
Abstract
For many years lactate was considered to be a waste product of glycolysis. Data are accumulating that suggest that lactate is an important energy substrate for neurons during activation. In severe traumatic brain injury (TBI) glutamate release and ischemic cerebral blood flow (CBF) are major factors for a mismatch between energy demand and supply and for neuronal cell death. Although ATP and behavior could be improved by lactate treatment after TBI, no histological correlate nor any linkage to better astrocytic glutamate uptake or CBF as possible mechanisms have been described. We subjected male rats to a controlled cortical impact (CCI; 5 m/sec, 2.5 mm). To study the effects of lactate treatment on lesion volume, glutamate release, and CBF, animals were infused with either NaCl or 100 mM lactate for up to 3 h. The role of endogenous lactate was investigated by inhibiting transport with α-cyano-4-hydroxy-cinnamic acid (4-CIN; 90 mg/kg). Lactate treatment 15 min post-CCI reduced lesion volume from 21.1±2.8 mm3 to 12.1±1.9 mm3 at day 2 after CCI. Contusion produced a significant three- to fourfold increase of glutamate in microdialysates, but there was no significant difference between treatments that began 30 min before CCI. In this experiment lesion volume was significantly reduced by lactate at day 7 post-CCI (23.7±4 to 9.3±1–2 mm3). CBF increased immediately after CCI and dropped thereafter below baseline in all animals. Lactate infusion 15 min post-CCI elevated CBF for 20 min in 7 of 10 animals, whereas 7 of 8 NaCl-treated animals showed a further CBF decline. Neuroprotection was achieved by lactate treatment following contusion injury, whereas blocking of endogenous lactate transport exerted no adverse effects. Neuroprotection was not achieved by improved glutamate uptake into astrocytes, but was supported by augmented CBF following CCI. Due to its neuroprotective property, lactate might be a beneficial pharmacological treatment for TBI patients.
Introduction
G
This coupling led to the “energy-on-demand” working hypothesis by Magistretti and co-workers (Magistretti et al., 1999). Restoration of ionic homeostasis is a major force that drives energy production to maintain function of ATP-dependent ionic pumps. According to the energy-on-demand hypothesis (Magistretti et al., 1999), glycolysis is turned on by re-uptake of glutamate that is accompanied by 3 Na+ ions, causing ionic imbalance in astrocytes and thus turning on ATP-dependent ion pumps. This cascade induces an overproduction of lactate that is shuttled to the extracellular space, and from there into neurons by monocarboxylate transporters (MCT). There is growing evidence that lactate can serve as an energy source for neurons (Alessandri et al., 2008; Schurr and Payne, 2007), and that it is even preferred over glucose under certain conditions (Larrabee, 1996). Once in neurons, lactate is metabolized for immediate energy production. This is associated with a simultaneous decrease of glucose uptake into neurons, leaving this energy substrate for astrocytes (Erlichman et al., 2008). Furthermore, electrophysiological function can be maintained for long periods by lactate as the sole energy source (Schurr et al., 1988).
Traumatic brain injury (TBI) imposes a massive increase in energy demand on neurons and astrocytes. This is partially due to a markedly increased glutamate level after TBI in the extracellular space (Palmer et al., 1993; Rose et al., 2002). A high glutamate concentration is known to be excitotoxic to neurons and should therefore be removed from the extracellular space by astrocytes as quickly as possible (Alessandri and Bullock, 1998). As a consequence of extracellular and intracellular ionic imbalance following neuronal activation and glutamate uptake into astrocytes, energy production has to take place. Unfortunately, TBI is also associated with regional hypoperfusion (Bryan et al., 1995; Kochanek et al., 1995; Reinert et al., 2000; Thomale et al., 2002), which leads to a mismatch between energy demand and supply. Thus, in addition to improving perfusion, an effective treatment regimen would also increase blood and brain glucose levels. Experimental and clinical studies indicate, however, that this approach could adversely affect injury sequelae following TBI. Post-traumatic infusion of 5% dextrose over 30 min worsens edema formation and neurological outcome following closed head injury in rats (Feldman, et al., 1995). Elevation of blood glucose by glucose infusion prior to, but not after, controlled cortical impact (CCI) injury in rats worsened the contusion significantly (Cherian et al., 1998a). If a secondary ischemic insult was added, even post-traumatic hyperglycemia augmented brain damage (Cherian et al., 1998b). Furthermore, Kinoshita and associates showed that although acute post-traumatic dextrose treatment increased lesion area only slightly, a significant infiltration of neutrophils was seen following fluid percussion injury (FPI; Kinoshita et al., 2002). In contrast, the results of several experimental studies investigating hyperglycemia following TBI did not support adverse effects on brain damage (Gurevich et al., 1997; Hill et al., 2010; Stover et al., 2002). Clinical studies, however, show that hyperglycemia at admission is associated with poor neurological outcomes and mortality following severe TBI, both in pediatric (Melo et al., 2010; Smith, et al., 2012) and adult patients (Jeremitsky et al., 2005; Pecha et al., 2011). In order to avoid the adverse effects of glucose, another tested treatment regimen might involve the infusion of lactate instead of glucose. Several in vitro and in vivo studies indicate that this strategy improves functional recovery after ischemia (Schurr et al., 1997,1988,1989), and TBI (Holloway et al., 2007; Levasseur et al., 2006; Rice et al., 2002). Venous infusion of 100 mM lactate for 3 h improved not only mitochondrial oxidative metabolism, but also ATP production following FPI in rats. A bolus injection of 100 mM lactate also enhanced the evoked reaction of CBF to light stimuli in the visual cortex of rats (Ido et al., 2004), and healthy volunteers (Mintun et al., 2004). Both of these mechanisms, improved energy production and increased CBF, could contribute to improved uptake of extracellular glutamate into astrocytes, and subsequently better outcomes post-TBI. On the other hand, an in vitro study by Levasseur and colleagues (Levasseur et al., 2006) indicated that lactate also increases oxygen consumption. This could hamper or reduce the neuroprotective effectiveness of lactate, especially if oxygen delivery is reduced after TBI. Thus it might be necessary to combine lactate treatment with an elevation of the fraction of inspired oxygen (F
Methods
Animals
All experiments were performed under animal welfare guidelines, and were approved by the local ethics committee. We used male Sprague-Dawley rats (weight 319–453 g) for all four experiments. They were housed in single cages after surgery, and had access to food and water ad libitum. The animal room had a 12-h:12-h light:dark cycle and 50% humidity.
Anesthesia and surgical preparation
All rats were anesthetized with chloral hydrate (36 mg/mL), at an initial dose of 1 mL/100 g body weight IP. Thereafter, 1 mL of chloral hydrate was given every hour or whenever necessary through an IP catheter. Before surgery, 1 mg/mL of atropine was injected. The rats were mechanically ventilated during surgical preparation, the infusion of NaCl or lactate, and acute monitoring of physiological parameters, CBF, and extracellular glutamate by microdialysis. Thereafter the animals were extubated. The rats were not intubated for the 4-CIN study.
The rats were placed on a temperature-controlled heating pad, and body temperature was maintained at 37±0.5°C using a rectal temperature probe. The femoral artery and vein were cannulated with polyethylene tubing (OD 0.96 mm) for continuous mean arterial blood pressure (MABP) measurement, blood gas, and lactate analysis, and saline or lactate infusion.
The animals were fixed in a stereotaxic frame. The skull was exposed and thoroughly cleaned and disinfected (3% H2O2). A craniectomy (7 mm diameter) was carefully made using a high-speed drill between the bregma and lambda sutures for placing the CCI device. Only animals with an intact dura mater were used for the experiment. Depending on the particular experiment, additional burr holes were drilled for the placement of microdialysis probes, or skull areas 2×2 mm in size were thinned out for CBF scanning (Fig. 1).

Picture of the CCI apparatus and schematic drawing of the rat skull. The main elements of the CCI device are: (1) a pneumatically-driven piston (0.6 mm diameter), (2) a photocell for measuring the velocity of the CCI, and (3) a screw with which injury depth can be adjusted. The entire CCI device is fixed to a movable arm (4), which is mounted on a stereotaxic frame (5). The enlargement shows the tip of the piston after passing though the screw, which is placed on the surface of the brain before injury. Circles on the schematic drawing indicate locations for craniectomy for CCI injury (A), and placement of burr holes for microdialysis probe insertion, or 2×2-mm areas that were thinned for CBF measurement (B and C). The semipermeable microdialysis tip was placed underneath the injury site. Microdialysis and CBF measurements were not performed in the same experiment (CCI, controlled cortical impact; CBF, cerebral blood flow).
Controlled cortical impact injury
CCI was performed using a pneumatically-driven impact device (produced by L. Kopacz, University Medical Center Mainz), with a concave tip 6 mm in diameter. The injury parameters were: 5 m/sec impact velocity, 2.5 mm injury depth, and 400 msec dwell time. Impact was applied perpendicularly to the brain surface and caused a small subdural hemorrhage in most animals. After each experiment the wounds were closed and the animals were returned to their cages for up to 7 days.
Microdialysis and high-performance liquid chromatography (HPLC)
Flexible microdialysis probes (CMA/20, 4 mm membrane; CMA/Microdialysis, Solna, Sweden) were manually implanted into the cortex of each hemisphere through two burr holes. The tip of each probe was placed in the gray matter directly underneath the craniectomy, and the probes were left in place during injury. Artificial cerebrospinal fluid (CSF) was perfused through the microdialysis probes at a flow rate of 2 μL/min. After an equilibration period of at least 60 min, four 10-min baseline samples were collected. After CCI, samples were taken every 5 min for 30 min, then every 10 min for another 30 min, and thereafter every 20 min until the end of the experiment. All samples were immediately frozen at −20°C until analysis. Only data from animals with probes placed in cortical tissue were used.
All samples were analyzed for glutamate by HPLC using a standard method (Stover et al., 1997). Briefly, 10 μL of dialysate was mixed with 10 μL ortho-phthaldialdehyde (Sigma-Aldrich, St. Louis, MO) and vortexed. After 60 sec the mixture was injected into the HPLC system, consisting of a pump connected to an injection valve with a 10-μL loop, a C18 column (3 μm), and a UV detector. Glutamate was detected at a wavelength of 270 nm and recorded and analyzed with ValueChrom software (v4.0; Bio-Rad, Hercules, CA). Values were compared to a 10-mM glutamate standard solution.
Cerebral blood flow scanning
A laser Doppler probe was attached to a micromanipulator (KM 10; Wagner Instrumentenbau, Schoffengrund, Germany), and connected to an LD monitor BPM 403 A (Laserflo; TSI Inc., St. Paul, MN). For CBF scanning 25 positions were predefined in a grid (1×1-mm squares) on the thinned out skull area over each hemisphere (Soehle et al., 2000). During each scan (180 sec) CBF was recorded at each position. After baseline scans over both hemispheres, post-trauma scans were started 1 min post-CCI on the ipsilateral and 5 min post-CCI on the contralateral hemisphere. Thereafter nine additional scans were performed every 10 min.
Histological analysis
Before removing the brains the animals were anesthetized with chloral hydrate and perfusion-fixed using heparinized saline (10 IU/mL) and 4% paraformaldehyde (pH 7.4) transcardially. The brains were carefully removed and post-fixed for 24 h. Thereafter, the brains were embedded in paraffin and 3-μm-thick sections were cut throughout the injury area spaced 250 μm apart. The sections were stained with hematoxylin and eosin, and the injured area was measured using an image analyzing system (Alessandri et al., 2006). Briefly, the injured tissue was analyzed in the cortex by encircling the area of damaged tissue on all digitized brain sections. The lesion border was verified by cell morphology and staining color. If a cavity had formed it was included in the lesion size calculation. In this case the gap between the intact medial and lateral cortical surface of the injured hemisphere was considered as the lesion border.
Experimental design
This study consisted of four experimental series. Three experimental series investigated the effects of lactate treatment (100 mM at 0.65 mL/h=5.9 mg/h/animal; pH 5.9) on neuroprotection (study 1), glutamate release (study 2), and cerebral blood flow (study 3). In study 4 the effect of increased endogenous lactate after CCI on brain damage was investigated by blocking the monocarboxylate transporter (MCT).
Lactate treatment and neuroprotection
Thirty-five rats were divided into four traumatized groups, receiving either physiological saline (0.9% NaCl, n=8), or lactic acid (100 mM in NaCl, n=9), at 21% F
Lactate treatment and glutamate release
Nineteen rats were divided into two traumatized groups, receiving NaCl (n=9) or lactate (100 mM; n=10M). All animals received microdialysis probes implanted into cortical tissues. In order to ensure that exogenous lactate was present at the time of injury, treatment was begun 30 min before CCI. NaCl and lactate infusions were continued for 3 h at 0.65 mL/h. Lactate-treated rats received 44.6±13 mg/kg body weight lactic acid. The final five animals randomized to each group were allowed to survive for 7 days, and the brains were then removed for histological assessment of lesion volume. The positions of the microdialysis probes in the brain tissue were checked histologically in these animals.
Lactate treatment and CBF changes
Eighteen rats were divided into two traumatized groups, receiving NaCl (n=8) or lactic acid (100 mM; n=10). In all animals a skull area frontal to the injury site was thinned out on both hemispheres. CBF was measured by laser Doppler scanning (Soehle et al., 2000). Treatment started 15 min after CCI. NaCl and lactate infusions were continued for 90 min at 0.65 mL/h. Lactate-treated animals received 23.4±1.2 mg/kg body weight lactic acid.
Endogenous lactate and neuroprotection
Rats were subjected to sham surgery (n=6) or CCI injury, in combination with either IP injection of vehicle (Tris-buffered saline; n=6), or 90 mg/kg 4-CIN (n=6), an inhibitor of the MCT. IP injections were given 60 min before CCI or sham surgery (Schurr et al., 2001). Sham animals received all surgical procedures but no CCI, and were treated with 4-CIN. The animals were euthanized on day 7 after injury, and the brains were removed and analyzed for injury volume.
Statistical analysis
All data are given as mean±standard error of the mean (SEM), and were statistically analyzed with SigmaStat for Windows 3.1 (Systat, San Jose, CA). Histological and physiological data of the series exploring lactate treatment and neuroprotection, and endogenous lactate and neuroprotection, were analyzed by a one-way analysis of variance (ANOVA), followed by a Student-Newman-Keuls post-hoc test for individual comparisons at each time point. Physiological and histological data of the two groups exploring lactate treatment and glutamate, and lactate treatment and CBF, were compared with Student's t-test. Glutamate and CBF values were compared using an one-way and a two-way ANOVA for repeated measures (Student-Newman-Keuls post-hoc test). Area under the curve (AUC) was calculated for post-CCI glutamate release and post-infusion percentage CBF changes, and compared by Student's t-test.
Results
Lactate treatment and neuroprotection
Except for blood oxygen levels all physiological parameters were within the normal range throughout monitoring (Table 1). Increasing inspired oxygen from 21% to 50% elevated P
p<0.001 versus saline.
Lactate treatment was given before (−5 min) and after (+10 and+120 min) CCI injury in rats. The animals were treated with either NaCl 0.9% or lactate (100 mM, 0.65 mL/h for 3 h) without (F
CCI, controlled cortical impact; P
Injury severity was similar in all groups, namely 5.07±0.04 m/sec (NaCl+21% F
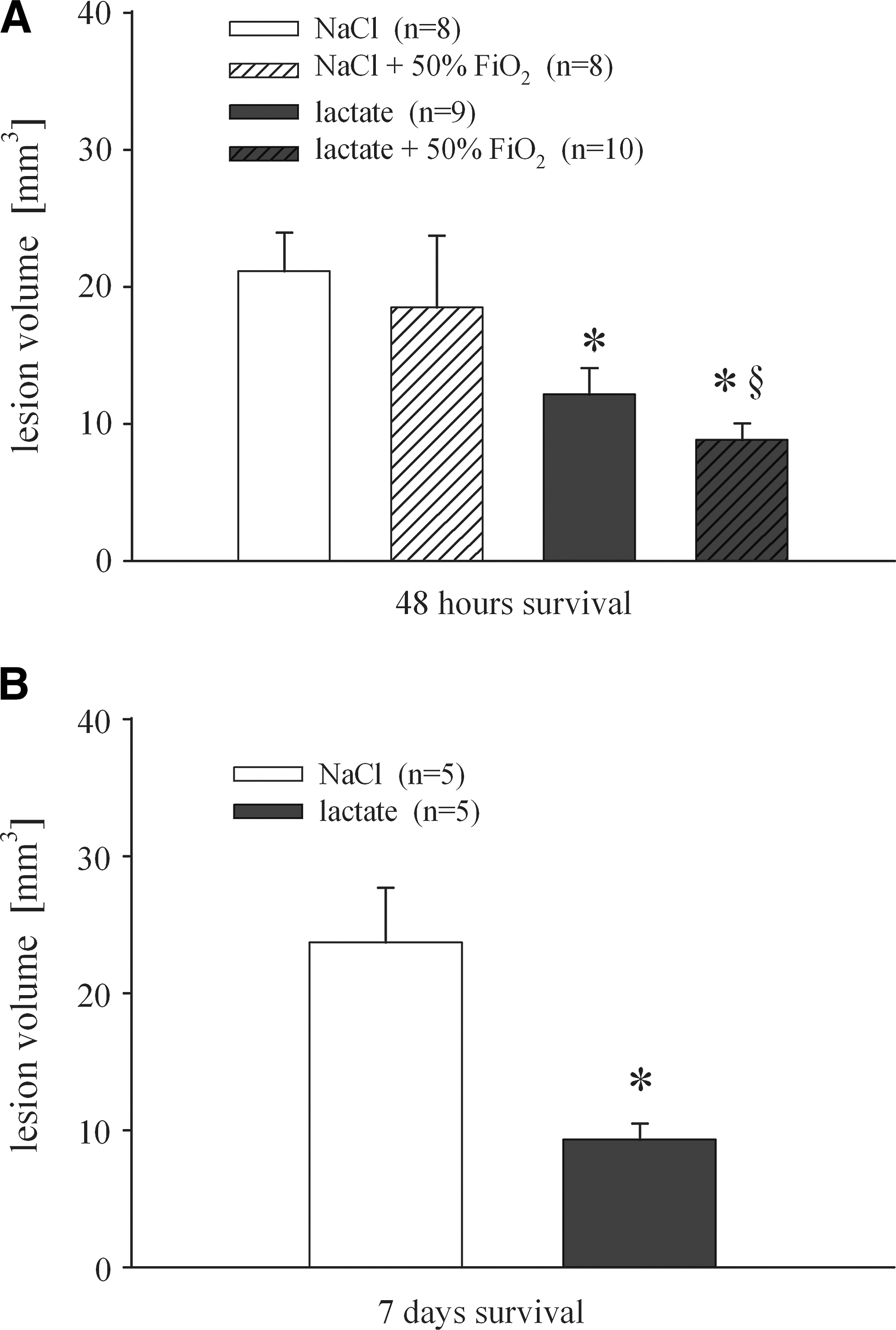
Lesion volume 2 days (
Lactate treatment and glutamate release
Most physiological values were within the normal range. MABP was significantly higher for the lactate-treated group at baseline (84.7±2.9 versus 92.8±0.8 mm Hg), and 15 min after injury (82.7±2.7 versus 90.3±0.6 mm Hg). Thereafter, no differences were found between groups (Supplementary Table S1; see online supplementary material at
As depicted in Figure 3, microdialysis glutamate levels were calculated as percent change from baseline in order to compensate for baseline differences and variations in probe performance. Ipsilateral measured glutamate concentrations in microdialysates at baseline were 1.68±0.2 μM for NaCl-treated and 1.47±0.4 μM for lactate-treated animals. CCI injury elevated dialysate glutamate to 5.56±1.6 μM and 6.08±2.2 μM in the NaCl and lactate groups, respectively. A two-way ANOVA of repeated measures showed a significant increase of glutamate by CCI (p<0.05; Fig. 3), and indicated a time effect (p<0.001), but no interaction of group×time (p=0.75). The groups did not differ from each other at any time point. Glutamate levels nearly reached baseline values within 15 min, and increased slightly toward the end of monitoring (2.4±0.8 μM and 4.0±2.4 μM; n.s.). Contralateral baseline values were 1.5±0.4 μM (NaCl) and 1.8±0.4 μM (lactate). Thereafter, glutamate was not affected significantly by CCI or treatment in the contralateral hemisphere (Fig. 3). Calculation of the area under the curve from the start of injury to the end of monitoring as an indicator of total glutamate release did not reveal a significant difference between groups in either hemisphere. Although injury severity was comparable (NaCl=5.10±0.04 m/sec; lactate=5.18±0.06 m/sec), lesion volume analyzed at day 7 post-CCI was significantly different between the two injury groups (NaCl=23.7±4 mm3; lactate=9.3±1–2 mm3; Fig. 2B).

Extracellular glutamate collected by microdialysis from the ipsilateral (top panel) and contralateral (bottom panel) hemispheres before and after CCI injury (arrow). NaCl and lactate (100 mM) infusion was started 30 min before CCI at a flow rate of 0.65 mL/h (gray bar). Glutamate values are expressed as percent change from baseline, and displayed as mean±SEM (§significant difference from baseline by one-way ANOVA for repeated measures, p<0.05). Note that there was no significant difference between groups at any time point of monitoring. The area under the curve from the start of CCI to end of monitoring was also not different between groups (ANOVA, analysis of variance; CCI, controlled cortical impact; SEM, standard error of the mean).
Lactate treatment and CBF changes
As depicted in Supplementary Table S2 (see online supplementary material at
One-way and two-way ANOVAs for repeated measures of percent CBF changes indicate a significant increase immediately after CCI (1 min post-CCI: p<0.05), which was followed by a significant drop below baseline for both groups (10 min post-CCI: p<0.05). CBF of NaCl-treated animals remained significantly below baseline throughout the monitoring period (Fig. 4). The statistical analysis also revealed an effect for time (p<0.001), and was near significance for the interaction group×time (p=0.061). As depicted in Figure 4, lactate but not NaCl caused an elevation of ipsilateral CBF immediately following the start of infusion. Seven of 10 lactate-treated animals showed an increase to 117–182% compared to the 10-min post-CCI value (the time point prior to start of treatment), whereas three animals had decreased CBF to 56%, 83%, and 97%, of pre-infusion values. On the other hand, CBF dropped in most NaCl-treated animals (7 of 8), to 94–66%, compared to the 10-min post-CCI time point. Only one animal showed an increase (129%). At 15-min post-infusion (30 min post-CCI) the same number of animals remained elevated in the lactate-treated group. This short-term effect is reflected by a significant difference in the AUC from the start of the infusion (two-tailed t-test: p=0.047; AUC for the entire time-course: p=0.198).
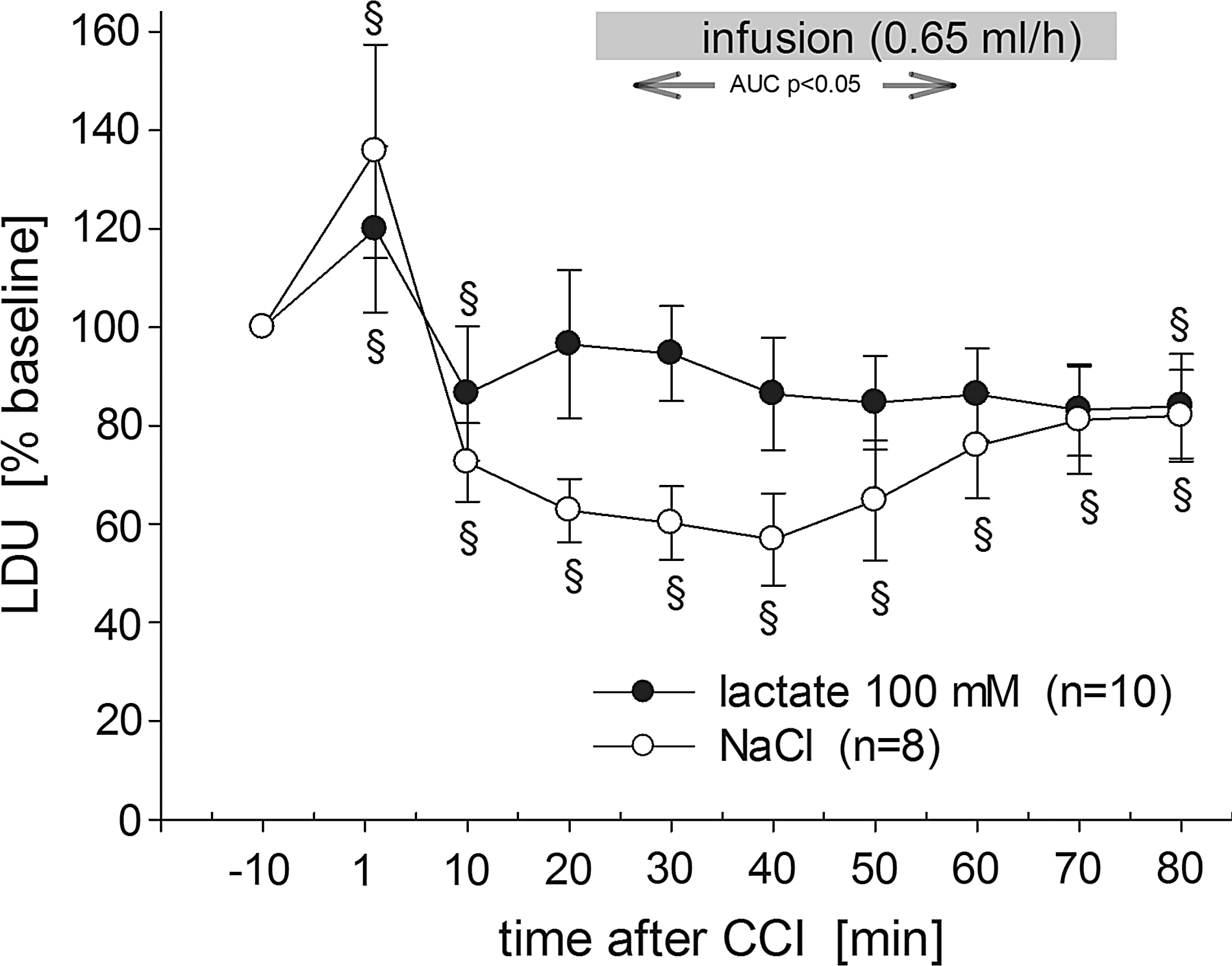
Percent changes of CBF (measured as laser Doppler units) before and during lactate or saline infusion (gray bar). LDU values were normalized to 100% of baseline and all values are given as mean±SEM (§significant difference from baseline by one-way ANOVA for repeated measures, p<0.05). Calculation of area under the curve during lactate and NaCl infusion revealed a short-term significant difference between groups (p<0.05; ANOVA, analysis of variance LDU, laser doppler units; CBF, cerebral blood flow; SEM, standard error of the mean).
Endogenous lactate and neuroprotection
Since the animals were not mechanically ventilated, arterial carbon dioxide was elevated in all groups compared to other studies. Although partial carbon dioxide pressure (P
Discussion
Lactate has long been known to be the end product of glycolysis. There is growing evidence that lactate is an important energy source for neurons during activation, and that treatment with lactate can improve behavioral and metabolic outcomes after TBI; however, a histological link has been missing. In this study we demonstrated clear histological neuroprotection by lactate treatment. Treatment for 3 h reduced lesion volume from 21.1±2.8 to 12.1±1.9 mm3 at 2 days after CCI (Fig. 2A). A similar difference in lesion volume of around 50% was seen after 7 days (23.7±4 and 9.3±1–2 mm3; Fig. 2B).
The histological results provide a strong link between the discovery of subtypes of monocarboxylate transporters (MCT) and lactate dehydrogenase (LDH) in astrocytes, neurons, and mitochondria, and the metabolism of lactate in brain cells and lactate as a neuroprotectant following TBI (Alessandri et al., 2008; Laughton et al., 2007; O'Brien et al., 2007; Schurr, 2006).
Evidence for lactate as an energy source in the brain is provided by in vitro and in vivo studies (Alessandri et al., 2008; Schurr and Payne, 2007). For instance, using radioactive-labeled glucose and lactate, Larrabee (Larrabee, 1995,1996) showed that a concentration of 1 mM of lactate suppresses CO2 production from glucose (i.e., oxidative phosphorylation in mitochondria). At in vitro concentrations above 2 mM a larger portion of CO2 is suppressed by lactate metabolism. The “energy on demand” hypothesis by Magistretti and co-workers further consolidated lactate as an energy source (Magistretti et al., 1999). Their theory suggests that neuronal stimulation activates astrocytic glycolysis, resulting in a high output of lactate. Lactate is then shuttled through MCT to neurons, where it is used as a preferred energy source. This is in accord with the fact that treatment of rats with lactate changes lactate/glucose dynamics in the extracellular space following TBI (Chen et al., 2000a), and that blocking lactate transport before global ischemia worsens histological outcomes (Schurr et al., 2001). Therefore, increased lactate production following traumatic events or treatment with lactate may be important for astrocytes, as well as for neurons, to support cell survival.
As for glucose, lactate metabolism can only occur in the presence of some oxygen and functional mitochondria. If TBI is accompanied by severe ischemia/hypoxia, lactate cannot be metabolized and becomes a marker of poor outcome (Goodman et al., 1999). In the peri-contusional area, however, hyperglycolysis is paralleled by high lactate output in the presence of oxygen and CBF adequate for neurons to use lactate for ATP production. Consequently, elevation of oxygen delivery or prevention of endogenous lactate to enter neurons most likely influences lactate-induced processes and neuronal metabolism. For instance, F

Effect of the monocarboxylate transporter inhibitor α-cyano-4-hydroxy-cinnamic acid (4-CIN) on lesion volume (mm3) 7 days after controlled cortical impact (CCI) injury. The animals were injected intraperitoneally with a single bolus of either vehicle (Tris-buffered saline), or 90 mg/kg body weight 4-CIN 60 min before CCI. Values are given as mean±standard error of the mean. Sham
Nevertheless, lactate stimulates oxygen consumption in vitro, which has to be considered if lactate is to be used for therapy. This stimulating effect can be blocked by 4-CIN, suggesting a strong effect of lactate on oxidative metabolism (Alessandri et al., 2008). If lactate cannot reach neurons due to blocking of MCT by 4-CIN, neuronal but not glial uptake of glucose is increased (Erlichman et al., 2008). Under pathophysiological conditions, blocking the MCT-2 transporter abolished neuroprotection by lactate in a model of global ischemia (Schurr et al., 2001). We could not replicate the findings of Schurr and associates (Schurr et al., 2001), that 4-CIN augmented brain damage following global ischemia in our CCI model (Fig. 5), although we used the same injection route and dose. It is possible that differences in the time course and degree of glutamate or lactate release influence the effects of 4-CIN differently in global ischemia and TBI. Our glutamate microdialysis levels changed briefly to elevated levels similar to those reported by Palmer and colleagues (Palmer et al., 1993). If the trauma is more severe, more pronounced and prolonged glutamate and lactate release occurs (Fukushima et al., 2009; Palmer et al., 1993; Rose et al., 2002). In a model of focal ischemia, reperfusion seems to further elevate and extend glutamate and lactate release (Lin et al., 2002), making an effect of 4-CIN more likely. In addition to the short-term effects of CCI on glutamate seen in our study (Fig. 3), factors such as the vehicle used (Tris-buffered saline versus saline), or the specificity of 4-CIN, may also have influenced 4-CIN in our experiment. For instance, 4-CIN has no effect on retinal ischemia, and disturbs trafficking of pyruvate into mitochondria to a higher degree than trafficking of lactate to neurons (Melena et al., 2003). Therefore, our 4-CIN data may indicate that CCI-induced endogenous lactate production was insufficient to support cell survival.
In contrast, treatment with 100 mM lactate for several hours was neuroprotective in a model of focal contusion (Fig. 2), and this seems to be an ideal treatment regimen for TBI (Holloway et al., 2007). The same treatment improved not only learning and memory performance in the Morris water maze (Rice et al., 2002), but partially restored oxygen consumption (Levasseur et al., 2006), and ATP (Holloway et al., 2007), following FPI. Rice and associates reported increased cell numbers in the hippocampal CA1 and CA3 by lactate compared to saline treatment, but the differences were not significant (Rice et al., 2002). A neuroprotective property of lactate has been shown in a model of glutamate neurotoxicity (Ros et al., 2001). Chen and colleagues demonstrated that systemically-infused lactate reaches tissue at risk after FPI (Chen et al., 2000b,2000c). They showed that radioactivity from IV-injected labeled lactate increased substantially underneath the injury site. This led to improved recovery of extracellular glucose (Chen et al., 2000a), and preservation of mitochondrial oxidative metabolism (Levasseur et al., 2006), and of ATP (Holloway et al., 2007) following FPI. In our CCI model the degree of energy demand and supply in the peri-contusional area is unknown. Severity of energy failure, however, can be estimated from the work of the Marklund group (Marklund et al., 2006), or CBF measurements (Engel et al., 2008; Friedrich et al., 2000). Mild CCI injury (1.5-mm injury depth) caused a temporary decline in ATP, which recovered to normal levels, whereas a severe CCI (2.5 mm) induced a longer-lasting change. In our study, regional CBF dropped by 20.7% at a distance of about 3 mm from the impact site. In other CCI studies, CBF dropped by 20–50% in the peri-contusional area within the first few hours post-CCI (Engel et al., 2008; Friedrich et al., 2000; Thomale et al., 2002,2004). Thus the energy failure is not severe, but a temporary mismatch between demand and supply could contribute to the pathophysiology seen in our model.
In this line of reasoning we assume that the neuroprotective effect of the lactate infusion was at least partially due to direct support of energy metabolism by lactate as a substrate. However, direct proof will only be evident if the neuroprotection by lactate infusion can be counteracted by blocking the lactate transporter.
Neuroprotection by increased glutamate uptake or stimulated CBF
As a consequence of an improved energy status by lactate treatment, we hypothesized that glutamate is removed from the extracellular space more efficiently in the lactate-treated group, and that this phenomenon is part of the neuroprotective property of lactate. We found that dialysate glutamate increased significantly over baseline, but dropped back to normal levels within 15 min. Lactate infusion also increased glutamate levels compared to NaCl-treated animals. This effect was rather surprising, but peak glutamate concentration as well as calculated overall glutamate release during monitoring were not significantly affected by lactate treatment. The peak levels of around 5 μM were comparable to those reported by Palmer and associates with a mild CCI injury (Palmer et al., 1993). Together with our CBF data, these extracellular glutamate concentrations suggest a mild to moderate injury. However, CCI-induced extracellular glutamate levels cannot easily be compared between studies, due to differences in injury severity, CCI model, and microdialysis probes. However, neuroprotection by NMDA-receptor antagonists suggests that released glutamate after CCI contributes to the pathogenesis of focal contusions (Kroppenstedt et al., 1998). Furthermore, changes from baseline should be large enough to detect effects of lactate treatment, but no significant difference between groups has been found. Improved glutamate uptake was therefore not involved in lactate neuroprotection. This is further supported by the fact that our treatment for neuroprotection started 15 min post-CCI in one experiment, and after glutamate had nearly reached baseline levels in another (15–20 min post-CCI). It would be necessary to use a more severe injury with a longer period of glutamate elevation to prove or disprove our glutamate hypothesis.
Ido and colleagues (Ido et al., 2004) showed that lactate as a bolus injection has the potential to stimulate CBF in the retina and visual cortex of rats. In their experiments, CBF was stimulated by a light pulse, and could further be increased by lactate but not by pyruvate. The authors hypothesized that this phenomenon may result from the influence of excessive lactate on the NAD+/NADH balance. An effect of a bolus injection of lactate on CBF was also recently confirmed in human volunteers (Mintun et al., 2004). A bolus injection of 1 mmol lactate/kg body weight had no effect on brain CBF before visual stimulation. Lactate augmented CBF in the visual cortex by 38–50% after visual stimulation. Whether such effects also occur following TBI is unclear. Analysis of TBI patients showed that CBF and the metabolic rate of oxygen (CMRO2) were generally higher in patients with good outcomes (Soustiel et al., 2005). CMR for lactate (CMRlact) did not show a clear trend over time, but correlation analysis indicated a positive relationship between CMRlact and Glasgow Coma Scale score. In patients with poor outcomes, CMRlact had a weak but positive relationship to CBF, which could be interpreted as an effect of lactate uptake on CBF. In our experiments, lactate infusion stopped the continuing deterioration of CBF such as that seen in NaCl-treated animals (Fig. 4). The weak and short-term effect seen does not single out lactate-induced CBF as the source of neuroprotection. A brief CBF elevation might have been sufficient to reperfuse the area at risk in the peri-contusional region. Using a model of subarachnoid hemorrhage (SAH) that induces a massive ipsilateral CBF depression, a 20-min augmentation of CBF by a bolus injection of hyperoncotic/hypertonic fluid was sufficient to induce neuroprotection. Treatment with mannitol and hypertonic solution did not affect CBF, and induced less neuroprotection following SAH (Bermueller et al., 2006), indicating that even a short-term CBF increase early after SAH contributes to neuroprotection. In this way the energy supply has been improved in areas at risk, and lactate may have supported protective processes such as oxidative metabolism (Holloway et al., 2007; Levasseur et al., 2006), free radical scavenging (Groussard et al., 2000), or upregulation of MCT-1 and COX (Hashimoto and Brooks, 2008). However, it remains unclear whether a slow infusion of lactate (1.0 mg/10 min lactic acid) is sufficient to affect CBF compared to a bolus injection. The same argument may account for the effect seen on energy metabolism and histology, especially since 1 mg/10 min lactic acid did not elevate blood lactate levels. Recently, Meierhans and co-workers analyzed TBI patients retrospectively and found that patients with blood lactate concentrations above 2 mM had higher brain lactate and lower glucose values (Meierhans et al., 2012). In a study by Prieto and colleagues (Prieto et al., 2011), only sodium lactate at 500 mM or higher influenced blood lactate (>2 mM). However, they did not find any effect of 100–1280 mM sodium lactate (394 mosm/l) on ATP, energy metabolites, peroxidation products, and NAD+, at 6 h after severe diffuse injury. These results are in contrast to a protective effect seen of 100 mM lactate on ATP (Holloway et al., 2007), and oxygen consumption (Levasseur et al., 2006) at 3 h after FPI. One might speculate that the injury model used, and particularly the severity of injury, influences the efficacy of lactate treatment. Furthermore, lactate is considered not only to be an energy substrate, but also a signaling molecule. Thus further studies will have to be conducted to better elucidate the neuroprotective properties of lactate.
In conclusion, in the present study we investigated the effects of lactate treatment on extracellular glutamate concentrations, local CBF, and on histological outcome, following CCI injury in rats. Our results showed that a lactate infusion of 100 mM for 3 h at a rate of 0.65 mL/h was neuroprotective for up to 7 days post-CCI. At 5 min after the start of the lactate infusion (i.e., 20 min post-CCI), a further CCI-induced CBF drop was prevented, whereas CBF in the control group kept declining. Lactate had no effect on extracellular glutamate. Since CBF changes were small, we assume that the effects of lactate on CBF, in combination with other lactate-induced mechanisms, led to the observed neuroprotection.
Footnotes
Acknowledgments
We thank A. Ehlert, L. Kopacz, and M. Malzahn for their technical support. Some data are part of the doctoral thesis of E. Schwandt.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.