatergic or GABAergic) differentiated from hNSCs were exposed to increasing intensities of stretch injury. RSI-induced damage in hNSC-neurons was intensity dependent, causing neuronal cell death at 40 psi (19±4.2%) and a loss of all cell types at 50 psi (reducing total cell numbers by 19.4±14.2% when compared to 0 psi). On the other hand, the lower degrees of stretch (10 to 30 psi) resulted primarily in shortening or loss of neurites 12 h after injury (Fig. 3A,B). To mimic the tissue damage in vivo, expression changes of several proteins (GSK3β and calcineurins) were compared between the CA3 regions exposed to 2.0 atm fluid percussion TBI in vivo and the hNSC-neurons after various intensities of RSI in vitro (data not shown). Subsequently, 30 psi was selected for the rest of the in vitro studies based on the similarity in the expression levels of those proteins as well as in consideration of neuronal death and neurite damage.
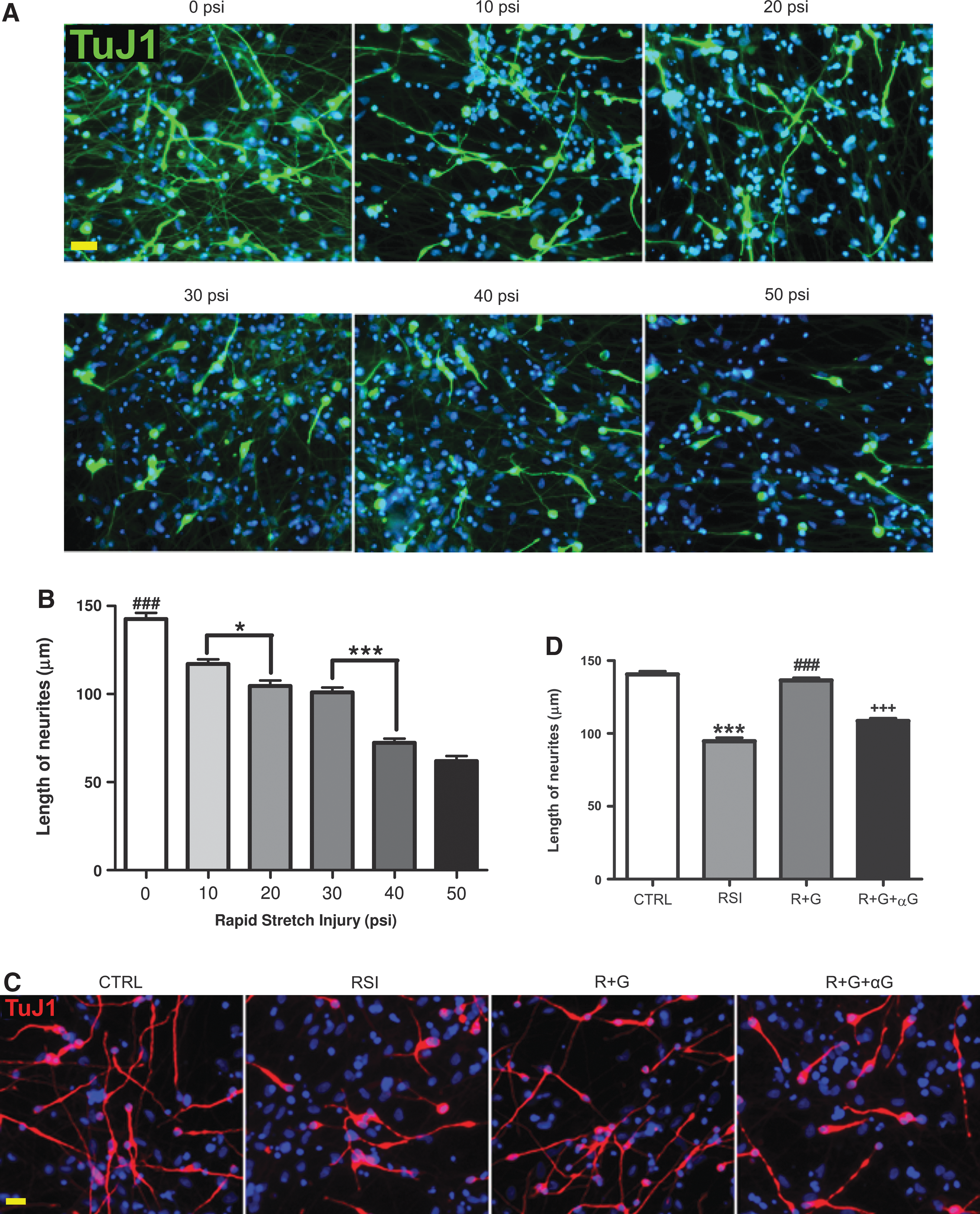
Changes in neurite length after rapid stretch injury and glial cell line-derived neurotrophic factor (GDNF) treatment.
RSI at 30 psi resulted in 30% (12 h) to 40% (4 days post injury) reductions in the average length of neurites labeled by the TuJ1 antibody against the type III β-tubulin, a neuron-specific protein located in somata, dendrites, and axons (Fig. 3). The RSI-induced neurite damage was completely inhibited by GDNF in cells treated 30 min after injury. This protective effect of GDNF on axons and dendrites was blocked by co-treatment with a GDNF neutralizing antibody (Fig. 3C,D).
GDNF reduces APP accumulation in neurons after stretch injury in vitro
To determine whether GDNF affected injury-induced abnormal axonal transport, we assessed APP expression and distribution in hNSC-derived neurons by Western blot and immunofluorescent analyses. As shown in Figure 4A, the expression level of APP in hNSC-neurons was significantly increased 4 days after RSI at 30 psi when compared to that of controls. The increased expression of APP was found in both cell bodies and neurites, including TuJ1-labeled axons (Fig. 4B) and MAP2-labeled dendrites (Fig. 4C). Furthermore, RSI resulted in the loss of microtubules (Fig. 4B, insets) and the appearance of axonal and dendritic swellings 4 days post-injury (Fig. 4B,C). The RSI-induced enhancement of APP expression and abnormal accumulation were significantly blocked by GDNF treatment at 30 min after injury, whereas the GDNF-neutralizing antibody significantly diminished the protective effect of GDNF (Fig. 4A, C).
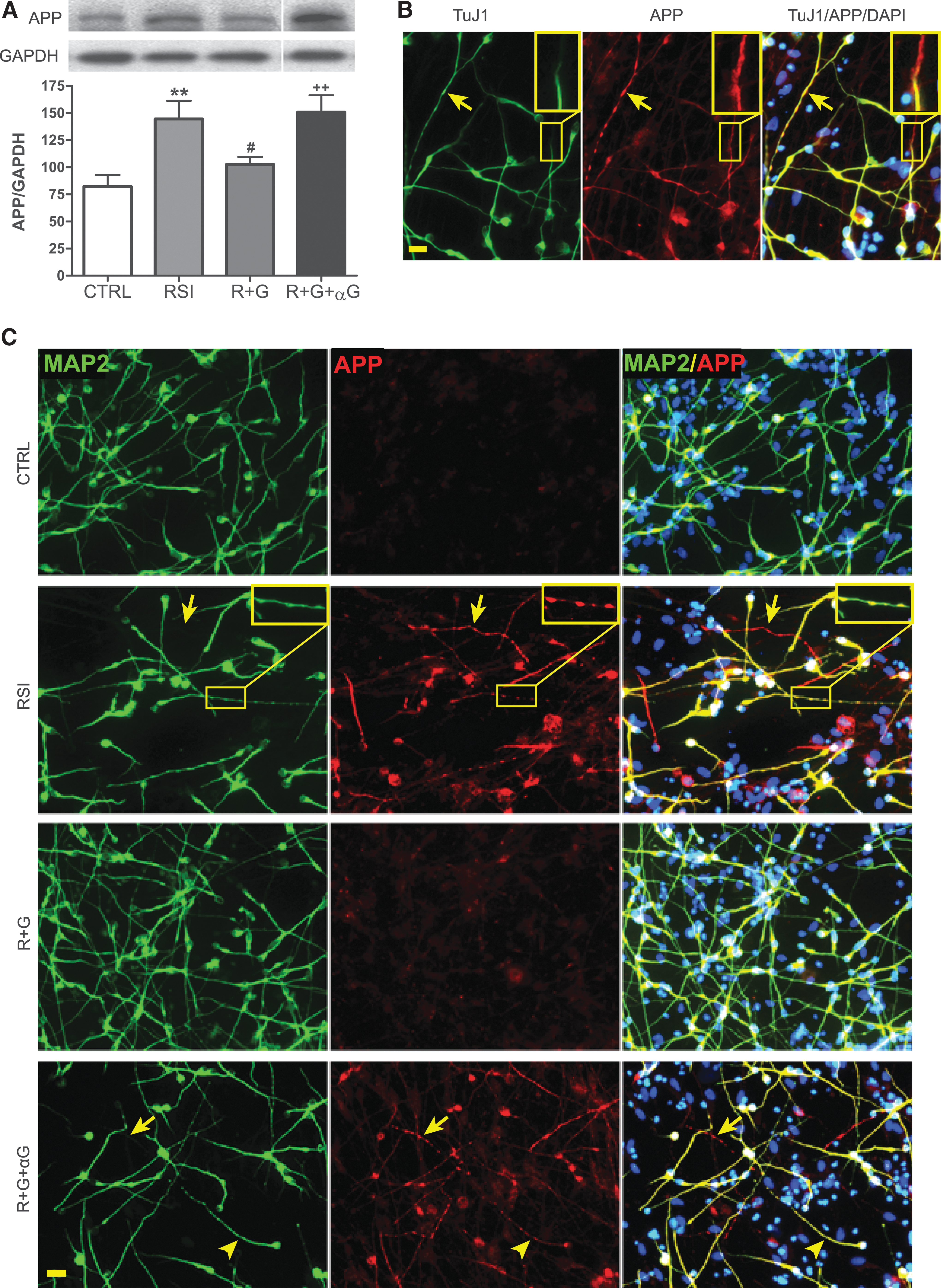
Altered amyloid precursor protein (APP) expression in human neural stem cell (hNSC)-derived neurons after rapid stretch injury in vitro.
GDNF reduces α-SMA expression and abnormal cytoskeletal changes in human neurons after stretch injury in vitro
Because cytoskeletal proteins play an important role in both structure and function of neurons and were dramatically changed after TBI and cell transplantation, we then focused on the effect of GDNF in modulating actins in the RSI in vitro model. Differentiated hNSCs expressed a very low level of α-SMA without injury. In contrast, RSI at 30 psi increased α-SMA expression by 1.7-fold (Fig. 5A) 4 days post-injury, mainly near and in the cell bodies of neurons (Fig. 5B). The F-actin patterns of normal neurons were visualized by an actin-specific binding peptide, phalloidin, showing a relatively even distribution throughout the cell body and neurites (Fig. 5C,D). RSI resulted in aggregated phalloidin-labeling in neurons near cell bodies (Fig. 5D). GDNF treatment at 30 min post-injury significantly reduced the magnitude of RSI-mediated α-SMA increase (Fig. 5A,B), and blocked the actin aggregation (Fig. 5D). GDNF-neutralizing antibodies, on the other hand, inhibited both effects of GDNF on hNSC-derived neurons.
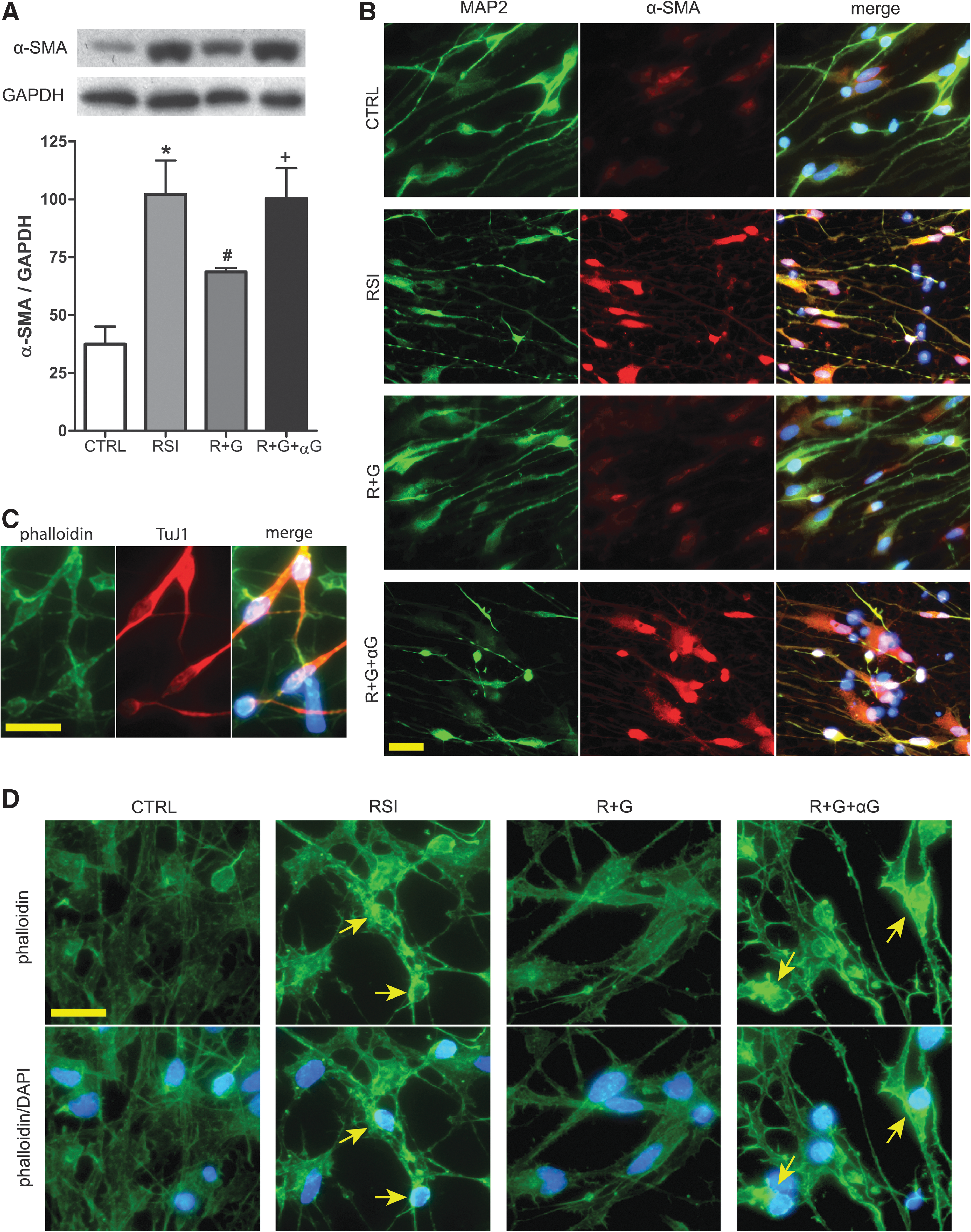
Enhanced expression of α- smooth muscle actin (α-SMA) in hNSC-derived neurons after rapid stretch injury in vitro. (
Neural stem cells and GDNF inhibit traumatic injury-induced calcineurin alterations
To further elucidate the molecular mechanisms underlying hNSC- and GDNF-mediated effects on neurites, we examined calcineurins based on our preliminary proteomics results (unpublished observations) and the literature regarding the role of calcineurin in TAI and axonal regrowth. Western blot analyses revealed that fluid percussion TBI at 2.0 atm significantly reduced the expression levels of the calcineurin regulatory subunit B type 1 (CANB1, PPP3R1 or CaNBα) by 55% (Fig. 6A), and the catalytic subunit alpha isoform (PP2BA, PPP3CA or CaNAα) by 24% in the CA3 region (Fig. 6B). No significant alterations were found in PP2BB (the beta isoform of the regulatory subunit, Fig. 6C) or CANB2 (the type 2 regulatory subunit B, Fig. 6D). Transplantation of hNSCs within 24 h post-TBI reversed the reduction of CANB1 and PP2BA (Fig. 6A,B).
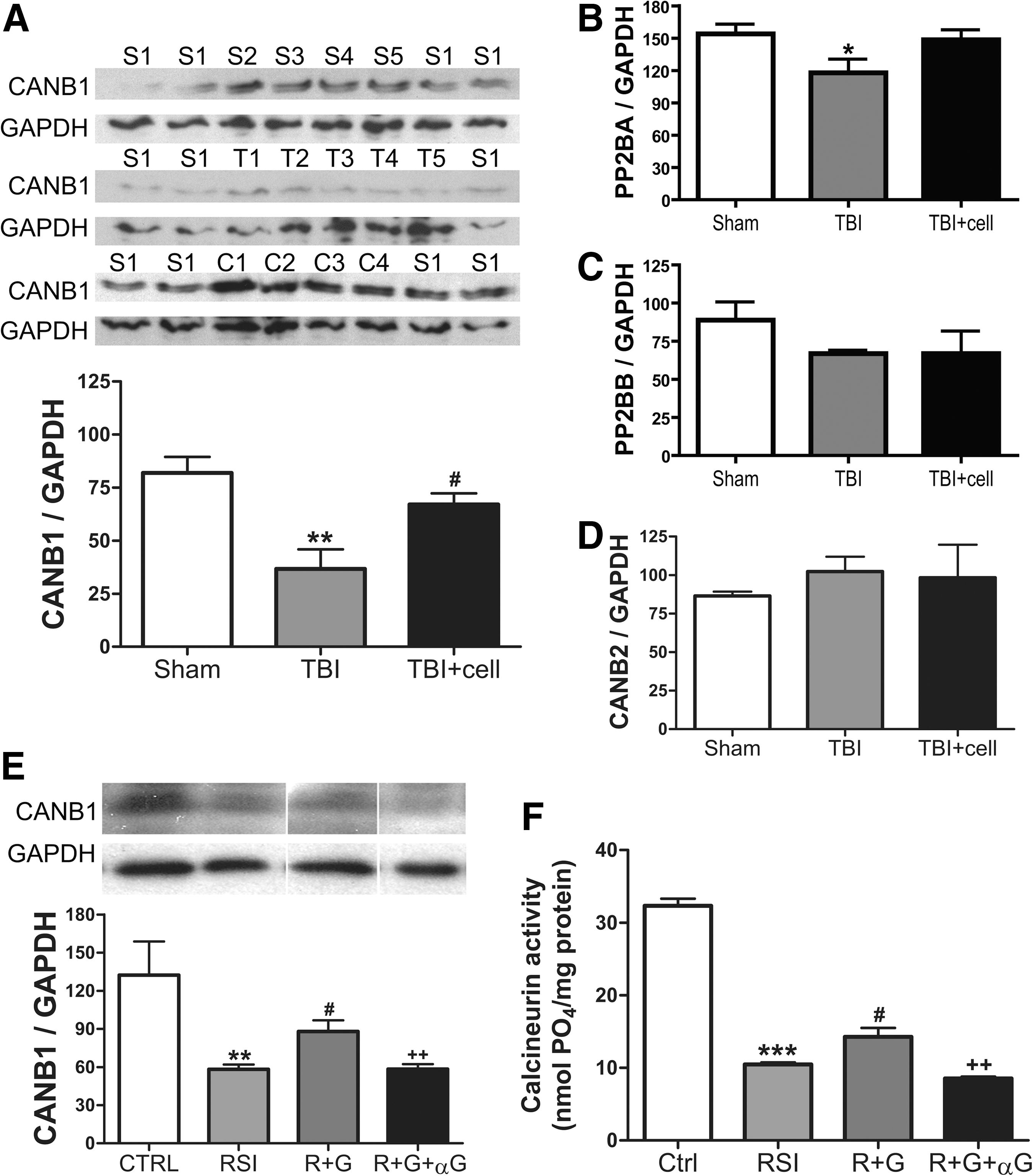
Changes of calcineurin after traumatic injury in vivo and in vitro.
Given the significant changes of CANB1 in TBI in vivo, we further examined its expression in the in vitro injury model. RSI at 30 psi resulted in a decrease of CANB1 expression in hNSC-derived neurons by 56% (Fig. 6E), a magnitude very similar to that in TBI tissues (Fig. 6A). Treatment with GDNF significantly decreased the CANB1 reduction after RSI, whereas GDNF-neutralizing antibodies significantly reduced the effects of GDNF (Fig. 6E). The activities of calcineurin in hNSCs assessed by a calcineurin enzyme activity assay also followed a similar trend: GDNF treatment significantly decreased the magnitude of RSI-induced reduction of calcineurin activity, which was blocked by the GDNF-neutralizing antibody (Fig. 6F).
Calcineurin is involved in the neurite-protective effect of GDNF without affecting α-SMA
To determine whether calcineurins play a role in GDNF-mediated neurite protection, neurons differentiated from hNSCs were subjected to RSI, RSI plus GDNF, or RSI plus GDNF and a calcineurin specific inhibitor (calcineurin autoinhibitory peptide). Inhibition of calcineurin activity significantly shortened the average length of neurites (including axons and dendrites), and thus blocked the protective effect of GDNF against traumatic axonal injury (Fig. 7A,B). Furthermore, calcineurin inhibition for either 4 h (data not shown) or 4 days reversed the inhibitory effect of GDNF on RSI-enhanced APP expression, accumulation, and swelling (Fig. 7C,D). However, specific inhibition of calcineurin did not significantly affect the expression of α-SMA (Fig. 7E,F)
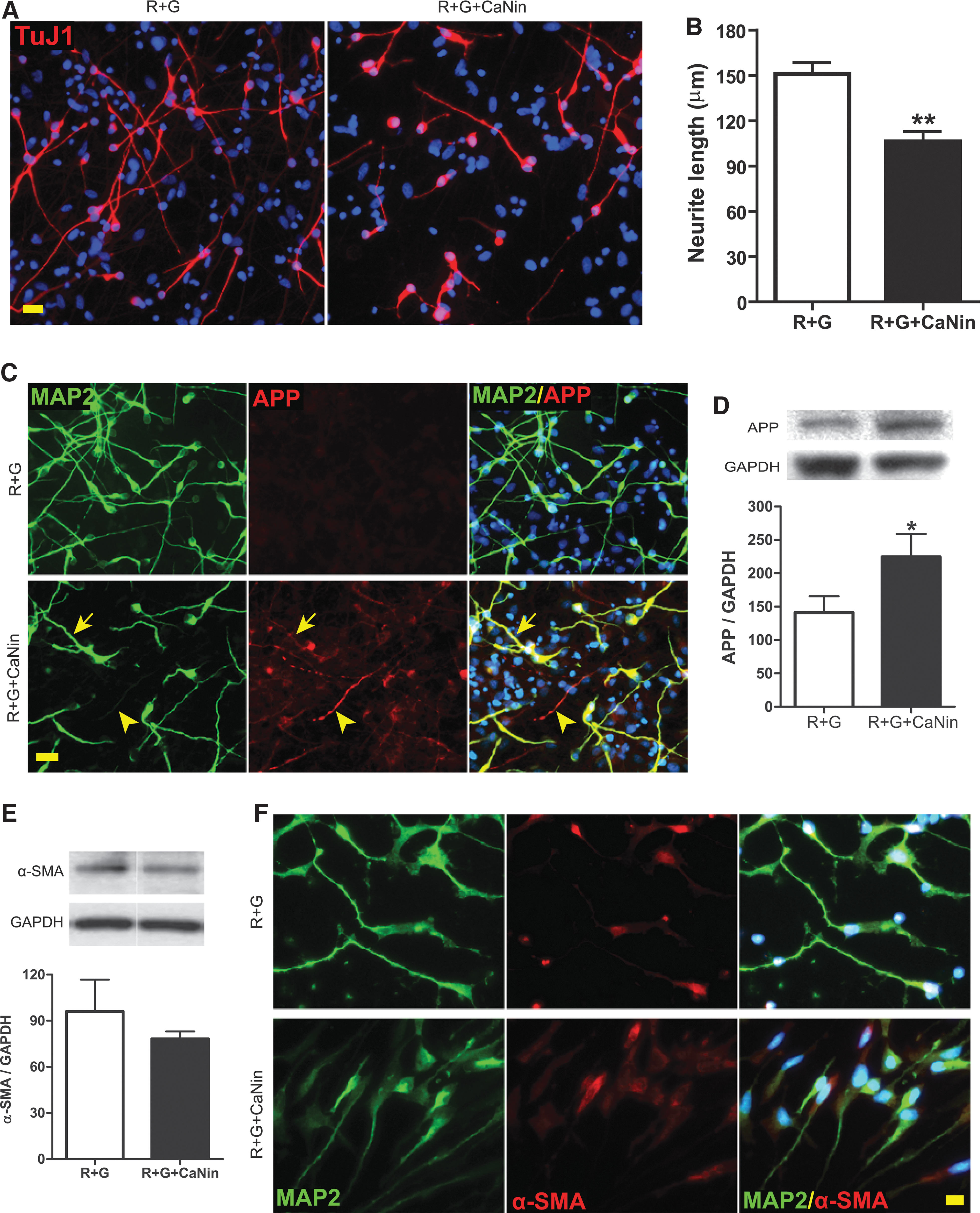
Effect of calcineurin inhibition on human neural stem cell (hNSC)-derived neurons after rapid stretch injury in vitro. (
Rho inhibition protects neurites from stretch injury by blocking abnormal α-SMA expression and cytoskeletal changes
RhoA, a small GTPase protein, is involved in blocking axonal outgrowth following injury (McGee et al., 2003). It is also known to regulate the actin cytoskeleton that forms stress fibers in peripheral cells such as fibroblasts (Nobes & Hall, 1995). However, it is unclear whether Rho regulates α-SMA expression and thus changes cytoskeletal structure in neurons after traumatic injury, particularly in the context of neurite regrowth. Here, we show that RSI shortened TuJ1+ axons and dendrites, whereas treating with a specific Rho inhibitor 4 days post-injury for only 4 h significantly increased the average length of neurites (Fig. 8A,B). This neurite-elongating effect of the Rho inhibitor was accompanied by significant reductions of α-SMA expression (Fig. 8C,D) and of actin aggregation (Fig. 8E).
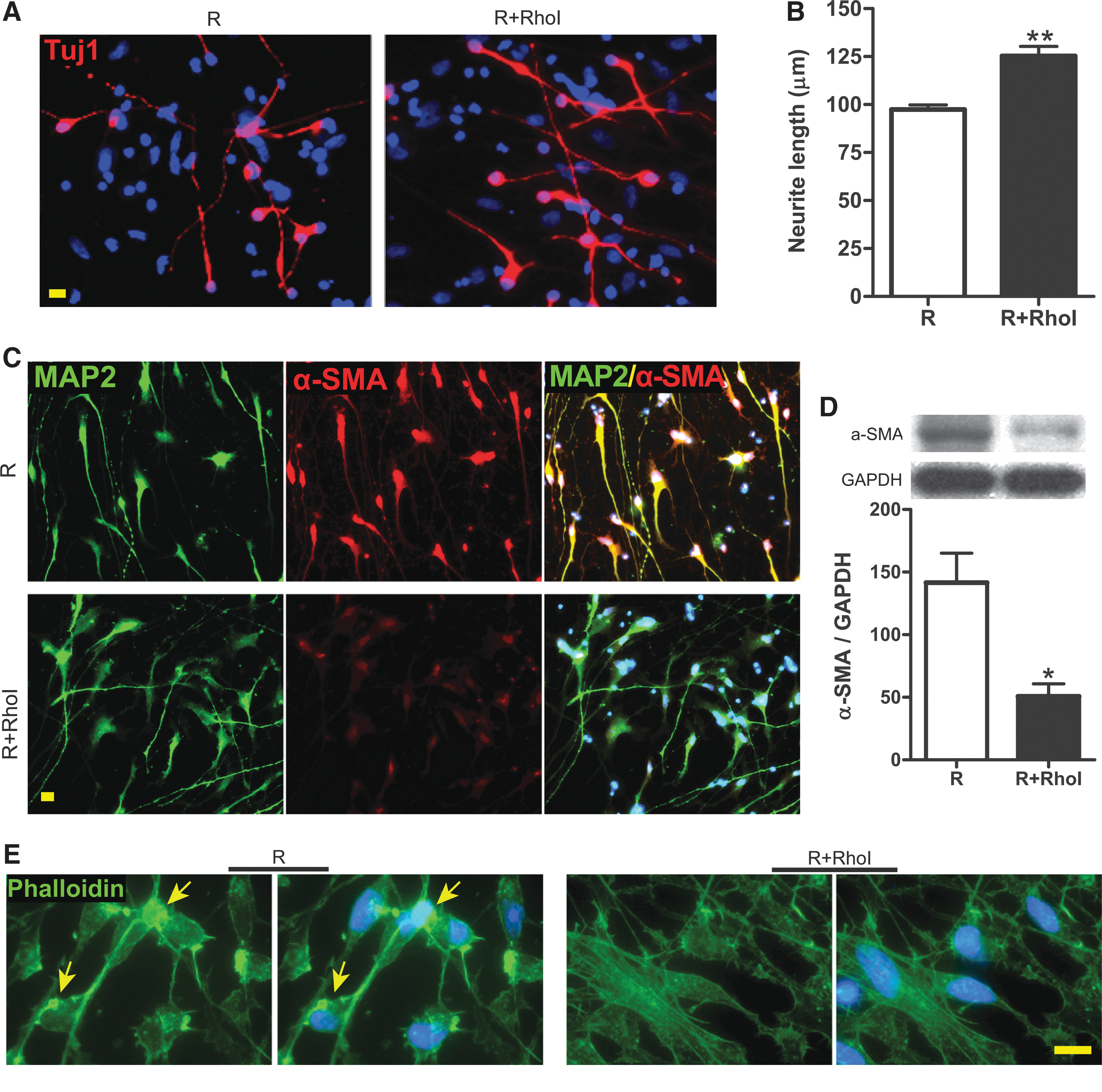
Effect of Rho inhibition on human neural stem cell (hNSC)-derived neurons after rapid stretch injury in vitro. (
Discussion
This study explored the cellular and molecular mechanisms underlying the beneficial effects of stem cell transplantation after TBI by using a parasagittal fluid percussion model in vivo and a rapid stretch injury model in vitro. We observed that human NSCs, via their secreted GDNF, blocked APP accumulation and TBI-induced α-SMA. The protective and regenerative effects of hNSCs and GDNF may result from balancing calcineurin functions and modulating cytoskeletal proteins, including α-SMA. The latter is reported for the first time in neurons.
Previously we have demonstrated that grafting hNSCs into injured hippocampi after parasagittal fluid percussion (2.0 atm) prevented cognitive deficits 2 weeks after TBI (Gao et al., 2006). The acute effects of hNSCs were then related to GDNF that was released from the grafted hNSCs rather than by neuronal replacement. To determine whether hNSC-secreted GDNF protects the remaining host hippocampal neurons, we monitored cell death 1–7 days after TBI by Fluoro-Jade and TUNEL. No differences were detected among control and TBI with and without cell implantation (unpublished observation), in agreement with the previous report of TAI without acute neuronal death after a certain degree of TBI (Singleton et al., 2002). Further, a powerful preliminary mass spectrometry proteomics study revealed significant changes of cytoskeletal proteins and APP after TBI alone that were normalized by hNSC transplantation (unpublished observations). Because many of the altered proteins, such as neurofilament and actin, are related to axonal structure and function, we then turned our attention to TBI-induced TAI that disrupts connections among different parts of the brain and is known as a factor in mortality and morbidity after TBI.
APP is a transmembrane protein that is expressed in the cell body and moves along axons by fast anterograde axoplasmic transport (Kang et al., 1987; Koo et al., 1990). TBI has been shown to increase APP expression (Ciallella et al., 2002) and disturb axonal transport, leading to APP accumulation or swelling as a hallmark of TAI (Bramlett et al., 1997; Gentleman et al., 1993; Kilinc et al., 2008; Sherriff et al., 1994). In agreement, we also found that the expression of APP increased in the CA3 region of hippocampi after fluid percussion TBI in vivo and in hNSC-derived neurons after rapid stretch injury in vitro. Furthermore, APP-immunoreactive swellings and bulbs were observed in the fimbria of the hippocampus and in stretch-injured neurons. Although APP was originally described and widely used as a marker for axonal injury (Gentleman et al., 1993), we found that APP swellings also appeared in the dendrites of neurons (as labeled by MAP2) after injury. In addition to abnormal APP transport, loss or shortening of axons and dendrites immunolabeled by MAP2 and TuJ1 indicated a loss of microtubules after traumatic injury, another key feature of TAI (Maxwell et al., 1997; Saatman et al., 1998). Given the recent discovery of APP being transported to axons and dendrites (Back et al., 2007), APP accumulation after injury may indicate a general transport deficiency of all neurites possibly caused by damage of microtubules. On the other hand, transplantation of hNSCs 24-h post-TBI or GDNF treatment at 30-min after stretch injury significantly reduced the abnormal accumulation of APP. Three possibilities may explain hNSC graft-mediated reduction of APP accumulation. First, hNSCs, via releasing GDNF, protect axons from trauma-induced pathological processes such as loss of microtubules and disturbance in intracellular levels of calcium, which then result in abnormal axonal transport. Second, grafted hNSCs may increase APP resolution; and third, they may delay APP accumulation. We believe that the first possibility is indicated by our in vivo data, and further supported by the evidence of GDNF-mediated protection against TAI in stretch injury in vitro, as this in vitro observation would seem to exclude the two latter possibilities. However, the single time point (4 days after injury) examined in our in vivo study, although allowing a detection of greater effects of hNSCs based on their time-dependent spreading, would not adequately distinguish the abovementioned possibilities. Therefore, further studies are required to assess the temporal effects of hNSC transplantation on APP accumulation.
In terms of the total expression levels of APP, cell transplantation did not significantly alter the TBI-induced APP increase in vivo, whereas GDNF treatment significantly reduced APP expression after stretch injury in vitro. Such a discrepancy may result from the nature of different experimental models, that is, a complex cell composition in the hippocampal CA3 tissue versus the relatively simple cell types of differentiated hNSCs in vitro. Alternatively, the difference of APP expression between in vivo and in vitro models may be caused by the different amounts of GDNF that neurons encountered, or the different time intervals after injury.
To explore the mechanisms of hNSCs and GDNF protective effects against TAI, we applied molecular and morphological analyses. We revealed that both in vivo and in vitro injuries increased the α-SMA expression in neurons. α-SMA is one of the six different actin isoforms, previously found primarily in peripheral cells, and it is widely used as a smooth muscle cell marker (Chaponnier et al., 2004). It is the major component of the stress fibers that are important for muscle contraction and cell migration (Goffin et al., 2006; Herman, 1993). However, expression and function of α-SMA have not been explored in neurons. Here, we report for the first time that α-SMA is expressed at a very low level in intact brains and neurons. Trauma, both TBI in vivo and rapid stretch injury in vitro, increased α-SMA expression in neurons, which was significantly reduced by hNSC transplantation and GDNF treatment. The latter was accompanied by elongation of neurites. Given the apparent injury-induced actin aggregation (visualized by phalloidin labeling) and the increased α-SMA expression accompanied by neurite shortening, we hypothesize that α-SMA may inhibit neurite regeneration through abnormal stress fiber formation. α-SMA upregulation may also represent one mechanism of innate neural protection against injury.
To understand how hNSCs and GDNF affect α-SMA expression and shortening of neurites, we focused on two downstream signaling molecules – calcineurin and Rho. Calcineurins, calcium, and calmodulin-dependent protein phosphatases are upregulated by GDNF in dopaminergic neurons (Consales et al., 2007), and required for axon outgrowth during development (Graef et al., 2003). Using Western blotting and mass spectrometry, we found that fluid percussion TBI significantly reduced the expression of the regulatory subunit CANB1 and catalytic subunit PP2BA in CA3 homogenates. This is different from previous reports that detected either no change or increased immunoreactivity of calcineurin subunits/isoforms in various regions of hippocampal formations after injury (Bales et al., 2010a,b; Kurz et al., 2005). Such discrepancies may be attributed to the different TBI models or different methods of detecting calcineurin expression. Using an in vitro stretch model, we confirmed that the injury decreased not only CANB1 expression, but also total calcineurin activity in neurons. Interestingly, transplantation of hNSCs in vivo and treatment by GDNF in vitro both reversed the trauma-induced CANB1 reduction, along with protecting axons and dendrites against injury. Exposing neurons to a specific calcineurin inhibitor, an autoinhibitory peptide, blocked the neurite protective effect of GDNF by maintaining injury-induced abnormal APP accumulations without affecting α-SMA. Therefore, our findings suggested that low calcineurin expression and activity contribute to the neurite injury. In contrast, several groups have previously observed that treatment with cyclosporine A or FK506, immunosuppressants known to block calcineurin activity, reduced APP+ profiles and secondary axotomy after traumatic injury (Buki et al., 1999; Okonkwo & Povlishock, 1999; Singleton et al., 2001; Staal et al., 2010). One possible explanation for this discrepancy is the different time points when administering the calcineurin inhibitors, i.e. post-injury in our case compared to pre-injury in other reports. Alternatively, the contradictory findings may be due to different calcineurin inhibitors. The inhibitor we used is an autoinhibitory peptide specifically targeting calcineurin. However, cyclosporine A and FK506, used by others not only block calcineurin, but also affect other targets, such as the mitochondrial permeability transition pore and intracellular calcium release channels, respectively (Hausenloy et al., 2009; Lanner et al., 2010). These effectors are known to regulate the intracellular levels of calcium that play critical roles in TAI. Taken together, these data indicate that protecting neurites against traumatic injury requires a certain level of calcineurin. A balanced calcineurin activity has also been suggested to be important in maintaining normal neuronal functions (Groth et al., 2003; Lee & Ahnn, 2004; Mansuy, 2003; Riedel, 1999).
Neurite sprouting has been reported in brains after TBI-induced TAI (Erb et al., 1991; Greer et al., 2011; Phillips & Reeves, 2001). However, such injury-induced sprouting is usually inefficient, but may be enhanced by applying various neurotrophic factors, including GDNF. In the context of our newly discovered α-SMA expression in the brain, we found that changing calcineurin activity had no effect on injury-induced α-SMA increases. We then focused on another type of downstream signaling molecule in the GDNF pathway, Rho GTPases. Particularly, RhoA is known to inhibit neurite outgrowth (Mackay et al., 1995), and such an inhibitory effect can be blocked by GDNF (Yoong et al., 2009). Indeed, blocking Rho activity, for only 4 h at 4 days post-injury, significantly reduced α-SMA expression and actin aggregation. This was accompanied by increased neurite length. Previous studies hypothesized that Rho activation in neurons may promote a redistribution of actin and/or formation of stress fiber-like actin bundles (Borisoff et al., 2003). Here, we provide evidence for the stress-fiber forming actin, α-SMA, being increased by traumatic injury, possibly through the enhancement of Rho activity. Therefore, blocking increases of α-SMA may be one of the mechanisms for GDNF to promote neurite outgrowth. Further studies are needed to explore the roles of α-SMA in the CNS, which may lead to a new avenue to facilitate recovery after TBI and other injures.
Conclusion
In summary, our results demonstrate that hNSC transplantation and GDNF treatment protect axons and dendrites against traumatic injury and promote neurite regeneration. GDNF reduces TAI via balancing calcineurin activities and maintaining normal intracellular transport. It also enhances neurite outgrowth by blocking Rho-mediated α-SMA expression and abnormal actin aggregation. The beneficial effects of GDNF against TAI (demonstrated in this study) and protecting host neurons reported by others (Bonde et al., 2000; Kim et al., 2001; Kitagawa et al., 1999; Minnich et al., 2010; Wang et al., 1997) indicate a clinical potential of GDNF in treating TBI patients. GDNF, as a large protein molecule, may be delivered into injured brains by direct protein infusion (Bonde et al., 2000; Kim et al., 2001), gene delivery via viral vector (Pertusa et al., 2008), and transplantation of NSCs as we report here. The advantages of cell-mediated GDNF delivery include a stable production of naturally bioactive GDNF, a targeted delivery caused by the nature of NSCs migrating toward the injury site, and the additional potential benefits from NSC-mediated replacement of neurons and oligodendrocytes. However, further methodological details need to be resolved before achieving an exact and well-controlled cell transplantation-mediated GDNF delivery system in both experimental and clinical settings.
Footnotes
Acknowledgments
We thank Sigmund J. Haidacher, Yaping Zeng, Wenru Zhang, and Jason R. Thonhoff for technical support. This work was supported by the Department of the Army (W81XWH-08-2-0137 to P.W.) via the United States Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick, MD 21702-5014. The content of this work does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred. Other support for this study came from the Coalition for Brain Injury Research (P.W.), the Moody Center for Traumatic Brain & Spinal Cord Injury Research/Mission Connect (P.W.), the TIRR Foundation (P.W.), the China Scholarship Council (E.W.), the John S. Dunn Research Foundation (P.W.), and the Cullen Foundation (P.W.).
Author Disclosure Statement
No competing financial interests exist.