
Abstract
Seizures are important neurological complications after traumatic brain injury (TBI) and are reported for up to 50% of patients with TBI. Despite several studies, no drug strategy has been able to alter the biological events leading to epileptogenesis. The glial water channel, aquaporin-4 (AQP4), was shown to facilitate cytotoxic cell swelling in ischemia and glial scar formation after stab wound injury. In this study, we examined post-traumatic seizure susceptibility of AQP4-deficient mice (AQP4–/–) after injection of pentylenetetrazole (PTZ) 1 month after controlled cortical impact (CCI) and compared them to wild-type sham injury controls. After PTZ injection, AQP4–/– mice demonstrated dramatically shortened seizure latency (120 ± 40 vs. 300 ± 70 sec; p < 0.001) and increased seizure severity (grade 7.5 ± 0.4 vs. 5.8 ± 0.4; p < 0.001) compared to their wild-type counterparts. Morphometric analysis demonstrated a significant 2-fold reduction in astrocytosis, with a concomitant increase in microgliosis in injured AQP4-null mice compared to their injured wild-type counterparts (44 ± 2 vs. 24 ± 3 cells per high power field [cells/hpf], respectively; p < 0.0001). Minocycline, an inhibitor of microglia, reversed the post-TBI epilepsy phenotype of AQP4-null mice. After minocycline treatment, AQP4–/– mice demonstrated similar latency of seizures evoked by PTZ (723 ± 35 vs. 696 ± 38 sec; p > 0.05) and severity of seizures evoked by PTZ (grade 4.0 ± 0.5 vs. 3.81 ± 0.30; p > 0.05) compared to wild-type counterparts. Immunohistochemical analysis demonstrated decreased immunostaining of microglia to levels comparable to wild-type (12 ± 2 vs. 11 ± 4 cells/hpf, respectively; p > 0.05). Taken together, these results suggest a protective role of AQP4 in post-traumatic seizure susceptibility by promoting astrogliosis, formation of a glial scar, and preventing microgliosis.
Introduction
Post-traumatic seizures (PTSs) are a well-known complication of traumatic brain injury (TBI), and post-traumatic epilepsy (PTE) accounts for 20% of all symptomatic epilepsy in the general population. 1 –3 PTSs are classified as immediate seizures (<24 h after injury), early seizures (<1 week after injury), and late seizures (>8 days after injury). 4 Current treatment of early PTS has been the use of antiepileptic medication; however, the ability to prevent the development of late seizures after TBI with early antiepileptic medication has not been demonstrated. 5,6 Additionally, the side effects of seizure medications often preclude its prolonged use. Therefore, it is important to investigate new approaches in the prevention of PTE.
Aquaporins are a family of membrane proteins that function as “water channels” in many cell types and tissues in which fluid transport is crucial. 7 There is evidence that water movement in the brain involves aquaporin channels. 8,9 Aquaporin-4 (AQP4) is expressed ubiquitously by glial cells, especially at specialized membrane domains including astroglial endfeet in contact with blood vessels and astrocyte membranes that ensheath glutamatergic synapses. 10,11 Activity-induced radial water fluxes in neocortex have been demonstrated that could be attributable to water movement through aquaporin channels in response to physiological activity. 12,13 Additionally, AQP4 promotes astrocyte migration, and AQP4-null animals have demonstrated delayed astrocyte movement. 14,15
Astrocytes in the healthy brain mediate glutamate reuptake, regulate the ionic environment and interstitial volume, and serve as a component of the neurovascular unit that controls blood–brain barrier permeability. 16 Although reactive astrocytes in the epileptic focus are known to undergo extensive morphological and physiological changes that modify these three overarching functions, the implications for epilepsy are just emerging. In models of TBI, conditional ablation of proliferating astrocytes increases neuronal injury and increases infiltration of damaged region by microglia. 17,18 These data strongly point to a protective function of proliferating astrocytes.
However, there is also evidence that suggests that reactive astrocytes might contribute to the hyperexcitable condition. During the epileptic state, there is a redistribution of AQP4 away from the perivascular membranes toward neuropil, which should create a situation allowing rapid astrocytic swelling in the neuropil with consequent shrinkage in extracellular space and enhanced ephaptic interactions among tightly packed neurons. 19 Additionally, neuroinflammatory signaling pathways engaged in reactive glia in epileptic foci may allow astrocytic glutamate signaling to become prominent enough to contribute to the hyperexcitable state. Together, these factors argue for a hyperexcitable role in reactive astrocytes.
In this study, we utilized AQP4-null mice, taking advantage of the known function of AQP4 in astrocyte migration and K+ buffering, to elucidate its role in mediating susceptibility to seizures. We demonstrate that susceptibility to seizures is significantly higher in mice lacking AQP4 in a pentylenetetrazole (PTZ)-induced seizure model. 20 We also demonstrate that seizure phenotype is related to decreased astroglia migration and, consequently, increased microglia formation. Last, we show that inhibition of microglia by minocycline treatment abolishes this seizure phenotype in AQP4-null mice.
Methods
AQP4–/– mice
All animal procedures were approved by the University of California, San Francisco committee on Animal Research. AQP4–/– mice were generated as previously described by Ma and colleagues. 21 These mice lacked detectable AQP4 protein and phenotypically have normal growth, development, survival, and neuromuscular function. A total of 40 wild-type mice and 40 AQP4-deficient mice were subjected to initial seizure testing, with 20 wild-type and 20 AQP4-null in the sham group and 20 wild-type and 20 AQP4-null in the CCI group. In the minocycline seizure experiments, a total of 24 wild-type and 24 AQP4-deficient mice were used, with 12 wild-type and 12 AQP4-null mice in the saline-treated group and 12 wild-type and 12 AQP4-null mice in the minocycline-treated group.
Controlled cortical impact
Mice (12 weeks of age) were anesthetized with 4% isoflurane (Anaquest, Memphis, TN) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH) and positioned in a stereotactic frame. Anesthesia was maintained using 2–3% isoflurane. A 5-mm craniotomy was made using a portable drill and trephine over the left parietotemporal cortex (the center of the coordinates of craniotomy relative to bregma: 1.5 mm posterior, 2.5 mm lateral), and the bone flap was removed. Mice were then subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder (E-CCI Model 6.3; Custom Design and Fabrication, New York, NY), velocity 6 m/sec, set to depth of 1.2 mm, and 100-ms impact duration. The bone flap was then replaced and secured with tissue sealant (Abbott Laboratories, North Chicago, IL). The scalp was sutured closed and mice returned to their cages to recover. Body temperature was monitored by a rectal probe and maintained with a heating pad throughout the procedure to a temperature of 38.0°C. There were two mortalities associated with CCI in the initial seizure cohort: one in the wild-type and another in the AQP4-null mice. Another mortality was present in a minocycline-treated wild-type mouse after CCI.
Assessment of seizure susceptibility
One month after initial CCI, seizures were induced by PTZ (Sigma-Aldrich, St. Louis, MO). PTZ was dissolved in phosphate-buffered saline (PBS) and delivered by intraperitoneal injection. PTZ was used at a concentration of 5 mg/mL and a dose of 40 mg/kg. For mice given PTZ, each mouse was placed in a cage and observed for 20 min after administration, with video recording. An investigator blinded to genotype analyzed the videotapes to quantify the time course, latency, 22 and severity of seizures according to published scales. 20 Seizure severity scores were: 0 = normal behavior; 1 = immobility; 2 = generalized spasm, tremble, or twitch; 3 = tail extension; 4 = forelimb clonus; 5 = generalized clonic activity; 6 = bouncing or running seizures; 7 = full tonic extension; and 8 = death. A pilot study showed that animals do not show increased seizure activity beyond 20 min past PTZ induction. It was also shown that seizures do not evolve further after this 20-min period, and length of seizures was unrelated to experimental group. Therefore seizures were scored as the greatest seizure activity within the 20-min observation period.
Immunostaining
Immediately after assessment of seizure susceptibility, mice were anesthetized by an intraperitoneal injection of Avertin (125 mg/kg) and perfused transcardially with 4% paraformaldehyde in PBS. Mice were decapitated, and then the brain was dissected and post-fixed for 24 h in paraformaldehyde at 20°C. Tissues were dehydrated with increasing concentrations of ethanol, treated with clearing agent, and embedded in paraffin. Some sections were deparaffinized and stained with cresyl violet (Nissl stain), using standard procedures. For immunofluorescence, epitope exposure was enhanced by incubation in citrate buffer (10 mM of sodium citrate, 0.05% Tween 20, pH 6.0; 30 min, 95°C–100°C). Sections were blocked with bovine serum albumin (3%), incubated with polyclonal anti-AQP4 antibody (1:200), monoclonal CD11b antibody (1:200), or polyclonal anti–glial fibrillary acidic protein (GFAP; 1:200) for 4 h at room temperature (Millipore, Billerica, MA), followed by Texas Red donkey/antirabbit (1:200) or biotinylated secondary antibody (1:200), and avidin horseradish peroxidase for GFAP and CD11b (Vector Laboratories, Burlingame, CA). Immunolabeling was visualized brown using diaminobenzidine.
Lesion volume was assessed by digital images (8-bit jpeg format) taken of five consecutive Nissl-stained coronal sections through the injury site (200-μm separation). We used ImageJ (v. 1.33u; NIH, Bethesda, MD) to arbitrarily set pixels with intensities 175–255 as white, and the remaining pixels (intensities 0–174) blue, and calculated the percentage of white pixels in each image.
A cell-counting procedure was performed with a 60 × objective to quantify positive cells to each antibody tested. Sections with the highest positive cell density were taken, and an average of five different coronal sections was used for each mouse (7 mice for each treatment group, 28 mice total). Data are presented as mean ± standard error. The experimenter performing the cell count was blinded to the genotype and treatment.
Scanning confocal laser microscopy
Immunostained sections were mounted in Vectashield Fluorescent Mounting Media (Vector Laboratories) and imaged using a Nikon Eclipse FN1 Upright Microscope or Nikon Eclipse C1si Confocal Microscope with a 20 × /0.75 Air-Plan Apo (DIC N2; Nikon Instruments Inc., Melville, NY) objective at a resolution of 512 × 512 pixels to 1024 × 1024 pixels. Nikon C1 acquisition software and NIS Elements software for three-dimensional (3D) reconstructions were used and sampled from a thickness of 50 μm with a Z step of 1 μm. Sections from each cohort (n = 6 in each) and condition were analyzed, and representative images are shown.
Minocycline treatment
Minocycline HCl (Sigma-Aldrich) was administered intraperitoneally 12 h before TBI at a dose of 45 mg/kg body weight. This dose was selected based on previous studies. 23,24 Beginning 30 min after trauma, animals received minocycline injections every 12 h, at a dose of 90 mg/kg for the first 24 h after trauma and then 45 mg/kg thereafter until mice were euthanized. Control animals received equivalent saline volume.
Statistical analysis
A value of p < 0.05 was considered significant. Error bars represent standard error of the mean. Genotype and treatment (CCI or minocycline) differences were analyzed by one-way analysis of variance, followed by Tukey-Kramer's test of post hoc comparisons.
Results
Increased seizure severity and decreased seizure latency in post-traumatic aquaporin-4-null mice
Seizure susceptibility after CCI was assessed by the well-established γ-aminobutyric acid type A (GABAA) receptor-antagonist PTZ-induced seizure assay. As expected, animals showed significantly increased seizure severity after CCI for both genotypes (Fig. 1A). Interestingly, brain-injured AQP4-null mice demonstrated a significantly increased seizure severity compared to wild-type mice (Fig. 1C).
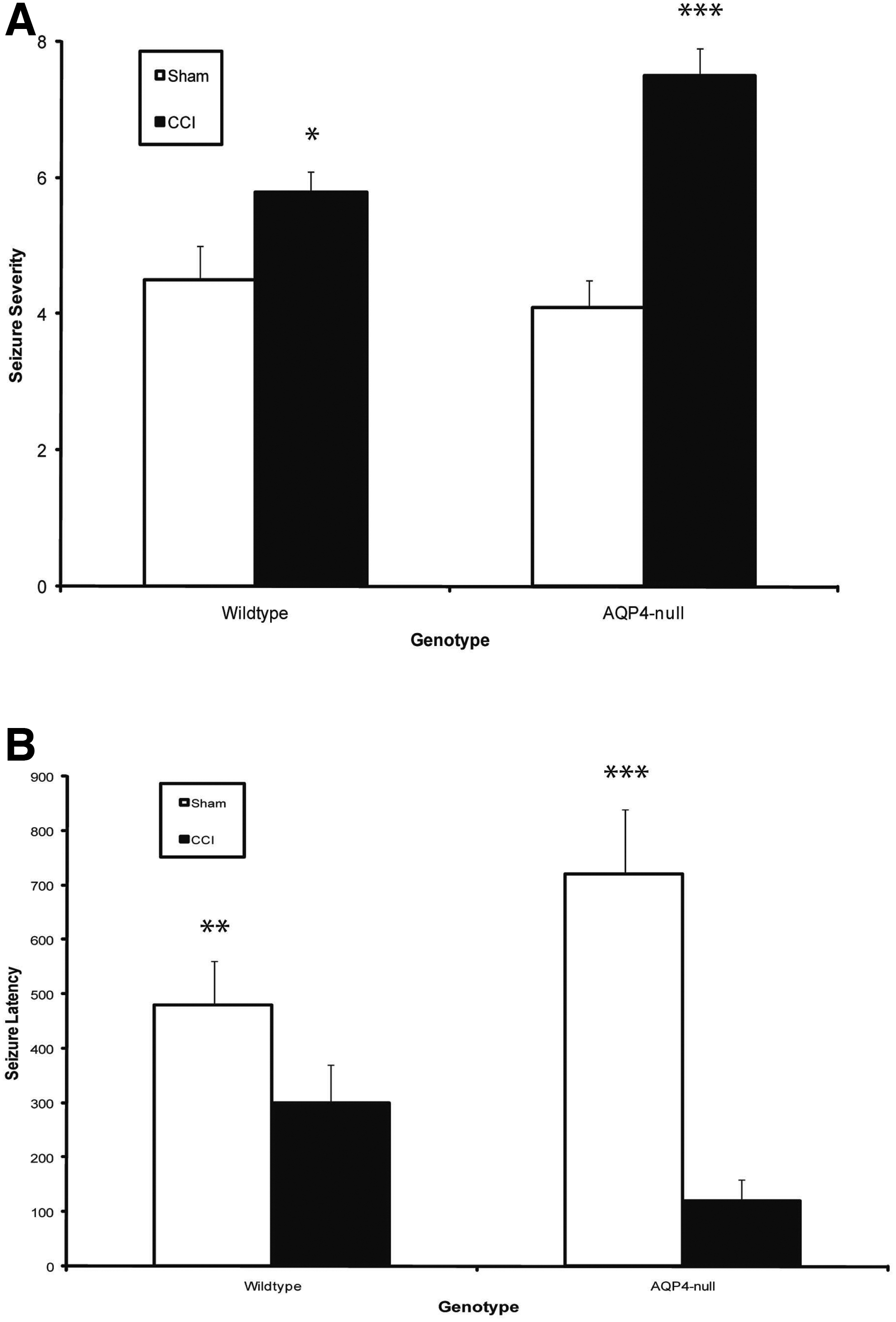
AQP4-null mice exhibited worse seizure profile after CCI. (
Seizure latency to stage 5 (generalized clonic activity) was measured in the animals above (Fig. 1B). Sham AQP4-null mice at baseline were resistant to PTZ-induced seizures compared to their wild-type counterparts. Additionally, CCI increased the sensitivity of mice to PTZ-induced seizures by decreasing eizure latency. Interestingly, AQP4-null mice had decreased latency after CCI as compared to wild type. These results establish that AQP4-null mice have a significantly worse seizure response after TBI. To investigate the cellular mechanisms responsible for these seizure findings, we conducted immunohistochemical studies to assess injury characteristics and specific markers pertinent to the known functions of AQP4 in glial scar formation.
Similar injury volume in aquaporin-4-null and wild-type mice after controlled cortical impact
To determine whether there was a difference in injury volume between wild-type versus AQP4-null mice, Nissl staining of coronal brain sections was performed (Fig. 2A). Grossly, there were no observed differences in the extent of injury between genotypes. The cortex beneath the impact site was obliterated with some perturbation to the underlying hippocampus. Quantifying the volume of injury demonstrated no genotype difference (Fig. 2B). Given that there was no difference in injury volume between the genotypes, we further investigate the mechanism of seizure phenotype by other markers.

Similar injury volume in wild-type and AQP4-null mice after CCI. (
Increased aquaporin-4 immunoreactivity after controlled cortical impact
To confirm the knockout genotype and expression pattern of AQP4 after CCI, we performed immunolocalization for AQP4 after CCI in wild-type and AQP4-null mice (Fig. 3A). As expected, there was no immunoreactivity in AQP4-null mice before or after injury, but a baseline diffuse immunostaining was present in wild-type mice. Interestingly, after injury, AQP4 immunoreactivity increases within the injury region, suggesting migration of AQP4 positive-cells within the region or upregulation of AQP4 within the reactive glial cells. This was assessed by quantitative cell count with high-power microscopic magnification (Fig. 3B).

Increased AQP4 immunoreactivity in injury zone after CCI. (
Decreased astrogliosis in aquaporin-4-null mice after controlled cortical impact
AQP4 has been implicated in glial migration, and astrogliosis is a common phenomenon after CCI. We therefore evaluated whether there is a difference in astrocytosis surrounding the impact zone between wild-type and AQP4-null mice after CCI. Indeed, we discovered a significant difference in astrocyte density between wild-type and AQP4-null mice (Fig. 4B). When compared, there were visibly fewer activated astrocytes in AQP4-null versus wild-type mice. After quantification, there was a 2-fold increase in the number of reactive astrocytes after CCI in wild-type mice compared to AQP4-null mice. As control, there were no baseline differences in reactive astrocyte count between sham wild-type and AQP4-null mice. These results confirm the decrease in the number of reactive astrocytes in AQP4-deleted animals after CCI. To evaluate the impact of reactive astrocytes on microglia, we stained these same sections for CD11b, a microglial marker.
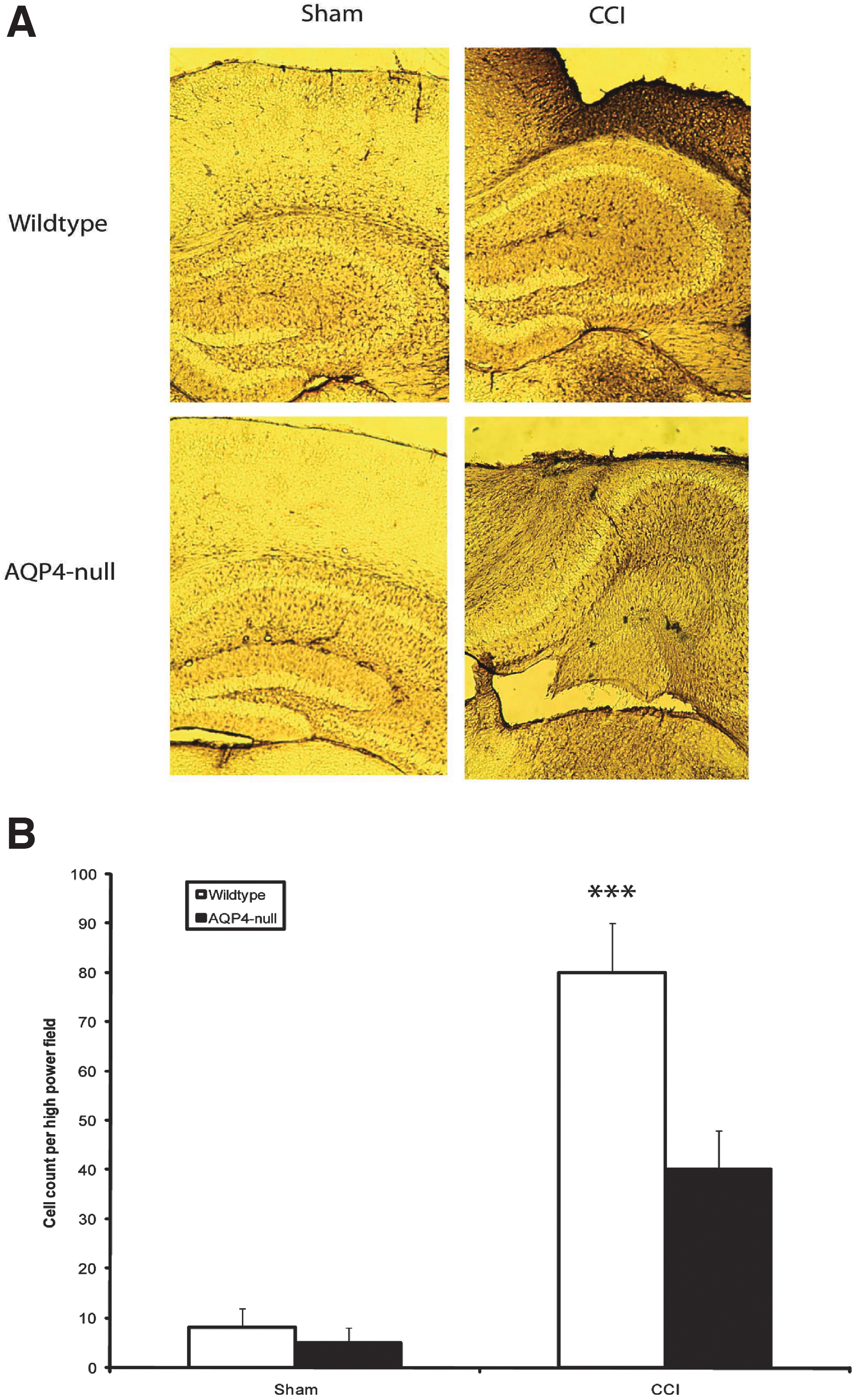
Reactive astrocytes in injury penumbra after CCI. (
Increased microgliosis in aquaporin-4-null mice after controlled cortical impact
Microglia are the immunoeffector cells of the brain. To assess microglia density, we utilized CD11b antibody. We discovered that indeed this difference in astrocytosis translates into differences in microglia scar density. Qualitatively, we observed an obvious difference in the sections (Fig. 5A), and found that wild-type mice had less CD11b reactivity compared to AQP4-null mice after injury. Quantitatively, we discovered a significantly increased microgliosis in AQP4-null mice compared to wild-type mice after CCI (Fig. 5B). There was minimal CD11b activation in non-injured controls. These results indicate that AQP4 deletion results in increased microglia accumulation in areas of injury.

Microgliosis in injury penumbra after CCI. (
Decreased astrocytic glial scar in aquaporin-4-null mice after controlled cortical impact
To look closely at the 3D morphology of the glial scar, we utilized scanning confocal laser microscopy on brain sections costained with GFAP and AQP4 (Fig. 6). Morphologically, in the wild-type mice, there is a robust astrocytic glial scar surrounding the injured cavity (white). This scar can be visualized as a barrier to the injured tissue; whereas in the AQP4-null mice, this scar formation was weak. To confirm AQP4 expression in the wild-type mice and lack of expression in AQP4-null mice, sections were costained with AQP4 antibody (red). As expected, AQP4-null mice did not have appreciable staining with AQP4 antibody. There appears to be a differential expression pattern of AQP4 in regions surrounding the injury cavity (somal pattern, A) as compared to regions near normal brain (pericapillary pattern, B).
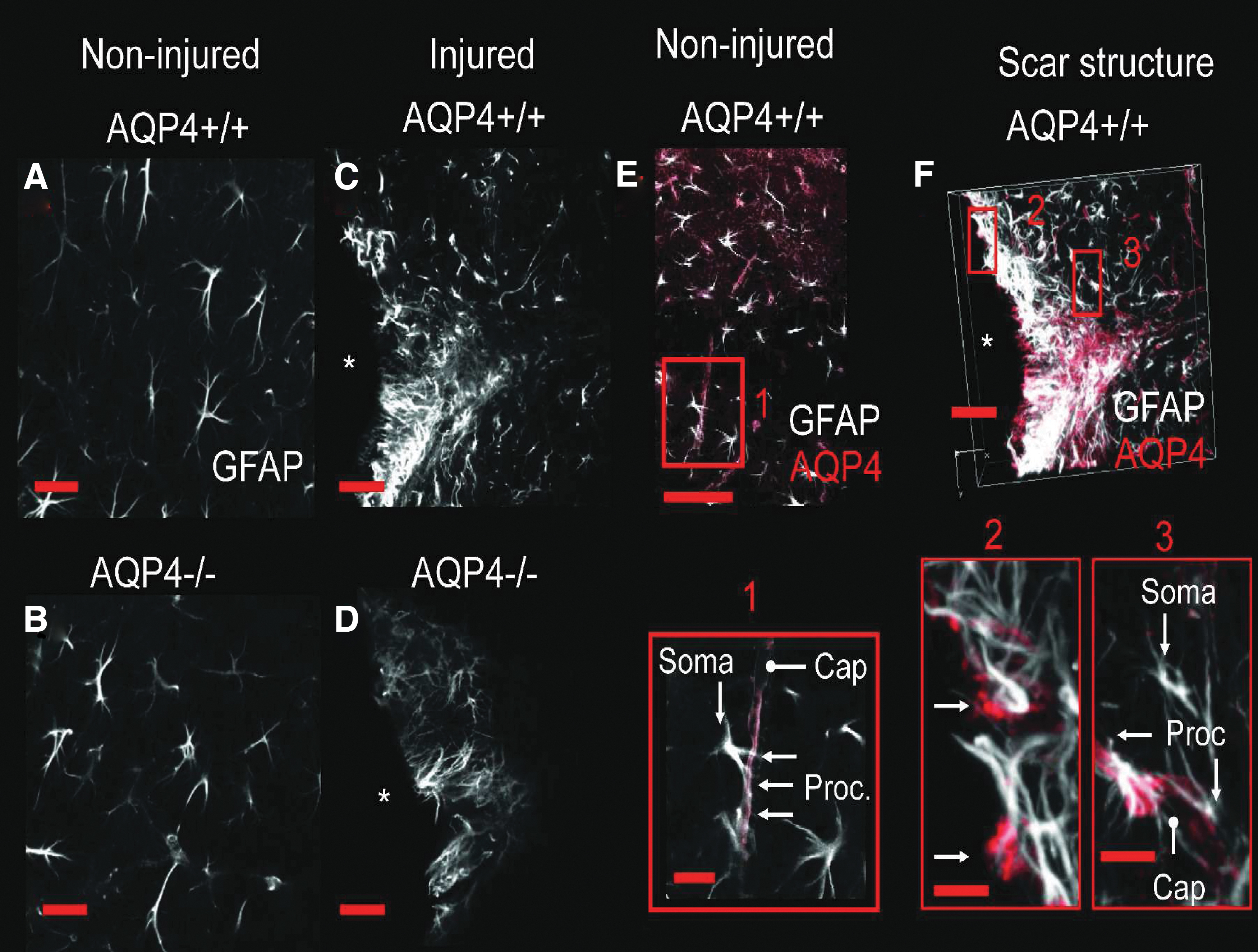
Astrocytic scar morphology after CCI. Expression pattern of GFAP and AQP4 after cortical contusion. Immunolabeling of GFAP (white) in the cerebral cortex of uninjured AQP4+/+ (
Minocycline treatment reverses seizure phenotype in aquaporin-4-null mice
To assess the effect of minocycline on seizure severity, AQP4-null and wild-type mice were subjected to PTZ-induced seizure assay. In the saline control cohort, as expected, AQP4-null mice demonstrated more severe seizures as compared to wild-type mice after injury (p < 0.0005); however, after minocycline treatment, seizure severity in AQP4-null mice decreased to a similar level as compared to its wild-type counterpart after injury (Fig. 7A). Further, minocycline had a significant inhibitory effect on seizure severity in wild-type mice as well. Seizure latency correspondingly increased with the treatment of minocycline after injury as compared to saline control in both AQP4-null and wild-type mice (Fig. 7B).
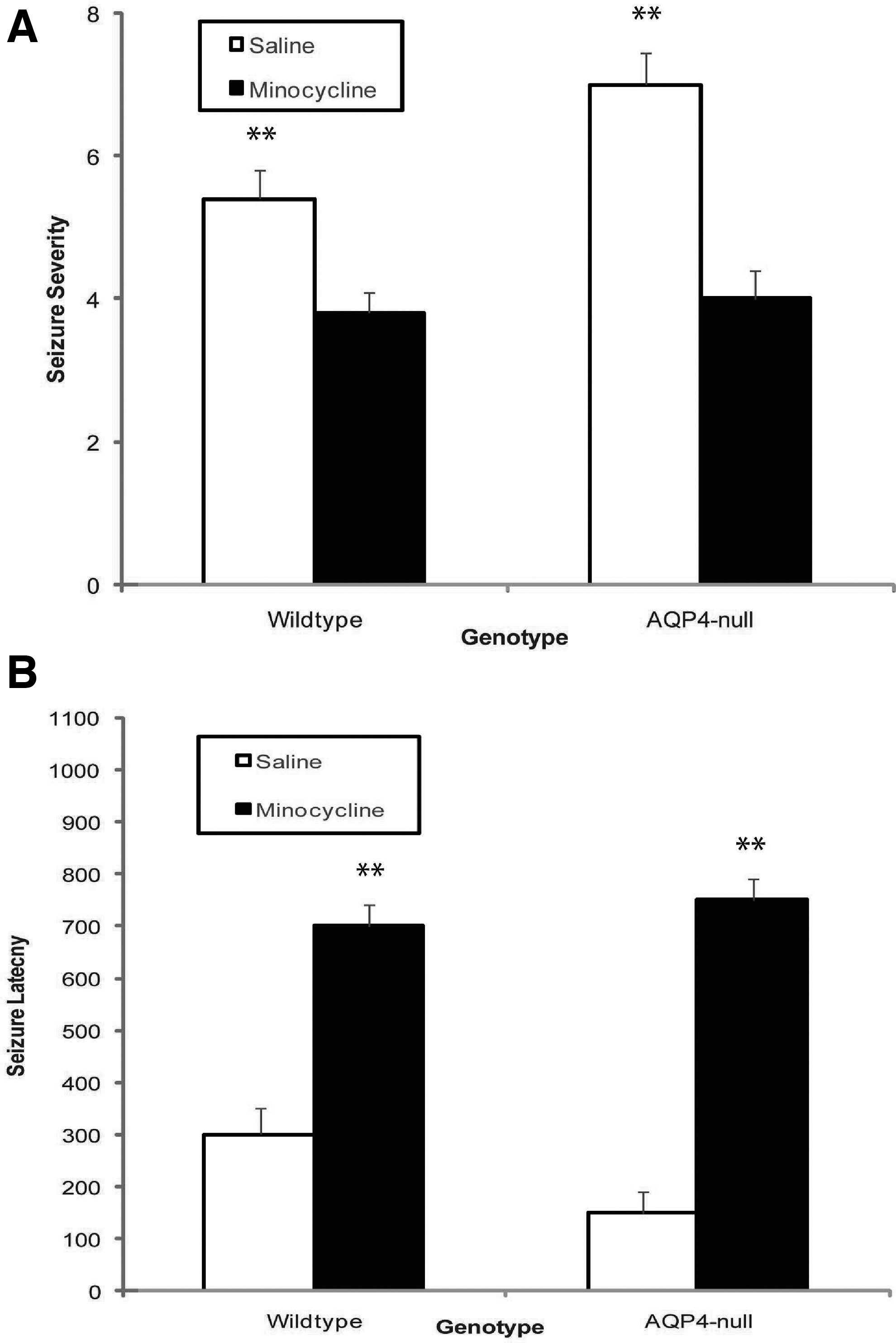
Minocycline treatment reverses seizure profile of AQP4-null mice after CCI. (
Minocycline treatment diminishes microgliosis without affecting injury volume, aquaporin-4 immunoreactivity, or astrocytosis
In looking for causes of the above seizure phenomenon, we subjected brain slices of the injured minocycline- and saline-treated mice to Nissl staining, in addition to AQP4, GFAP, and CD11b antibodies to assay injury volume, AQP4 immunoreactivity, astrocytosis, and microgliosis, respectively (Fig. 8). Minocycline treatment did not have an appreciable effect on injury volume (Fig. 8A), AQP4 immunoreactivity (Fig. 8B), nor GFAP immunoreactivity (Fig. 8C) as compared to saline control. In contrast, minocycline-treated animals had substantially decreased CD11b immunoreactivity as compared to saline-treated controls, such that there was no statistical difference between the two genotypes (Fig. 8D). This inhibitory effect of minocycline on microgliosis was present in both wild-type and AQP4-null mice.

Minocycline treatment inhibits microgliosis. (
Discussion
The principal finding of this study is an increase in seizure susceptibility in AQP4-deficient mice after CCI injury, with reduced astrocytes and increased microglia within the injury region. Inhibition of microgliosis by minocycline reversed the seizure phenotype in these AQP4-deficient mice. These observations imply that glial scar formation is protective against post-traumatic seizures by a microglia-dependent mechanism.
Immunohistochemical studies demonstrated similar injury volume, decreased reactive astrocytes, and increased microglia in AQP4 knockout animals after CCI. Further, strongly AQP4-positive astrocytes were present within the glial scar of wild-type animals. These findings suggest that reactive astrocytes play a therapeutic role in the repair of the injured area by displacing microglial cells. Several lines of evidence show that the deletion of AQP4 results in reduction of the astrocytic component of the glial scar in various pathologies. 14,15 It has been proposed that, as a water channel, AQP4 polarizes at the leading edge of the migrating astrocytes and thus enhances cellular water uptake and cell volume changes to facilitate cell movement. 25 Consistent with this hypothesis, AQP4 expression was shown to enhance the reactive astrocytes of the glial scar. Microglia are leukocytes of the central nervous system (CNS) and perform phagocytic functions. In higher vertebrates, leukocytes gain little entry into normal CNS parenchyma; therefore, CNS inflammatory response to injury differs from that of other tissues. 26,27 After injury, blood-borne monocytes and lymphocytes gain delayed and limited access to CNS parenchyma. 27 –29 However, in the absence of astrocytes, leukocytes gain increased and prolonged entry into CNS parenchyma. 17 Previously, the peripheral inflammation model of microglia activation has been linked to CNS excitability. Microglia inhibition by minocycline prevented the increase in seizure susceptibility. 30 Recently, minocycline has been demonstrated to be a selective inhibitor of microglia activation in various disease models from cerebral ischemia to intracerebral hemorrhage. 31 –34
In our study, the prolonged and increased presence of microglia may present a pathogenic mechanism for inflammatory disturbance that could alter seizure susceptibility. Indeed, such a mechanism is supported by our observation that microglia inhibition with minocycline reverses the seizure phenotype of AQP4-null mice.
Reactive astrocytes are known to demonstrate enhanced inwardly rectified K+ currents, 35 which suggest their role of enhancing K+ uptake during neuroexcitation.
The differential expression pattern of AQP4 within the injury penumbra as we observed may impact K+ buffering given that AQP4 is associated and colocalizes with the inwardly rectifying K+ channel, Kir4.1, 36 and within this association, AQP4 is proposed to act as an ion-water trafficking protein to facilitate K+ clearance. Absence of such a mechanism may lead to lower seizure threshold 37 –39 in AQP4-null mice and contribute to the AQP4-null seizure phenotype after CCI. However, in our study, given that inhibition of microglia activation reverses much of the seizure phenotype in AQP4-null mice, the contribution of K+ uptake in this injury model may be modest.
Our present study is the first to demonstrate in vivo the beneficial role of glial scar formation in post-traumatic seizures using a transgenic mouse model. Clinically, glial scarring has been determined as a contributor to the pathogenesis of epilepsy, and treatment consists of surgical resection, as in the case of mesial temporal sclerosis, after failing medical treatment (both in traumatic and non-traumatic cases). However, failure rates are high, and our current study may provide a mechanistic understanding of the failure. During the repair phase of the surgical site, the proepileptic effects of microglia may predominate.
Further, our findings may be relevant not only in the TBI setting, but also to surgical treatment of the CNS disorders where surgical trauma is introduced and post-operative antiepileptic medications are used. This study suggests that inhibition of microglia or reinforcement of AQP4 function in reactive astrocytes may serve as a potential therapeutic target to mitigate epileptogenesis after brain injury.
Footnotes
Funding Information
This work was supported by a grant from the National Institutes of Health (R01 NS050173-01A1).
Author Disclosure Statement
No competing financial interests exist.