
Abstract
The family of the collapsin response mediator proteins (CRMPs) plays a significant physiological role in neuronal cell bodies and axons within the integrated mammalian central nervous system (CNS). Trauma-induced damage to the CNS results in variable degrees of axonal degeneration, and this may lead to neuronal cell death in key grey matter regions. Site-specific phosphorylation of certain CRMPs has been associated with trauma-induced axonal degeneration. Moreover, recent data implicate the pro-apoptotic, calcium-dependent protease calpain as a key initiator of CRMP cleavage. The primary cleavage product of injury-induced neuronal calpain activation is a C-terminus truncated 55- to 58-kDa form of CRMP, which may exert its effects within the cytoplasm and axonal core, or alternatively through its translocation into the nucleus, initiating neuronal cell death. The precise structure of cleaved CRMP has yet to be elucidated, as is the reason for nuclear translocation. Once the crystal structure of the cytoplasmic and nuclear-translocated forms of CRMPs is determined, a greater molecular understanding of why these forms can initiate neurodegeneration following CNS injury will be established. Such information will be particularly informative in the design of inhibitors of specific protein-protein interaction sites between cleaved CRMP and vital cytosolic or nuclear molecules.
Introduction
T
Discussion
The CRMPs are a family of dihydropyrimidinase-related phosphoproteins, occasionally referred to in the literature as dihydropyrimidinase-like (DPYSL) proteins, which share sequence homology with the Unc-33-like phosphoprotein of Caenorhabditis elegans (Goshima et al., 1995). Mutations in the unc-33 gene in C. elegans produces axonal abnormalities and uncoordinated body movements (Tsuboi et al., 2005). The CRMPs, however, share their major homology with the uracil and thymine catabolism enzyme dihydropyrimidinase (DPYS), but although CRMPs maintain a dihydropyrimidinase domain (Fig. 1), they lack enzymatic activity (Wang and Strittmatter, 1996). There are 5 members of the CRMP family, as defined from screening rat brain cDNA libraries, and they are designated as CRMP 1, CRMP 2, CRMP 3, CRMP 4, and CRMP 5 (also known as CRAM in the rat; Wang and Strittmatter, 1996,1997). These proteins display 50–70% homology with their mature proteins, differentiating between 60 and 66 kDa on a SDS gel by electrophoresis (Fig. 1; Wang and Strittmatter, 1996,1997). All CRMPs are expressed intracellularly at high levels during development in the central nervous system (CNS), being responsive to extracellular guidance/inhibitory cues, but it is CRMP 2 that remains at high levels in the adult mammalian CNS within neurons and specific oligodendrocytes (Ricard et al., 2001). Alternative splicing at the mRNA level renders multiple variants of CRMPs, primarily at the N-terminus of the molecule, to form either a 75-kDa CRMP-A or a 62-kDa CRMP-B isoform (Fig. 1; Majava et al., 2008). In their native unphosphorylated form they can interact to produce homo- or hetero-tetramers (Fig. 1), and play an important role in associating with key regulatory proteins of the cytoskeleton, the axonal transport mechanism (vesicle trafficking), and the neuronal ion channels (Brittain et al., 2009; Kawano et al., 2005; Kimura et al., 2005; Wang and Strittmatter, 1997). However, in their phosphorylated or cleaved state they have been clearly linked with neurodegeneration, including neuronal cell death, during injury or disease (Mimura et al., 2006; Zhang et al., 2007).

The role of the collapsin response mediator proteins (CRMPs) in neuronal physiology and pathology. (

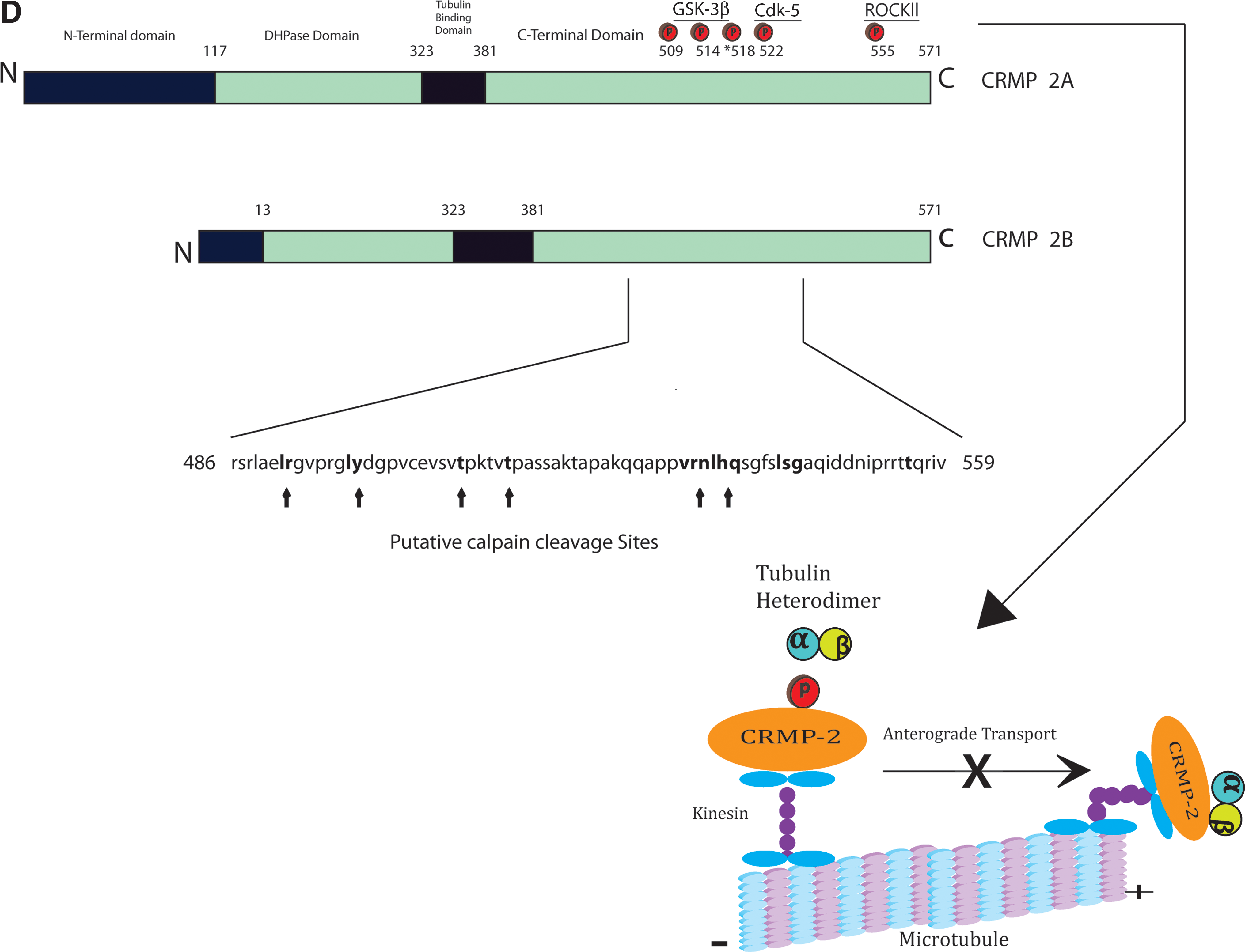
In experimentally-induced spinal cord injury models, there is suggestion that CRMP 2 specifically becomes phosphorylated in axons as they undergo degeneration (Mimura et al., 2006). The Cdk-5-dependent phosphorylated form of CRMP 2 (pSer522 CRMP 2) was shown to be increased in axons attempting regrowth in the chick spinal cord following a cervical transection injury at E11 and E15 in ovo (Gögel et al., 2010). However, ROCKII-dependent phosphorylation of CRMP 2 (pThr555 CRMP 2) was shown to be the predominant mechanism limiting regrowth of axons following an acute transection of spinal cord white matter tracts in rodents, and is initiated through myelin-associated inhibitory factors (MAIFs), myelin-associated glycoprotein (MAG), and Nogo-A (Mimura et al., 2006). In fact, these investigators showed that if these spinal cord-transected rats received the Rho kinase-specific inhibitor Y-27632, this abrogated the phosphorylation of CRMP 2, preserving axonal microtubules and limiting their degeneration. These data suggest that the phosphorylation of CRMP 2 is related to axonal degeneration following acute transection of spinal cord axons, which may well be induced through MAIF signaling.
Recently, Touma and associates (2007) demonstrated that CRMP 2 and CRMP 4 protein patterns were altered during the process of the “protrusion-like” beading formation, a classic sign of axonal and neuronal degeneration. The significance of this demonstration is that any alterations in the expression or cleavage of CRMP 2 and CRMP 4 will have an adverse effect on its ability to bind cytoskeletal proteins, thereby affecting the integrity of the axon. Moreover, the axonal abnormalities demonstrated in cell culture can be seen under neuronal cell stress conditions, which can be further illustrated by two-dimensional gel electrophoresis of neuronal protein fractions. Such techniques enable a comparison between those neurons that were exposed to nerve growth factor (NGF) and those deprived of NGF. Subsequent Western blotting analysis was also conducted and demonstrated that calpain-mediated cleavage of the 64-kDa form of CRMP 2 resolved into a truncated 58-kDa form upon growth factor withdrawal (Touma et al., 2007). The results indicated that an elevated amount of the cleaved 58-kDa form of CRMP 2 corresponded to the observed pattern of beading formation. As this cleaved form is devoid of the C-terminus, it will not enable CRMP 2 to bind to actin filaments and kinesin, disabling these molecules, thereby affecting the neuronal internal structure and transport mechanisms (Touma et al., 2007). It should be noted that due to the high proteolytic susceptibility of the C-terminus, the investigators were unable to determine its exact amino acid sequence. Moreover, the process of cleavage implicates calpain in the early stages of neuronal degeneration, possibly under calcium-independent conditions (Touma et al., 2007).
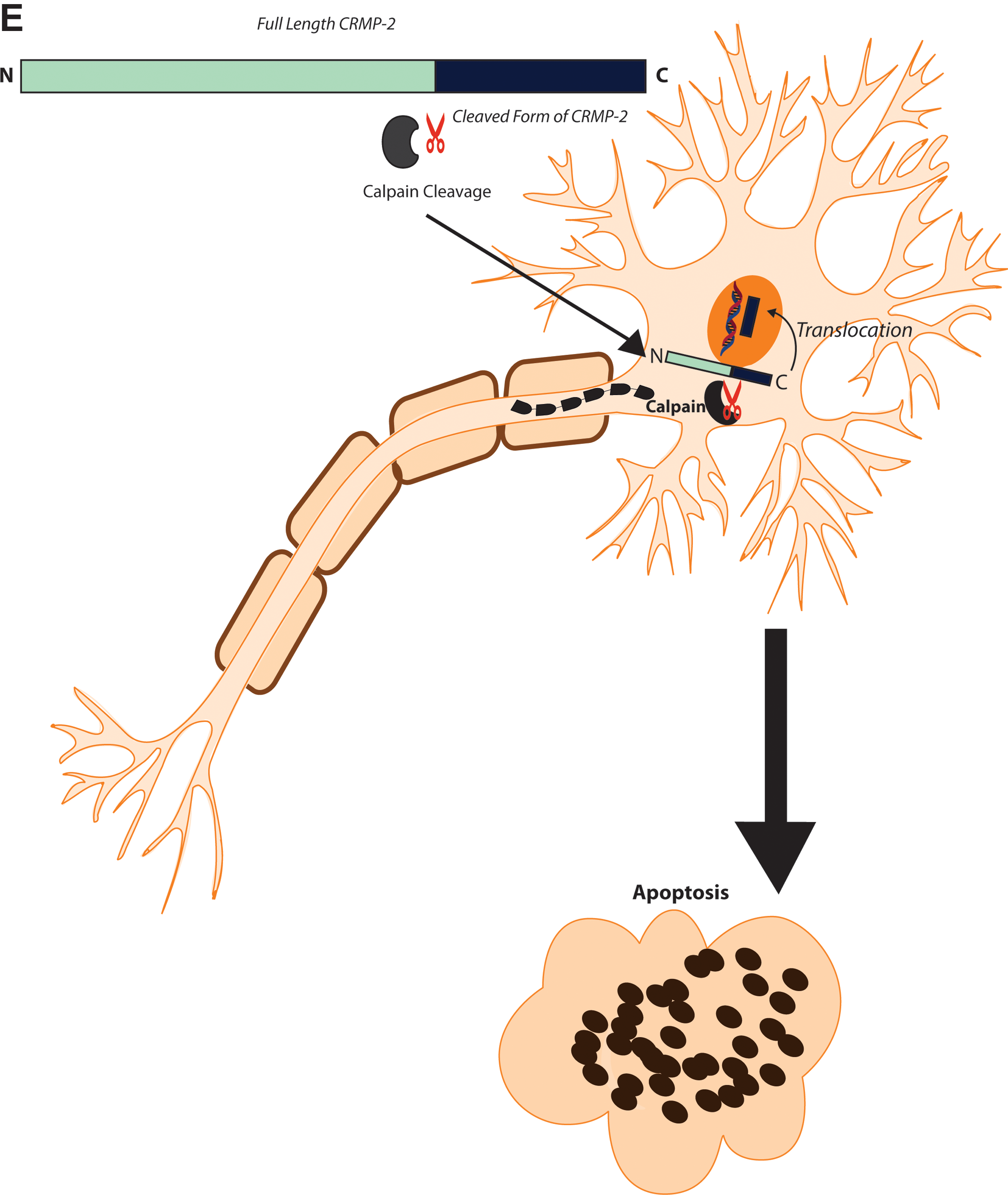
However, studies have implicated the role of calcium as a catalyst in initiating cleavage of CRMPs (Jiang et al., 2007). It has been observed that the accumulation of toxic levels of intracellular calcium ions activates calpains, which break down critical structural proteins affecting synaptic functions and causing neuronal cell death. Increased levels of the various CRMPs in ischemic brains were determined by immunofluorescence and Western blotting. Ischemic brains showed that CRMPs were cleaved from a band of 65-kDa to produce a smaller band of about 54-kDa. These data support an association between calpain activation and CRMP cleavage in cerebral ischemia. Moreover, the calpain cleavage product of CRMP 3 translocated into the nucleus, causing axonal retraction and neuronal death. Although not clearly defined, CRMP 2, CRMP 4, and CRMP 5 may undergo cleavage towards the C-terminus. It was noted by the authors that synaptosomal CRMPs are more sensitive to calpain cleavage than those from the cytoplasm (where less calcium was required for the process). These synaptosomal-processed proteins transmit ischemic injury signals from the distal axons to the cell body to cause neuronal degeneration. However, this is less likely to occur in physically-disrupted axons, which lose synaptic contact and therefore any retrograde transport of these cleaved CRMPs to the nucleus. This study highlights the significance of axonal degeneration regulated by calcium-dependent calpain-mediated CRMP cleavage.
The study by Zhang and colleagues (2009) extrapolated the role of calcium in neuronal cell death in relation to a calmodulin (CaM) concentration-dependent mechanism. Here it was postulated that when intracellular calcium levels increase, the calcium-sensitive protein CaM binds to CRMP 2 in the region of the C-terminal helix, between residues 491 and 572. This region entails hydrophobic amino acid residues of Phe 475, Ala 484, Leu 488, and Leu 491. The binding of CaM to CRMP 2 limits the ability of calpain to mediate proteolysis of CRMP 2, despite the proximity of the C-terminal region to the calpain cleavage site. The C-terminus binding site is also shared by CRMP 1–4, with CRMP 5 being the exception. This study proposes the hypothesis that CaM may facilitate CRMP 2 functions of growth cone formation and neurite outgrowth.
The phenomenon of translocation of cleaved CRMP fragments mentioned above has been identified as an important mechanism of cell death. Hou and associates (2006) investigated the role of CRMP 3 in excitotoxic induced neuronal death, mediated by cleavage of the protein by calpain, and the subsequent translocation of the cleaved fragment into the nucleus. Again by Western blot analysis, CRMP 3 appeared as a single band of 63-kDa, yet was shown to break down to a p54 fragment in glutamate-induced cerebellar granule neurons (CGNs) and focal cerebral ischemic brains. Further Western blotting and mass spectrometry studies confirmed the p54 fragment as being that of CRMP 3. The cleavage product of CRMP 3 functions as a positive injury signal to cause neuronal death after cerebral ischemia and excitotoxicity, most likely by spatial translocation into the nucleus. The translocation of the cleaved product of CRMP 3 would seem to be independent of a nuclear translocation signal, since a typical amino acid sequence for this physiological function could not be found by screening the p54 fragment (Hou et al., 2006). However, a plausible mechanism for nuclear localization of p54 may relate to a vimentin-dependent signal, as demonstrated for nerve injury-activated mitogen-activated protein kinase (MAP kinase; Perlson et al., 2004,2005).
These findings were subsequently supported by another study undertaken by Aylsworth and colleagues (2009). These investigators found that full-length CRMP 3 inhibited tubulin polymerization and neurite outgrowth in mature cerebellar granule neurons. Here, upon measurement of the lengths of microtubules, it was observed that the full-length CRMP 3 reduced the average lengths of microtubules, while the N-terminal-truncated CRMP 3 (p54) did not. Concerning the nuclear translocation of truncated CRMP 3, it was found that the D-domain played a significant role in mediating this process. The D-domain is a core region observed in all CRMPs, comprised of 57–413 amino acids (Fig. 1), exhibiting a strong degree of sequence homology to dihydropyrimidinase (DHP), an aminohydrolase (Aylsworth et al., 2009).
Evidence to support the importance of calpain-dependent CRMP cleavage has been presented through the utilization of a rat model of controlled cortical impact (CCI, an accepted model of TBI), which demonstrated the time-dependent appearance of a 55-kDa breakdown product in the rat hippocampus and cortex (Zhang et al., 2007). This study also simulated excitotoxic injury through the in vitro application of the glutamate receptor agonist N-methyl-D-aspartate (NMDA), and the calcium channel opener maitotoxin (MTX), in primary cortical neurons. The results revealed a marked reduction in intact CRMP 2 (66- and 62-kDa), and the appearance of a 55-kDa band. It is believed that this 55-kDa band is a breakdown product of CRMP 2, as observed in the rat TBI model, which may also be occurring in CRMP 1 and CRMP 4. The results also revealed the application of calpain inhibitors (SJA6017) in the maintenance of the 62-kDa fragment, prevention of the redistribution of CRMP 2 from neurites to the cell body, and the preservation of neuronal architecture. These data provide evidence concerning the role of calpain in mediating neuronal degeneration, possibly during the sequelae of TBI. The suggested calpain cleavage sites in this study were postulated to be between residues 486 and 551/9 (Fig. 1).
A more robust analysis of the CRMP breakdown products was performed using a differential neuroproteomics analysis, whereby differentiated proteins from pooled naïve or CCI-injured rat brains were analyzed with reversed-phase LC-MSMS (Kobeissy et al., 2006). The data clearly show a reduction in the level of full-length CRMP 2 (62-kDa) 48 h post-CCI in the rat brain, with a corresponding rise in the 55-kDa breakdown product. Similar breakdown products of αII-spectrin were observed post-TBI (Kobeissy et al., 2006), a common substrate for cysteine-proteases during cell death (Pineda et al., 2004; Czogalla and Sikorski, 2005). Very recently, a critical investigation of the role of cleaved CRMP-2 in neuronal death post-TBI has demonstrated that by therapeutically targeting the Ca2+ channel-binding domain (CBD3) of CRMP 2 after injury, this can have a neuroprotective effect, presumably by limiting Ca2+-sensitive, calpain-dependent cleavage of neuronal CRMP 2 (Brittain et al., 2011). These investigators showed that the CBD3 peptide of CRMP 2 fused to the HIV TAT protein (TAT-CBD3) could prevent calpain-mediated cleavage of CRMP 2, prevent glutamate-dependent excitotoxic neuronal cell death in vitro, and protect against ipsilateral hippocampal neuronal cell death following moderate TBI, after intraperitoneal infusion with TAT-CBD3. These data bring us closer to defining a central role for cleaved CRMP 2 in regulating neuronal cell death following TBI, and promotes the targeting of this mechanism as a neuroprotective therapy.
An alternative function of calpain is also evident in programmed cell death. Liu and colleagues (2009) investigated the role of calpain in potassium deprivation-induced apoptosis of CGNs. Apoptosis is a cellular process known to occur under certain physiological and neurological conditions (Kerr et al., 1972). In apoptosis of CGNs, CRMPs are cleaved by calpain, where specifically, it is the truncated forms of CRMP 3 and CRMP 4 that promote neuronal apoptosis (Hou et al., 2006). This is not the case with CRMPs 1 and 2 (Arimura et al., 2005). Thus proteolytic processing is an important regulatory mechanism of apoptosis. Proteolysis of structural proteins by calpain leads to the loss of membrane structural integrity and contraction of the cell body (Fischer et al., 2003). Calpain also cleaves other structural proteins, thereby affecting cell signal transduction, leading to cell death (Auth and Brawerman, 1992). This study also mentioned that the C-terminus of CRMP 1 can be phosphorylated, contributing to the inactivation of CRMP 1 in performing its normal function of actin and tubulin assembly.
The contribution of phosphorylation to inactivating CRMPs underscores the importance of this biochemical process to neuronal viability. The study by Hou and associates (2009) aimed to examine axonal changes following glutamate toxicity. It was found that blocking the phosphorylation of CRMP 2 by Ca/calmodulin-dependent protein kinase (CaMKII) was effective in protecting axons and neurons from damage. Additionally, CaMKII and calpain inhibitors were effective in reducing the appearance of varicosities and the thickness of axons. Thus it was deduced that the actions of calpain and CaMKII were essential in inducing axonal changes in response to glutamate toxicity. To answer the question of whether phosphorylation of CRMP 2 affects calpain cleavage, it was found that CRMP 2 is independently targeted by calpain and CaMKII in response to calcium activation. However, phosphorylation of CRMP 2 at Thr555 increases its resistance to calpain-induced cleavage (Hou et al., 2009).
Investigating the role of calpain in neuronal cell death may be extended to a close examination of the relative forms of the protease. The study by Bevers and associates (2009) aimed to evaluate the relative roles of m-calpain and mitochondrial μ-calpain in cultured primary hippocampal neurons following the induction of NMDA-mediated excitotoxicity. These isoforms exist as heterodimers, and differ in the level of calcium required for activation, where μ-calpain requires 400–800 μM of calcium, while μ-calpain requires 3–50 μM of calcium (Bevers et al., 2009). The knockdown of m-calpain, which improves survival of hippocampal neurons following NMDA toxicity, provides evidence that m-calpain can play a causal role in neuronal pathology. Study of this injury model also revealed that only CRMP 1 translocated to the nucleus following NMDA exposure.
Finally, Bretin and colleagues (2006) conducted a study that utilized neurons from mouse cultured cerebral cortex as an in vitro model to investigate the development of resistance by neurons following a secondary exposure to NMDA. Additionally, the study demonstrated that all CRMPs are cleaved in response to NMDA mediated by calpain. The physiological activation of calpain occurs secondary to NMDA receptor activation (Adamec et al., 1998). Antibody-specific experiments revealed a reduction in high-molecular-weight forms of CRMP 2 after NMDA treatment, with a corresponding increase in lower-molecular-weight forms. This observation supports the cleavage of CRMP 2 from the C-terminal sequence.
In conclusion, phosphorylation or cleavage of CRMPs and the subsequent translocation of the truncated fragments have been observed in experimental models of TBI/SCI. The cell culture evidence linking such cleaved fragments with axonal degeneration and eventual neuronal cell death, emphasize the need for systematic investigations of CRMP cleavage as a causative mechanism of axonal injury and neuronal death in CNS trauma. The literature provides strong evidence concerning the interaction between the family of CRMP proteins and calpain, and the latter mediates a site-specific cleavage with the onset of axonal damage. Consequently, events such as nuclear translocation of a given truncated segment have been observed to occur, thereby leading to irreversible neuronal cell death. The findings of the aforementioned studies raise the future prospect of directing pharmacological interventions to inhibit calpain-mediated cleavage of CRMPs in order to prevent ensuing axonal degeneration post-trauma.
Footnotes
Acknowledgments
S.P. was funded by a National Multiple Sclerosis Society (USA) Project Grant. K.T. was funded by a Multiple Sclerosis Research Australia Student Scholarship.
Author Disclosure Statement
No competing financial interests exist.