
Abstract
Traumatic brain injury (TBI) and intracerebral hemorrhage (ICH) are leading causes of neurological mortality and disability in the U.S. However, therapeutic options are limited and clinical management remains largely supportive. HMG-CoA reductase inhibitors (statins) have pleiotropic mechanisms of action in the setting of acute brain injury, and have been demonstrated to improve outcomes in preclinical models of ICH and TBI. To facilitate translation to clinical practice, we now characterize the optimal statin and dosing paradigm in murine models of ICH and TBI. In a preclinical model of TBI, mice received vehicle, simvastatin, and rosuvastatin at doses of 1 mg/kg and 5 mg/kg for 5 days after the impact. Immunohistochemistry, differential gene expression, and functional outcomes (rotarod and Morris water maze testing) were assessed to gauge treatment response. Following TBI, administration of rosuvastatin 1 mg/kg was associated with the greatest improvement in functional outcomes. Rosuvastatin treatment was associated with histological evidence of reduced neuronal degeneration at 24 h post-TBI, reduced microgliosis at day 7 post-TBI, and preserved neuronal density in the CA3 region at 35 days post-injury. Administration of rosuvastatin following TBI was also associated with downregulation of inflammatory gene expression in the brain. Following ICH, treatment with simvastatin 1 mg/kg was associated with the greatest improvement in functional outcomes, an effect that was independent of hemorrhage volume. Clinically relevant models of acute brain injury may be used to define variables such as optimal statin and dosing paradigms to facilitate the rational design of pilot clinical trials.
Introduction
T
HMG-CoA reductase inhibitors (statins) have been demonstrated to improve outcomes in preclinical models of TBI (Wang et al., 2007; Wu et al., 2008a,2008b), ICH (Jung et al., 2004; Seyfried et al., 2004; Yang et al., 2011), subarachnoid hemorrhage (Bulsara et al., 2006; McGirt et al., 2002; Takata et al., 2009), and stroke (Chen et al., 2003). Statins have pleiotropic mechanisms of action, and have been suggested to exert beneficial effects in both the acute and subacute period after acute brain injury (Wible and Laskowitz, 2010). For example, statins have been associated with a downregulation of glial activation and a reduction in the release of mediators of inflammation and oxidative stress (Erdos et al., 2006; Wang et al., 2007). Modulation of these neuroinflammatory responses may lead to a reduction in secondary neuronal injury, reduction of blood–brain barrier (BBB) breakdown, and development of cerebral edema. Statins have also been demonstrated to attenuate post-traumatic hypoperfusion, likely through nitric oxide-mediated mechanisms (Wang et al, 2007). In the subacute to chronic period after injury, statin administration has been demonstrated to enhance neurogenesis (Lu et al., 2007), synaptogenesis, and angiogenesis (Chen et al., 2003; Lu et al., 2004a).
Although the pleiotropic mechanisms of action, long history of clinical use, and favorable side-effect profiles make statins attractive candidates for translation to clinical trials, a number of variables, including optimal statin type and dosing regimen, have not been systematically defined. For example, although a number of preclinical studies have demonstrated as proof of principle that statins exert robust neuroprotective effects after acute brain injury, the majority of early studies used supra-physiological doses that are not FDA approved, and may be associated with increased risks of liver and muscle toxicity (Chauhan and Gatto, 2011; Chen et al., 2009; Karki et al., 2009; Seyfried et al., 2004; Wang et al., 2007). Moreover, differences in potency, CNS penetration, and metabolism exist between different members of the HMG-CoA reductase family, and there is a paucity of data defining the optimal statin for a particular type of injury.
In the current study, we examine the relative efficacy of simvastatin and rosuvastatin following acute brain injury. Although both drugs are widely used in clinical practice, simvastatin is more lipophilic, and might be expected to more efficiently cross the BBB, whereas rosuvastatin has higher potency and favorable metabolic characteristics (McKenney, 2003). These practical issues of optimal drug, dosing, and timing would play an important role in rationally informing early translational trials. Therefore, in order to facilitate translation into clinical practice, we now characterize the optimal statin and dosing in murine models of ICH and TBI.
Methods
Closed head injury model
Following approval by the Duke University Animal Care and Use Committee, closed head injury was induced in 8- to 12-week-old C57BL/6J male mice (Jackson Laboratories, Bar Harbor, ME). This murine closed head injury model (Lynch et al., 2005a) was adapted from a previously described model of closed cranial trauma for the rat (Foda and Marmarou, 1994). After anesthesia induction with 4.6% isoflurane, the trachea was intubated and the lungs were mechanically ventilated with 1.6% isoflurane in 30% O2/70% N2. Rectal temperature was maintained at 37°C. The animal was positioned in a stereotactic device, a midline scalp incision was made, and the skull was exposed. A concave 3-mm metallic disc was affixed to the skull immediately caudal to the bregma along the midline to distribute the impact. A 2.0-mm-diameter pneumatic impactor (Air-Power, Inc., High Point, NC) was used to deliver a single midline impact to the disc surface. The impactor was discharged at 6.8±0.2 m/sec with a head displacement of 3 mm. Sham-operated mice had scalp incision without pneumatic impact. After injury, anesthesia was discontinued, the animals were allowed to recover spontaneous ventilation, and the trachea was extubated. Following recovery, mice were allowed free access to food and water.
Intracerebral hemorrhage model
Our murine ICH model (James et al., 2007) was adapted from a previously described rat model of ICH (Rosenberg et al., 1990). Male C57BL/6J mice (8–12 weeks of age; Jackson Laboratory, Bar Harbor, ME) were used in these experiments. The trachea was intubated after anesthesia induction with 4.6% isoflurane and the lungs were mechanically ventilated with 1.6% isoflurane in 30% O2/70% N2. Rectal temperature was maintained at 37±0.2°C by an underbody circulating warm waterbed. The animal's head was secured in a stereotactic frame, local anesthetic was injected, and the scalp incised. After exposure of the skull, a burr hole was created 2.2 mm left lateral to the bregma, and a 1-μL syringe needle (Hamilton, Reno, NV) was advanced to a depth of 3 mm below the cortical surface. Type IV-S Clostridial collagenase (Sigma-Aldrich, St. Louis, MO) was injected over 5 min (0.075 U in 0.4 μL NS). Sham-operated mice had burr-hole incision, but not collagenase injection. The incision was then closed, and the animals were allowed to recover spontaneous ventilation and then extubated with free access to food and water.
Statin dosing and randomization
The mice were number-coded and randomized to treatment arms before surgery. All rosuvastatin (AstraZeneca, Wilmington, DE) and simvastatin (UDL, Rockford, IL) treatments were administered within 1 h after injury, with the exception of data presented in Figure 1D, which was 3 h post injury, and then twice daily in divided doses of 1 mg/kg/day or 5 mg/kg/day with an oral gavage (in 100 μL normal saline) until day 5 post-injury. Normal saline was used as vehicle control.
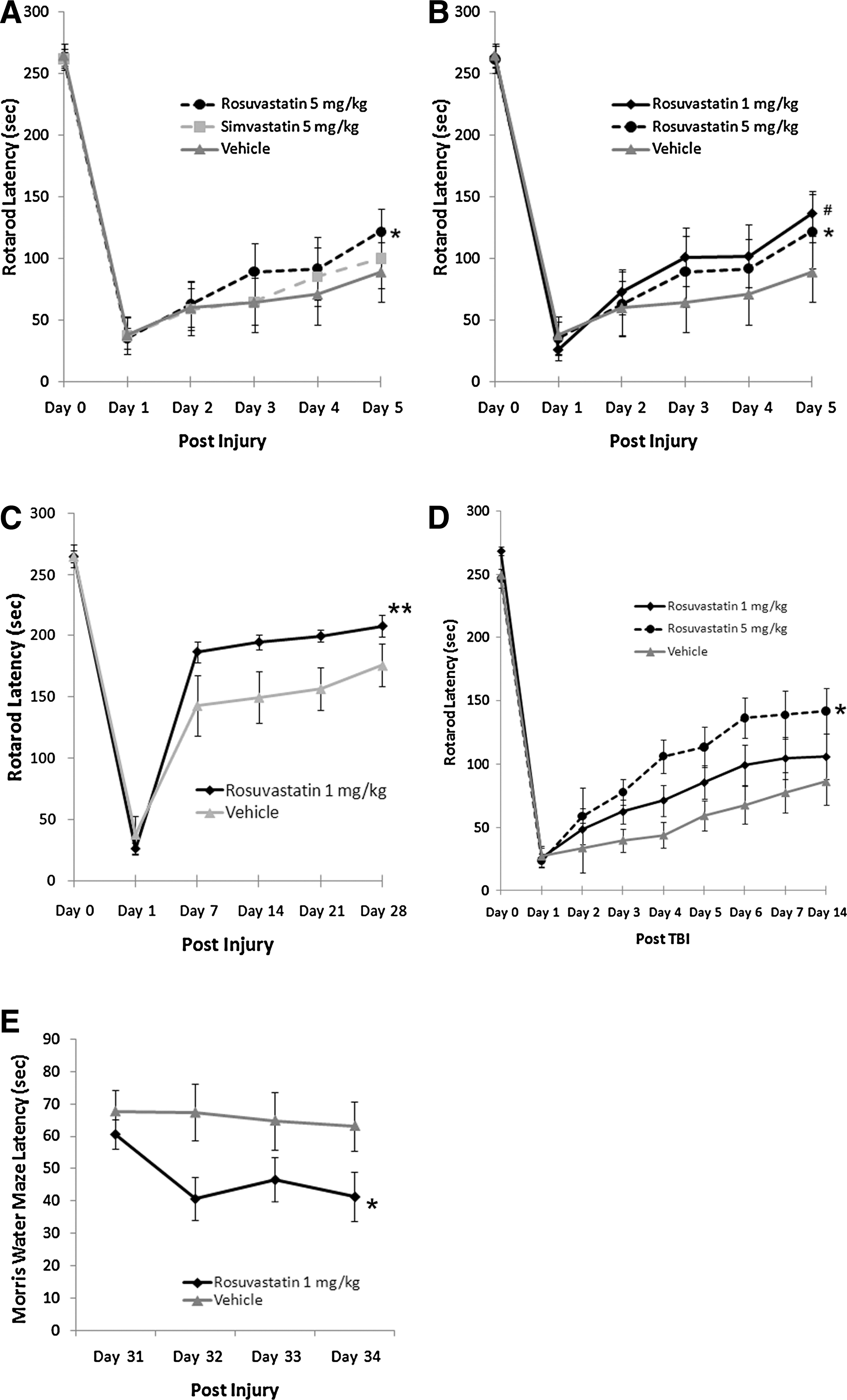
Rosuvastatin, but not simvastatin, improved short-term and long-term functional outcome after traumatic brain injury (TBI). Injection of statins occurred 1 h (
Neurobehavioral tests (rotarod and Morris Water Maze)
An investigator blinded to group assignment assessed behavioral outcomes. For neurobehavioral experiments in TBI models, the mice were randomized into vehicle (n=12), simvastatin 5 mg/kg (n=12), and rosuvastatin 5 mg/kg (n=12) groups to establish which statin was more effective. Additional experiments were then performed using the most effective statin to establish the lowest effective dose (n=12 animals/arm), and whether a treatment delay of 3 h after injury was still associated with functional improvement. In the ICH model, mice were randomized into three groups: vehicle, rosuvastatin, and simvastatin (n=24 mice/arm), with the lowest effective dose defined from previous experiments in TBI.
An automated rotarod (Ugo Basile, Comerio, Italy) was used to assess vestibulomotor function (Hamm et al., 1994; Lynch et al., 2005a). On the day prior to injury, mice underwent two consecutive conditioning trials at a set rotational speed (16 rpm) for 60 sec, followed by three trials with an accelerating rotational speed to obtain baseline latency. For post-injury data collection, TBI mice were tested on days 1–7, 14, 21, and 28, while ICH mice were tested on days 1–7 using three trials with accelerating rotational speed (intertrial interval 15 min). The average latency to fall from the rod was recorded.
Neurocognitive function was characterized with the Morris water maze (MWM) test (Morris, 1984; Lynch et al., 2005a), which was initiated at 31 days following TBI. An aluminum pool (105 cm in diameter and 60 cm in depth) was painted black and filled to 17 cm with water (25–27°C, opacified with powdered milk). The maze was kept in a room dedicated to behavioral testing, with light, sound, and visual cues held constant. The goal was a hidden plastic platform, 7.5 cm in diameter, submerged 0.5 cm below the water's surface. Mice were tested for 4 consecutive days with 4 trials per day (intertrial interval 1 h). The mice were placed in one of four different quadrants for each trial. Starting quadrants were randomly defined each day. Mice were allowed to search for the platform for a maximum of 90 sec. If the mouse found the platform within 90 sec the latency was recorded. If the mouse could not escape onto the platform it was guided to the platform where it remained for 15 sec, and the latency was counted as 90 sec. Swim speed was recorded for all trials. Animal movement was recorded using a computerized video tracking system (MazeTracker; KeilSoft LLC, Chapel Hill, NC). The mice were kept in heated cages between trials.
Immunohistochemistry: Fluoro-Jade B, F4/80, NeuN, and IgG
To assess the effects of therapeutic interventions on early inflammation, immunohistochemical staining was performed using Fluoro-Jade B (a marker of degenerating neurons), F4/80 (a microglial marker), and NeuN (a neuronal marker), on days 1, 7, and 35 after TBI, respectively. Except on day 35, immunohistochemistry was done on separate cohorts of mice from those used in neurobehavioral tests. For Fluoro-Jade B histology, there were 6 mice in the treated and vehicle groups, with 5 sections/mouse. For the F4/80 staining, there were 8 mice in the treated group, and 7 mice in the vehicle group, with 6 sections/mouse. For the NeuN quantitative stereology experiments, there were 7 mice in the treated group and 6 in the vehicle group with 6 sections/mouse. To assess BBB integrity, the mice were examined on day 1 post-injury for IgG immunohistochemistry (n=6/group). In all cases the mice were anesthetized, euthanized, and perfused with 30 mL phosphate-buffered saline (PBS) via transcardiac puncture. The brains were then immersed in buffered formalin solution for 24 h, then changed to 30% sucrose buffer for at least 24 h. Sagittal or coronal sections (40 μm) of fixed brain were cut on a freezing microtome and collected in cryoprotectant solution containing ethylene glycol, sucrose, and sodium phosphate. Every eighth section was mounted onto a charged slide. For assessment of neuronal degeneration, the slides were stained with Fluoro-Jade B using previously described protocols (Schmued and Hopkins, 2000; Wang et al., 2007). For F4/80, NeuN, and IgG immunohistochemistry, free-floating sections were incubated in 1% H2O2 for 5 min, then transferred to 0.1% saponin for 1 h. Next the sections were incubated for 30 min in 10% goat serum. Primary antibody was applied overnight at 4–C at its working dilution. After washing with PBS, biotinylated IgG secondary antibody (1:3000; Vector Laboratories, Inc., Burlingame, CA) was applied for 2 h, followed by avidin-biotin-peroxidase complex treatment for 1 h (ABC kit; Vector Laboratories, Inc.). Staining was visualized with diaminobenzidine (DAB; Vector Laboratories, Inc.). The sections were mounted onto slides and allowed to dry overnight. Following immunostaining, all sections were counterstained with hematoxylin (Fisher Scientific, Fair Lawn, NJ) for 4 min. After dehydration the sections were cover-slipped using DPX. For immunohistochemistry, the following antibodies were used: anti-mouse F4/80 antibody (rat monoclonal, 1:10,000; Serotec, Raleigh, NC), anti-neuronal-specific nuclear protein (NeuN) antibody (mouse monoclonal, 1:30,000; Chemicon, Temecula, CA), or biotinylated goat anti-mouse IgG (1:100; Vector Laboratories, Inc., Burlingame, CA).
Cell quantification and image analysis
Before counting, all slides were coded to avoid experimenter bias. Stereological analysis was performed on microglia that were positive for F4/80 immunostaining, and neurons that were positive for NeuN staining (the dorsal hippocampal pyramidal layer of CA1–CA3). Stereological analysis was performed using the optical fractionator method on a Nikon 218912 light microscope interfaced with the StereoInvestigator software package (MicroBrightField, Williston, VT) as described previously (Dawson et al., 2007, 2010). Results are presented as objects per unit volume to allow comparability between animals. For Fluoro-Jade B, sections containing hippocampus were examined for degenerating neurons using an epifluorescence microscope (Nikon, Tokyo, Japan), with a medium band blue excitation (Nikon B-2A, 450–490 nm) filter set. Degenerating neurons were quantified at 20× by counting the total number of Fluoro-Jade B-positive neurons present in the dorsal hippocampus of every eighth section throughout the brain hemisphere. For IgG immunohistochemistry, images were taken at 2× magnification and analyzed using ImageJ software (
RNA extraction and RT-PCR
For differential gene expression experiments, the brains were flash-frozen in liquid nitrogen. Frozen, pulverized tissue from half of the TBI brains was processed for RNA extraction (n=4/group). RNA quantity and quality was assessed with the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE), and by agarose gel electrophoresis. Only samples with a 260:280 ratio between 1.9 and 2.1, and a 260:230 ratio greater than 2.0, were further processed. First-strand complementary DNA (cDNA) was generated from 2 μg total RNA using the RT2 First Strand kit (SABiosciences, Frederick, MD) according to the manufacturer's instructions. Gene expression was measured using the Mouse Inflammatory PCR Array (SABiosciences), which profiles the expression of 84 genes related to inflammation. RT-PCR was performed according to the manufacturer's instructions using the 384-well plate format (4 samples, 96 wells per sample). One sample from each experimental group was run per plate to minimize any potential batch effect between RT-PCR runs. Quality of the cDNA and PCR efficiency was verified by housekeeping genes and RT-PCR controls included in the PCR array.
Gene expression data analysis
Raw RT-PCR data were analyzed using the Web-Based PCR Array Data Analysis software (SABiosciences). ΔCt values and ΔΔCt-based fold-change were calculated from raw threshold cycle data, using β-actin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as internal standards for normalization. Fold changes were then normalized against sham-operated controls. Gene pathway analysis was conducted using Gene Network Central Pro (SABiosciences) (
Hemorrhage volume measurement
On days 1, 7, and 14 after collagenase injection, hemorrhage volume was measured (n=6 animals per treatment group per recovery interval). The mice were anesthesized with 5% isoflurane. The brains were harvested, then frozen in 2-methylbutane at −30°C. Standardized cryostat-cut coronal sections (20 μm thick) were taken at 400-μm intervals from 11.2 mm to 5.2 mm anterior to the interaural line using a stereotaxic brain atlas (Paxinos and Watson, 1986). Brain sections were stained with hematoxylin and eosin, as previously described (Beray-Berthat et al., 2010). Images were taken at 2× magnification. Hemorrhage volume was outlined and measured using an image analyzer and lesion volume was computed.
Statistical analysis
Neurobehavioral data and hemorrhage volumes were analyzed using two-way analysis of variance (ANOVA) tests. Dunnet's post-hoc method was used to correct for repeated measures and to define between-group differences. Histological results were analyzed using the t-test. Survival data were analyzed by the Mantel-Cox log rank test. Statistical significance was set at p<0.05. All values were expressed as mean±standard deviation, and were analyzed using GraphPad Prism 5 software (La Jolla, CA).
Results
Rosuvastatin improves short-term and long-term functional outcome following TBI
Both rosuvastatin 5 mg/kg and simvastatin 5 mg/kg were associated with improved vestibulomotor performance (rotarod latencies) as compared to vehicle; however, only the group treated with rosuvastatin 5 mg/kg performed significantly better than the vehicle group (Fig. 1A). We next performed a dose-response analysis for rosuvastatin. Mice treated with rosuvastatin 1 mg/kg performed significantly better than those treated with vehicle, with no significant difference between the 1- and 5-mg/kg doses (Fig. 1B). To establish the durability of this effect, we next tested the effect of rosuvastatin 1 mg/kg and found that treatment was associated with sustained functional improvement throughout the 28-day testing period compared to the vehicle group (Fig. 1C). Long-term neurocognitive deficits are also common sequelae after injury; to evaluate this, neurocognitive assessment was performed with the MWM. Mice treated with rosuvastatin 1 mg/kg had shorter latency to escape to the platform (Fig. 1E). Given the impracticality of initiating therapy within 1 h after injury in a clinical trial, we next assessed whether the functional benefit of rosuvastatin would be maintained if the initial dose were delayed for 3 h after injury. When initial treatment was delayed, the 5-mg/kg but not the 1-mg/kg dose of rosuvastatin was associated with improvement in functional outcome (Fig. 1D). In aggregate, these data demonstrate that rosuvastatin improves long-term vestibulomotor and neurocognitive deficits following TBI to a greater extent than simvastatin. When administered within 1 h after injury, a dose as low as 1 mg/kg conferred durable benefit, whereas in a paradigm where initial treatment is delayed, higher doses were associated with optimal benefits.
Rosuvastatin decreases neuronal degeneration and microgliosis, and is associated with increased neuronal density following TBI
To evaluate whether there was a histological correlate to the functional improvement associated with rosuvastatin administration, we quantified neuronal degeneration in the hippocampus by staining with Fluoro-Jade B at 24 h after injury (Fig. 2). The hippocampal CA3 has been reported to be most susceptible to neuronal degeneration following closed head injury (Deng and Xu, 2011; Tashlykov et al., 2007), and Fluoro-Jade B-positive cells were predominantly found in the CA3 and the polymorphic layer of our model. Rosuvastatin 1 mg/kg was associated with a reduction in Fluoro-Jade B-positive hippocampal neurons (Fig. 2).

Rosuvastatin 1 mg/kg was associated with decreased neuronal degeneration in the hippocampus at day 1 post-injury, as demonstrated by Fluoro-Jade B neuronal staining (
One mechanism by which statins may reduce secondary neuronal injury is by attenuating endogenous neuroinflammatory responses. At day 7 post-injury, 1 mg/kg rosuvastatin was associated with decreased microgliosis as depicted by F4/80 immunostaining in the dorsal hippocampus (Fig. 3B and D), relative to vehicle-treated mice (Fig. 3A and C). Quantification with formal stereology showed that the reduction of F4/80-positive microglia was statistically significant (Fig. 3E). To establish whether the subacute reduction in neuronal injury and microgliosis was associated with a longer-term preservation of neuronal density in susceptible areas of the hippocampus, unbiased stereology of NeuN-positive hippocampal neurons was performed at 35 days post-injury. Administration of rosuvastatin 1 mg/kg was associated with a decrease in neuronal loss in the CA3 region, although no difference was noted in the dentate gyrus polymorphic layer (Fig. 4E). In aggregrate, these data indicate that improved functional outcomes were associated with histological evidence of a reduction in microgliosis, and both acute and long-term reductions in hippocampal neuronal injury.
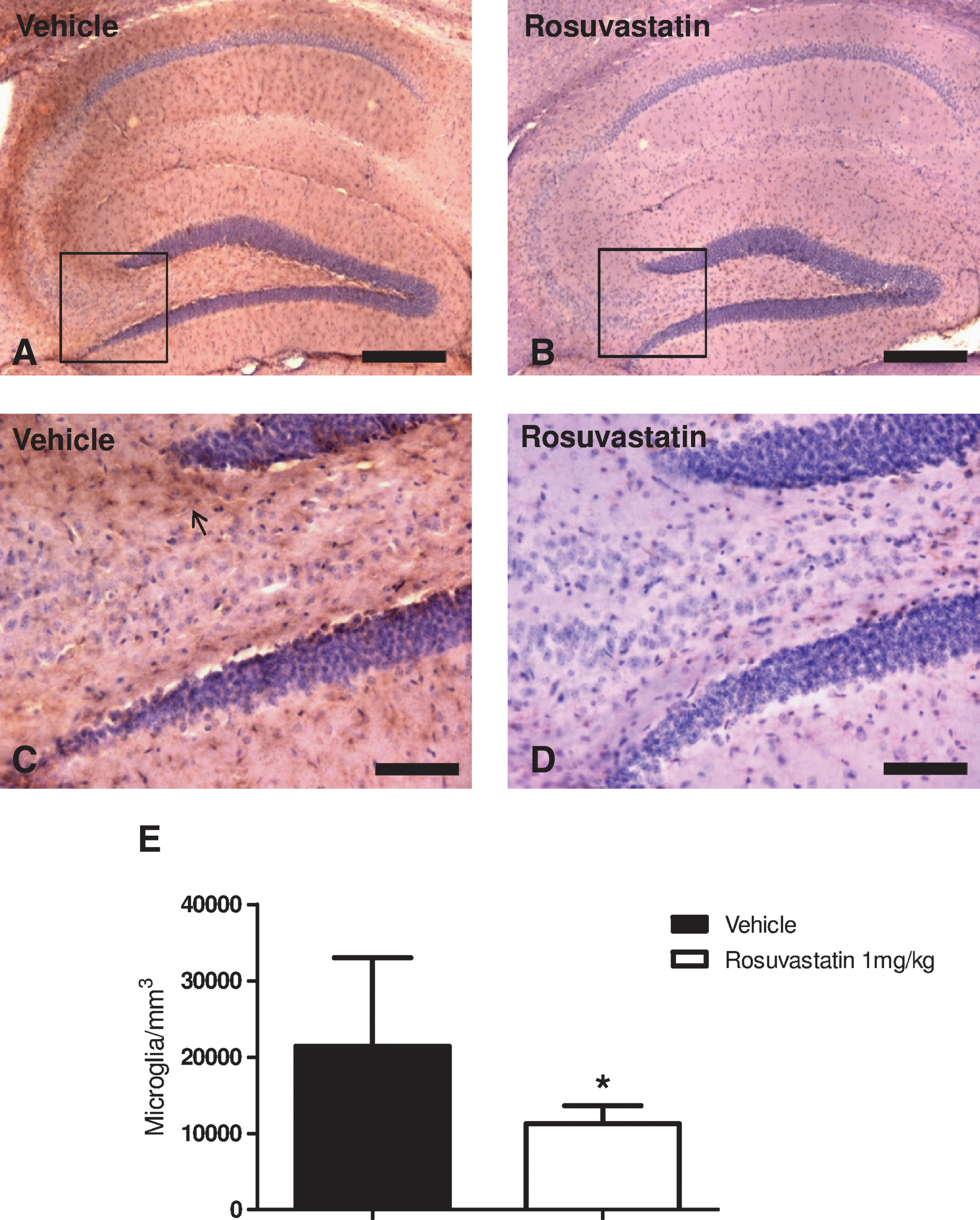
Rosuvastatin 1 mg/kg decreased microgliosis (as demonstrated by F4/80-positive cells) in the hippocampal area (CA3 and dentate gyrus polymorphic layer) at day 7 post-injury. (
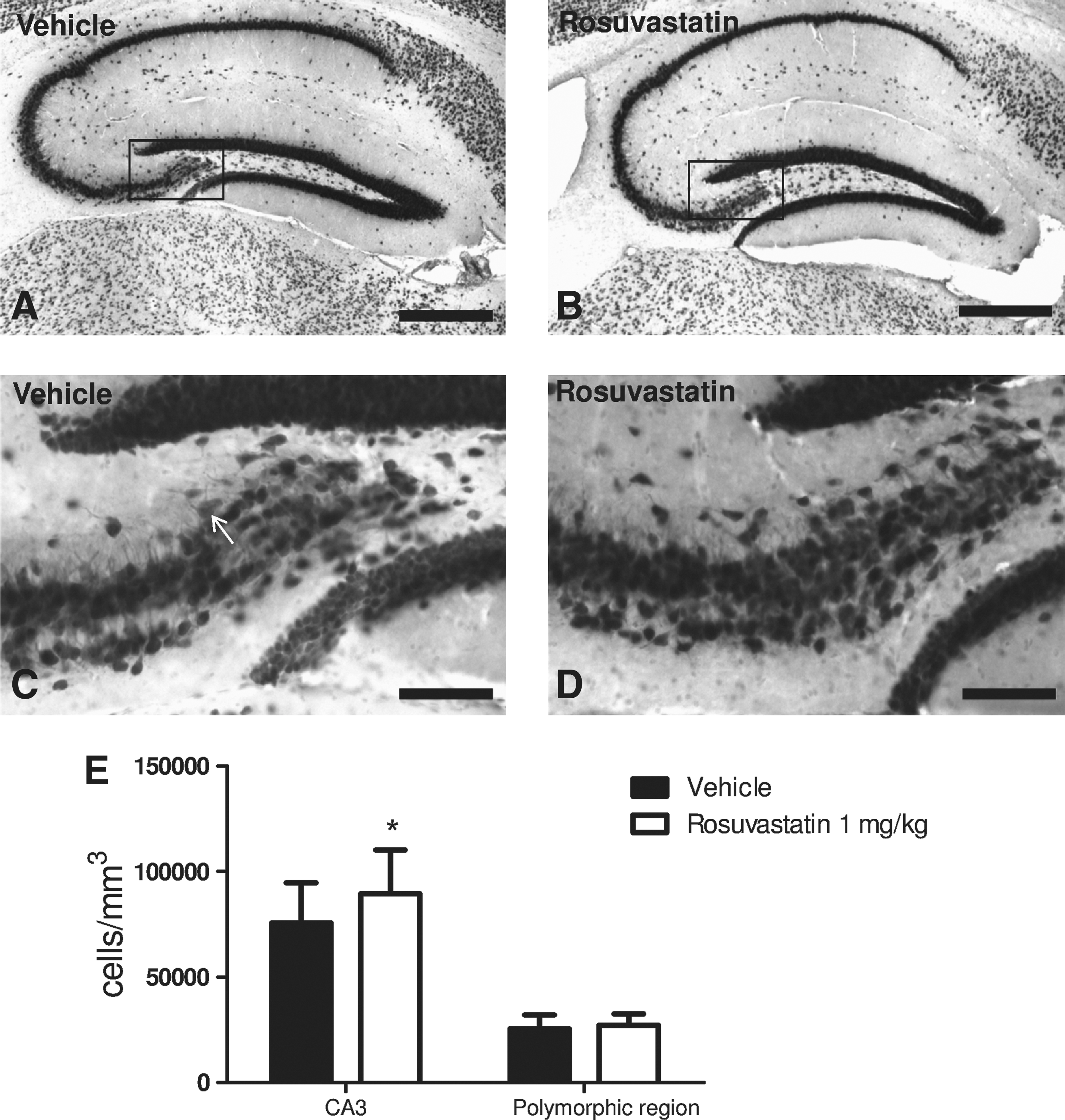
Neuronal-specific nuclear protein (NeuN) staining demonstrates that animals treated with rosuvastatin 1 mg/kg had improved neuronal density in the hippocampus at 35 days post-injury. (
Rosuvastatin attenuates the CNS inflammatory gene profile after TBI
To further evaluate the mechanisms by which rosuvastatin exerted its neuroprotective effects, differential gene expression was performed at days 1 and 7 post-injury in vehicle- and statin-treated mice. Genes that were downregulated >2.5 times after rosuvastatin treatment are shown in Figure 5A–C. TBI was associated with robust upregulation of a number of inflammatory genes in brain tissue at day 1 post injury, including representative toll-like receptors, chemokines, inflammatory cytokines, and apoptotic genes, which were markedly attenuated by 1 mg/kg of rosuvastatin. TBI also resulted in the downregulation of a cluster of genes associated with anti-inflammatory and trophic effects; in many cases, rosuvastatin resulted in a relative upregulation of these candidate genes (Fig. 6). The description of each gene and the relative fold changes against sham-operated mice are presented in the supplemental table (Supplemental Table 1; see online supplementary material at
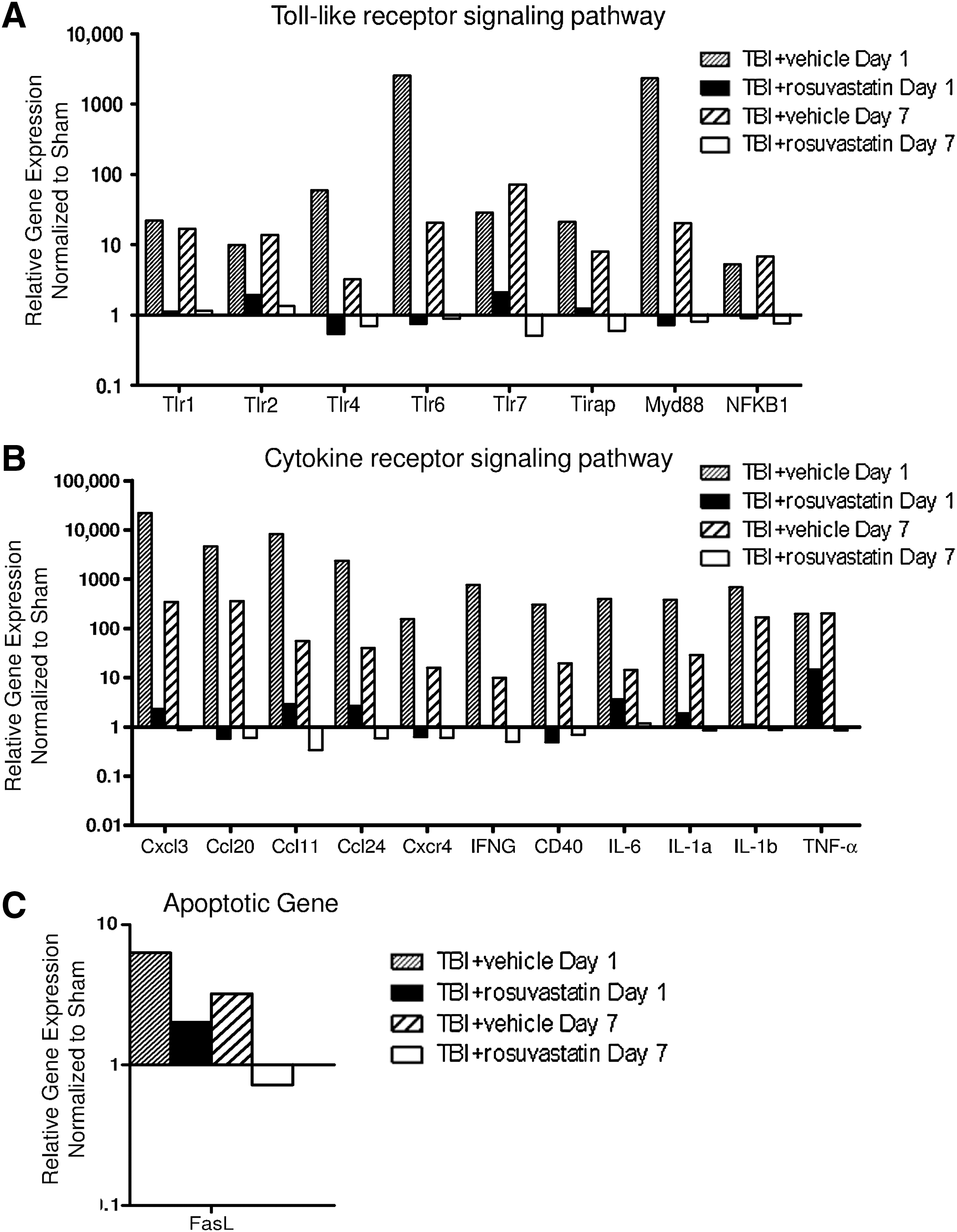
Differential gene expression assessed by microarray analysis showed that rosuvastatin 1 mg/kg markedly downregulated proinflammatory markers at day 1 and day 7 post-injury (
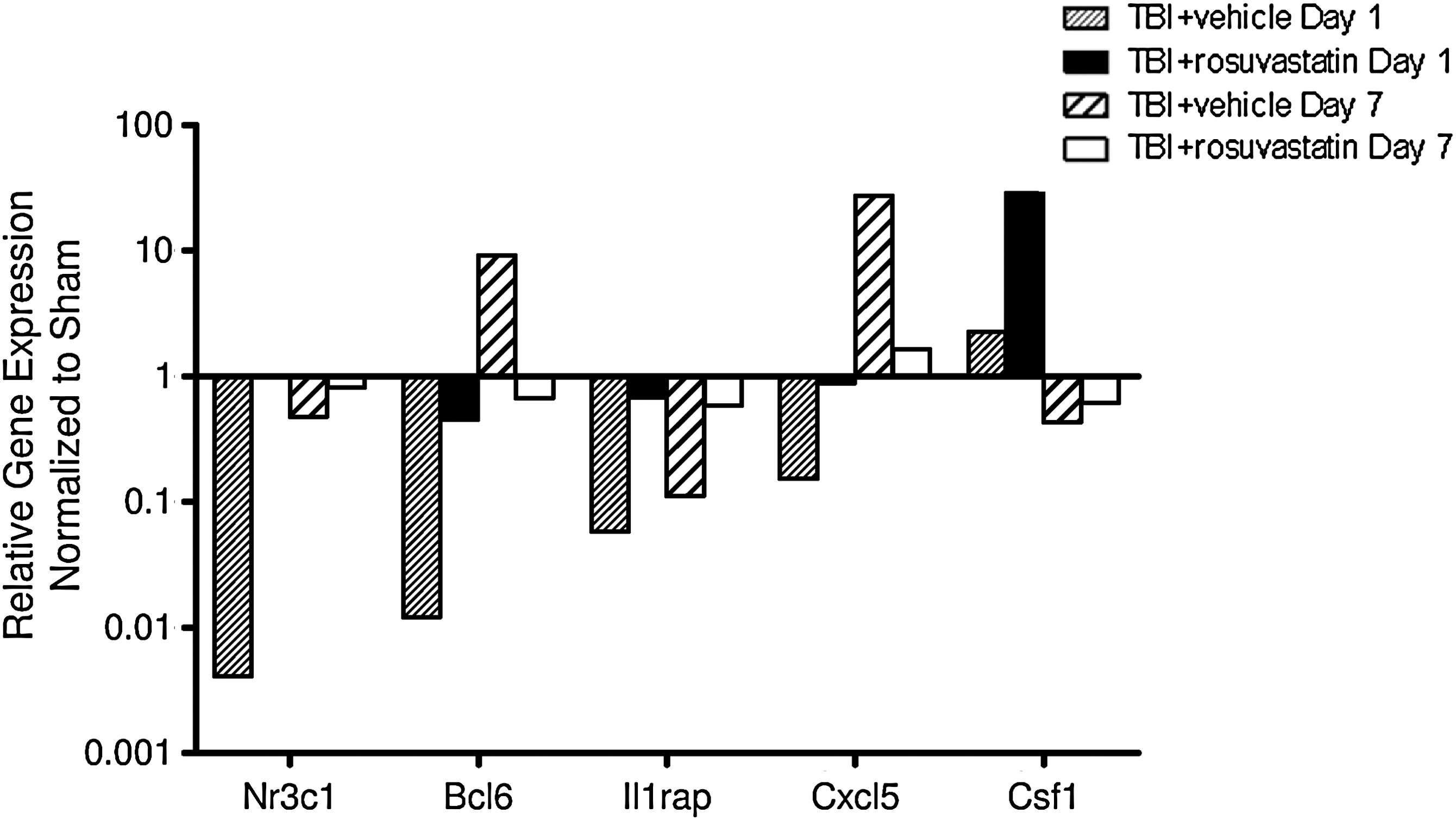
Genes that were upregulated >2.5 times after rosuvastatin treatment, relative to the vehicle group at day 1 post-injury and day 7 post-injury. Fold changes were normalized against sham-operated mice and expressed in log scale (n=6/group for day 1 post-injury, and 7/group for day 7 post-injury; TBI, traumatic brain injury).
Simvastatin improves functional outcome and survival in ICH
We next assessed the effect of statin administration in ICH in order to determine the optimal type and dose of statin for this injury paradigm. Administration of simvastatin at 1 mg/kg for 5 days improved rotarod function and survival, whereas no effect of rosuvastatin was observed (Fig. 7). No differences were noted in hemorrhage volumes between groups measured at days 1, 7, and 14 post-injury (Fig. 8B and C), suggesting that statin treatment acts via subacute mechanisms involving neuronal survival and CNS inflammatory responses, rather than by influencing hemostasis or clot resorption.
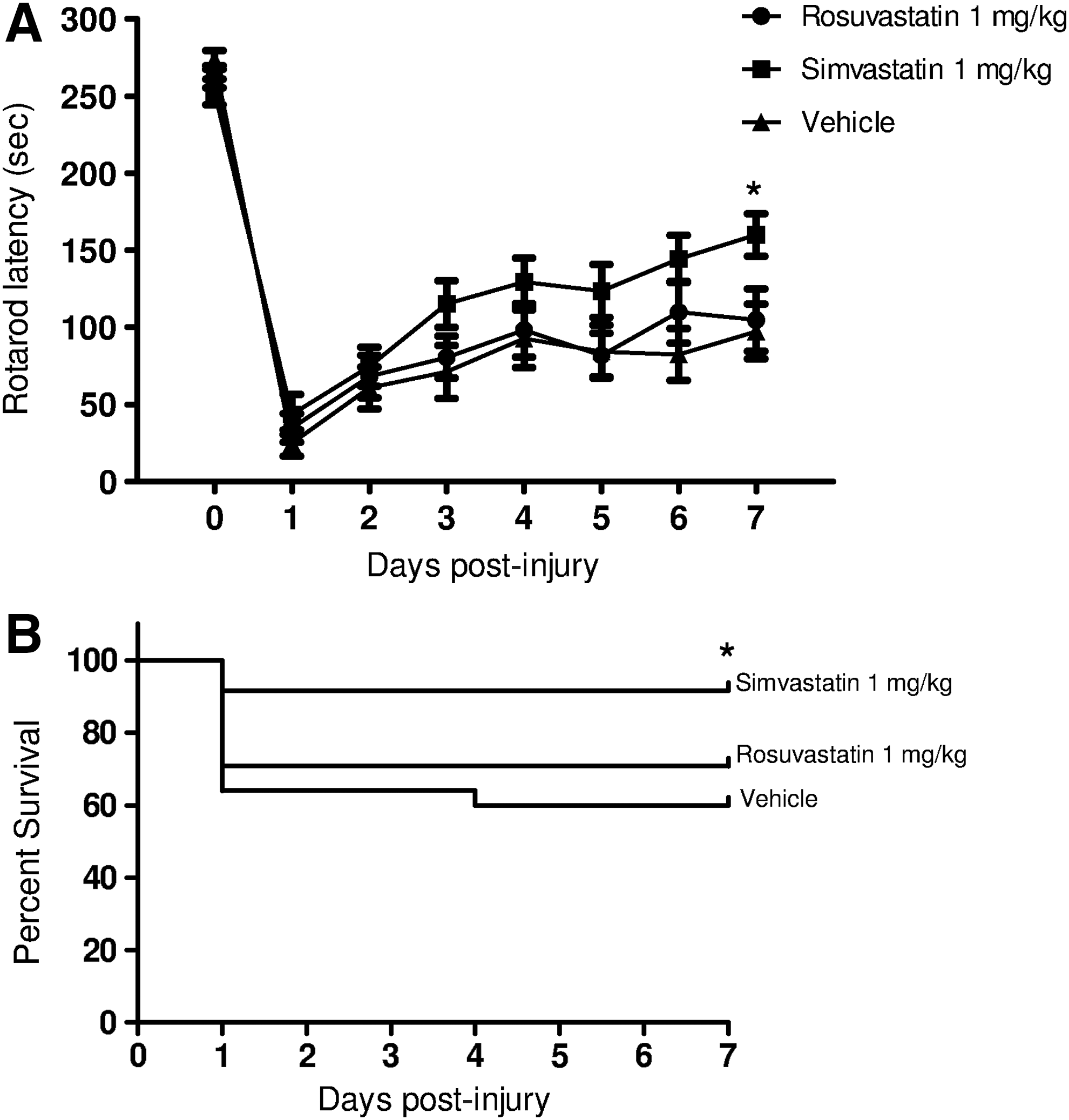
(
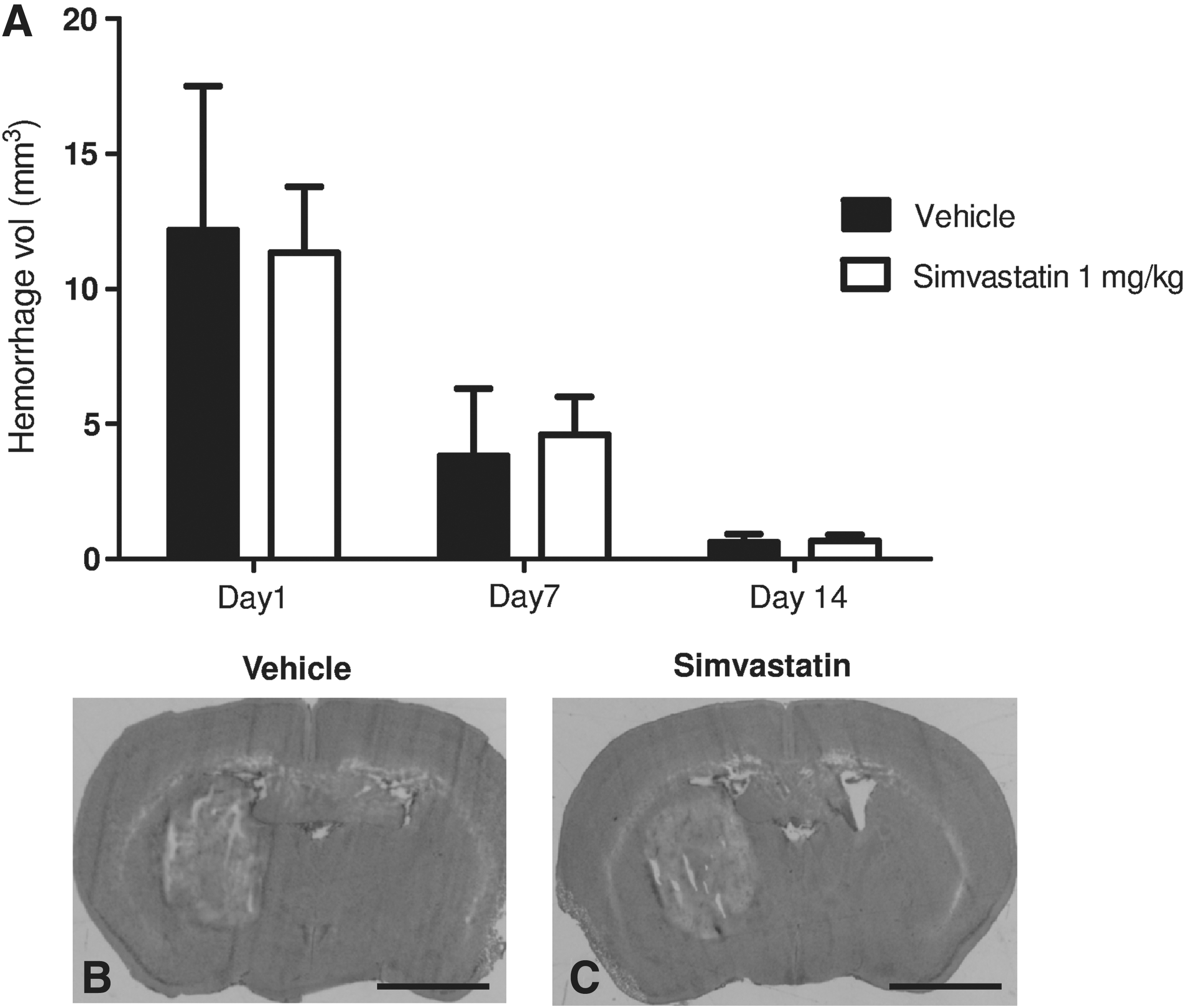
(
TBI and ICH cause differential breach of the blood–brain barrier
One possible explanation for the differential effects of simvastatin and rosuvastatin in the TBI and ICH models was their difference in lipophilicity and CNS penetration. To establish whether TBI and ICH were associated with differential breakdown of the BBB, IgG immunohistochemistry was performed (positive staining indicates IgG extravasation from the vasculature due to BBB breakdown). As illustrated in Figure 9A and B, both TBI and ICH were associated with significant BBB breakdown compared to sham mice. However, TBI was associated with a more diffuse breach in the BBB, whereas ICH had more focal disruption of the BBB. Representative images are presented in Figure 9C, D, and E.
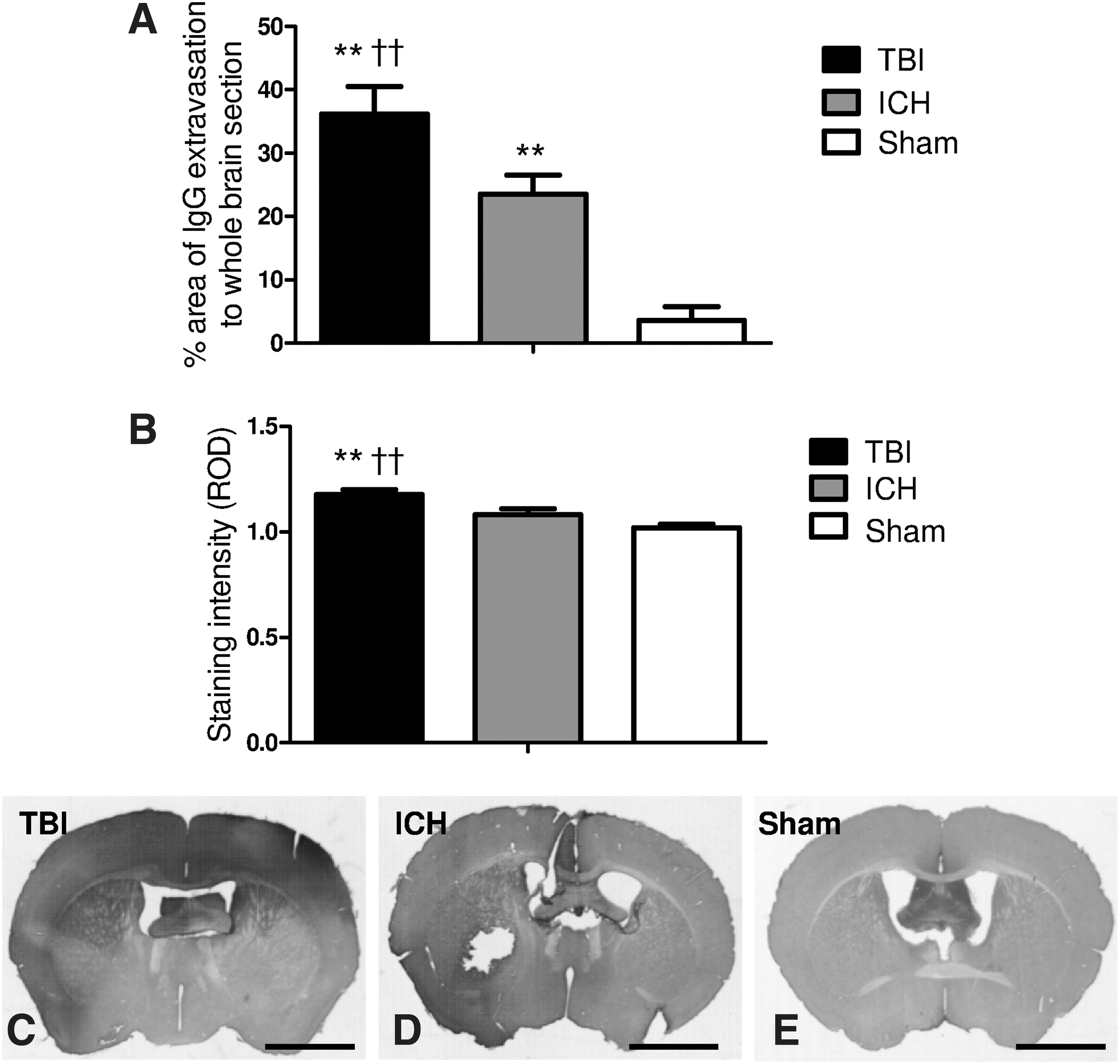
Immunohistochemical analysis of blood–brain barrier (BBB) integrity of traumatic brain injury (TBI), intracerebral hemorrhage (ICH), and sham-operated mice. As shown in
Discussion
In this study, statins improved vestibulomotor and neurocognitive outcomes following TBI and ICH. This functional improvement was associated with a reduction of acute neuronal degeneration and long-term preservation of neuronal density following head injury. Statin treatment was also associated with attenuation of neuroinflammatory responses, as demonstrated by a decrease in microgliosis and downregulation of inflammatory genes. In our TBI model, higher doses of statin were not associated with increased efficacy when administered within 1 h of injury, although a higher dose proved more effective when treatment was delayed. In addition to optimizing dosing, another important consideration for planning clinical translation is our finding that statin efficacy was dependent on injury model. Simvastatin worked better than rosuvastatin in ICH, perhaps due to better CNS penetration following a more focal breach of the BBB.
Statins influence multiple mechanisms of acute and secondary neuronal injury, such as improvement of cerebral blood flow through upregulation of epithelial nitric oxide synthase (Williams et al., 1998), reduced excitotoxic cell death (Zacco et al., 2003), and reduced apoptosis by downregulation of caspases (Wu et al., 2008a). Statins have also been associated with a decrease in oxidative stress by attenuating NADPH oxidase-dependent superoxide production (Erdos et al., 2006), reduced thrombogenic responses (Lu et al., 2004b), downregulation of inflammatory cytokines (Chen et al., 2007) and attentuated microgliosis (Li et al., 2009). In addition to these acute and subacute effects, statins may also play a beneficial role in more chronic responses, such as promoting neurogenesis and plasticity by upregulating the PI3K/Akt pathway (Wu et al., 2008a), and upregulating angiogenesis via a VEGF-mediated pathway (Wu et al., 2008b). These pleiotropic effects of statins are partly mediated by inhibition of isoprenoids, which serve as lipid attachments for intracellular signaling molecules (Liao, 2005).
The acute neuroinflammatory response associated with brain injury may exacerbate cerebral edema and secondary neuronal injury via release of free radicals, and excitotoxic and inflammatory mediators. Microglia are the resident histiocytes of the CNS, and the cornerstone of the endogenous neuroinflammatory response. Our current findings indicate that statins decrease microgliosis and attenuate inflammatory gene upregulation following TBI. Although the inflammatory cascade that occurs following injury remains incompletely defined, the genes that were downregulated by rosuvastatin after TBI are in the toll-like receptor (Tlr) pathway, thus leading to proinflammatory cytokine production and pro-apoptotic responses. Tlr6 shares homology with other toll-like receptors such as Tlr1 and Tlr2, and upon activation, activates NF-κB through an adaptor protein myeloid differentiation primary response protein (Myd88) (Takeuchi et al., 1999), causing production of proinflammatory cytokines and pro-apoptotic proteins.
Interestingly, administration of statins also upregulated pro-survival anti-inflammatory genes that are downregulated by brain trauma. B-cell lymphoma 6 protein (Bcl-6) represses the transcription of proinflammatory genes that are activated upon Tlr4 and NF-κB stimulation (Barish et al., 2010). Similarly, an isoform of IL-1 receptor accessory protein (IL-1rap) in the CNS attenuates the local inflammatory response and decreases neuronal degeneration (Smith et al., 1995). Nr3c1, a glucocorticoid receptor, induces expression of anti-inflammatory genes (Rhen and Cidlowski, 2005). CXC motif chemokine 5 (Cxcl5) is associated with villus angiogenesis after small bowel resection (McMellen et al., 2010), while colony stimulating factor 1 (Csf1) was shown to enhance arteriogenesis and ameliorates cerebral damage in a mouse model of ischemic stroke (Sugiyama et al., 2011).
A number of preclinical and clinical studies have demonstrated neuroprotection by a number of different members of the HMG-CoA reductase inhibitor class, suggesting a class effect for neuroprotection (Liao, 2005; Wible and Laskowitz, 2010). However, important differences exist between potency, absorption, distribution, and metabolism in the target organ. For example, simvastatin is relatively lipophilic and is able to penetrate into the CNS compartment, whereas rosuvastatin, by nature of its polar methane sulfonamide moiety, is relatively hydrophilic (McKenney, 2003), and therefore would be expected to have less CNS penetration in the setting of an intact BBB. However, rosuvastatin may have other advantages in the setting of CNS injury. For example, as compared to simvastatin, rosuvastatin is more potent, possesses a longer elimination half-life, and has greater bioavailability than simvastatin (McKenney, 2003). Moreover, unlike simvastatin, rosuvastatin is not metabolized via the CYP3A4 enzyme system, which decreases the potential for drug-drug interactions. This may be particularly important in the intensive care setting, where polypharmacy is common.
The current data indicate that the choice of optimal statin for clinical translation may not be “one size fits all,” but may depend on the type of brain injury. For example, in the ICH paradigm, but not the TBI model, simvastatin was superior to rosuvastatin. Our results suggest that ICH is associated with a more localized breach of the BBB than closed head injury. In the setting of a focal injury, it may be advantageous to administer a lipophilic statin with greater CNS penetration, whereas this characteristic may be less important in the setting of global breach of the BBB, such as may occur following closed head injury. Animal models may also be helpful in defining the dosing regimen in translational protocols. Most early studies have demonstrated therapeutic effects of statins at relatively high doses (Chauhan and Gatto, 2011; Chen et al., 2009; Karki et al., 2009; Seyfried et al., 2004; Wang et al., 2007), which may result in a higher incidence of adverse effects when translated clinically. In contrast, this study has shown no benefit of 5 mg/kg rosuvastatin as compared to 1 mg/kg when administered within 1 h after TBI, although higher doses may be necessary when there is a more prolonged latency to treatment.
There are several limitations to this study that should be addressed. The age of the animals used (8–12 weeks) corresponds to young adulthood. Clearly, injury responses vary as a function of age, and it is possible that our results may not be applicable to juvenile or aged animals. Although every attempt has been made to employ clinically-relevant models, these paradigms do not entirely recapitulate the clinical scenario. For example, ICH was induced by stereotactic collagenase injection, as opposed to the spontaneous rupture of small penetrating vessels (Qureshi et al., 2009) that is often associated with hypertensive ICH. Thus, extrapolating pathological features in clinical ICH, such as the extent of BBB breakdown, should be performed with caution. It is also possible that collagenase may directly contribute to CNS inflammatory responses that would not otherwise be present (Lema et al., 2004). Although we have focused on the anti-inflammatory mechanisms by which statins may reduce brain injury, there is also clear evidence of late effects on neurogenesis, angiogenesis, and synaptogenesis (Chen et al., 2003), which would be an important consideration when determining length of treatment. In addition to modulating acute injury responses, it is also possible that statins may directly affect learning during the repeated rotorod testing, although it is notable that the improved performance associated with statins was retained after the drug was discontinued.
In conclusion, administration of statins in patients with acute brain injury represents an attractive strategy for clinical translation. Our current work is consistent with a growing body of literature suggesting that statins improve outcomes after acute brain injury. Animal models may be helpful in defining the clinical parameters of optimal statin timing and dosage to facilitate the rational design of pilot clinical trials.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Acknowledgments
The authors would like to thank Brenden Dawson for his assistance with some of the histological work. This work was partially supported by an investigator-initiated grant from AstraZeneca, which also supplied rosuvastatin.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.