
Abstract
Neurogenesis is stimulated following injury to the adult brain and could potentially contribute to tissue repair. However, evidence suggests that microglia activated in response to injury are detrimental to the survival of new neurons, thus limiting the neurogenic response. The aim of this study was to determine the effect of the anti-inflammatory drug minocycline on neurogenesis and functional recovery after a closed head injury model of focal traumatic brain injury (TBI). Beginning 30 min after trauma, minocycline was administered for up to 2 weeks and bromodeoxyuridine was given on days 1–4 to label proliferating cells. Neurological outcome and motor function were evaluated over 6 weeks using the Neurological Severity Score (NSS) and ledged beam task. Microglial activation was assessed in the pericontusional cortex and hippocampus at 1 week post-trauma, using immunohistochemistry to detect F4/80. Following immunolabeling of bromodeoxyuridine, double-cortin, and NeuN, cells undergoing distinct stages of neurogenesis, including proliferation, neuronal differentiation, neuroblast migration, and long-term survival, were quantified at 1 and 6 weeks in the hippocampal dentate gyrus, as well as in the subventricular zone of the lateral ventricles and the pericontusional cortex. Our results show that minocycline successfully reduced microglial activation and promoted early neurological recovery that was sustained over 6 weeks. We also show for the first time in the closed head injury model, that early stages of neurogenesis were stimulated in the hippocampus and subventricular zone; however, no increase in new mature neurons occurred. Contrary to our hypothesis, despite the attenuation of activated microglia, minocycline did not support neurogenesis in the hippocampus, lateral ventricles, or pericontusional cortex, with none of the neurogenic stages being affected by treatment. These data provide evidence that a general suppression of microglial activation is insufficient to enhance neuronal production, suggesting that further work is required to elucidate the relationship between microglia and neurogenesis after TBI.
Introduction
I
The pathophysiology of brain injury is very complex and involves the activation of an overwhelming array of molecular cascades that could either attenuate or enhance neurogenesis. As part of the brain's innate inflammatory response to injury, microglia are rapidly activated and recruited to the pericontusional cortex, where they release numerous pro- and anti-inflammatory cytokines, chemokines, and reactive nitrogen and oxygen species, but also many growth factors (Rock et al., 2004). Recently, a number of studies have begun to reveal that activated microglia have a complex, multifaceted impact on injury-induced neurogenesis, with both beneficial and deleterious actions. While microglial-derived factors may stimulate the early stages of neurogenesis, their ultimate consequence may be to create an environment that is detrimental to the survival of new neurons (Das and Basu, 2008; Ekdahl et al., 2009). For example, it has been shown that increased microglial activation through over-expression of the proinflammatory cytokine interleukin-6 (IL-6), or injection of lipopolysaccharide, was associated with a reduction of newborn neurons in the rodent hippocampus, predominantly due to attenuated survival of the new cells (Ekdahl et al., 2003; Monje et al., 2003; Vallieres et al., 2002). Moreover, anti-inflammatory treatment with minocycline and indomethacin decreased microglial activation, resulting in enhanced hippocampal neurogenesis after cranial irradiation, epilepsy, and ischemic stroke (Ekdahl et al., 2003; Hoehn et al., 2005; Liu et al., 2007; Monje et al., 2002), which was associated with improved functional outcome (Liu et al., 2007). Together, these results support the hypothesis that activated microglia inhibit the endogenous neurogenic response in the injured brain.
Neurogenesis has become an emerging field within neurotrauma research due to its potential for endogenous regeneration, yet the mechanisms regulating neurogenesis after TBI are largely unexplored. We and others have extensively characterized post-traumatic cerebral inflammation in a focal closed head injury (CHI) model, and we have recently demonstrated that a robust microglial response occurs in the injured cortex, peaking from 4–7 days after injury (Bye et al., 2007; Semple et al., 2010; Ziebell et al., 2011). Based on the studies described above indicating a detrimental function of activated microglia towards hippocampal neurogenesis in models of acute inflammation and stroke, we proposed that microglia may play a similar role in hippocampal neurogenesis after TBI. Furthermore, activated microglia may also affect the ability of SVZ precursors to produce new neurons within the damaged cortex. Therefore, in this study we aimed to determine whether attenuating microglial recruitment and activation by treatment with the anti-inflammatory drug minocycline could enhance specific stages of neurogenesis arising from hippocampal and SVZ precursors, and whether this corresponds to an improvement in neurological outcome in a CHI mouse model of focal TBI. We used the anti-inflammatory tetracycline derivative minocycline because of its well established ability to specifically inhibit microglial activation in the injured brain (Bye et al., 2007; Das et al., 2011; Liu et al., 2007; Tikka et al., 2001; Wasserman and Schlichter, 2007; Wu et al., 2009).
Methods
Mouse model of focal closed head injury
All experimental procedures were approved by the Alfred Medical Research and Education Precinct (AMREP) Animal Ethics Committee. In this study, adult C57BL/6 mice aged 12–14 weeks (28–34 g), were provided by the AMREP Precinct Animal Centre (Melbourne, Australia). The mice were housed with food and water ad libitum under a 12-h dark/light cycle.
Unilateral focal brain injury was induced to the left cortex using a mouse model of CHI as described previously (Bye et al., 2007; Chen et al., 1996; Semple et al., 2010). Briefly, the mice were ether-anesthetized and the skull was exposed by a 2-cm longitudinal scalp incision. A 333-g metal rod with a silicone tip was dropped from a height of 2 cm onto the intact skull of the left hemisphere, 2 mm lateral to the midline in the mid-coronal plane between the bregma and lambda to induce a skull fracture and a localized underlying cortical contusion. Only mice that sustained a skull fracture were included in the study. After the impact, the scalp incision was sutured and the mice were monitored during the first hour of recovery, and then housed individually on heat pads at 37°C overnight. Sham control mice underwent anesthesia and scalp incision, but did not receive traumatic impact. Following CHI, the mice were randomly assigned to treatment groups: CHI+minocycline, CHI+vehicle (sterile phosphate-buffered saline [PBS]) and sham+vehicle, for survival until 1 week or 6 weeks post-trauma (n=6–7 per treatment per time point).
Bromodeoxyuridine labeling
To label proliferating endogenous precursors, mice were given a single daily IP injection of bromodeoxyuridine (BrdU, 200 mg/kg in 0.007 M NaOH/0.9% NaCl; Sigma-Aldrich, St. Louis, MO), beginning 24 h post-CHI for 4 consecutive days.
Administration of minocycline
Minocycline (5 mg/mL in 20 mM NaOH/PBS; Sigma-Aldrich) was administered by IP injection initially at 30 min post-trauma (45 mg/kg), and subsequently every 12 h (22.5 mg/kg per injection) until the mice were sacrificed at 1 week. Mice surviving up to 6 weeks post-injury received twice-daily minocycline injections for 2 weeks. Vehicle-control CHI and sham groups received equivalent volume IP injections of sterile PBS. The mice were assigned randomly to treatment groups.
Evaluation of neurological impairment
Neurological Severity Score (NSS)
As adapted from Chen and colleagues (Chen et al., 1996), the 10-point Neurological Severity Score (NSS) was used to assess post-traumatic neurological outcome of the mice according to 10 different parameters to investigate motor function, alertness, and physiological behavior. One point was awarded for failure of each task, such that a maximum of 10 points represents severe neurological dysfunction, whereas 0 points indicates normal neurological function. NSS is considered a good measure of recovery after the induction of TBI (Flierl et al., 2009; Semple et al., 2010). The NSS was assessed by a researcher blinded to treatment, at 1 h post-CHI, and every 24 h thereafter, for the first week, then twice weekly for the 6-week cohort of mice.
Ledged Beam Test
A ledged beam test, described previously by our group for use in this model (Bye et al., 2007), was used to assess hemiparesis after trauma. Briefly, mice traversed a 1-m-long inclined (30o) tapered beam with underhanging ledges, from the widest point of the beam to the narrowest end. The numbers of foot faults—defined as steps taken on the lower ledge—were quantified for the forelimbs and hindlimbs contralateral to the side of brain injury, through blinded analysis of videotape recordings. The mice underwent three consecutive trials per session, and the average score of the three trials was generated per mouse for statistical analysis. The ledged beam test was performed at times corresponding to NSS assessment, beginning at 24 h following CHI.
Immunohistochemistry
Tissue sections
The mice were perfused at either 1 week or 6 weeks post-CHI/sham with heparinized saline (5 units heparin/mL) followed by 4% paraformaldehyde (PFA). The brains were post-fixed in 4% PFA at 4°C overnight, and cryoprotected in 30% sucrose in PBS for at least 24 h before snap freezing. Approximately 300 serial coronal cryostat sections (20 μm) were collected between bregma +1.7 mm and −4.04 mm, spanning the lateral ventricles, hippocampus, and the entire lesioned region of the cortex, and stored at −80°C until use.
Immunohistochemical staining was performed to investigate the post-traumatic inflammatory response of macrophages and microglia (F4/80), as well as several stages of neurogenesis, including precursor proliferation and survival (BrdU), neuronal differentiation and migration (double-cortin; Dcx), and mature phenotype of new cells (BrdU/NeuN/GFAP). Endogenous peroxidases were blocked in 1% H2O2/PBS (F4/80, NeuN, and Dcx), or 10% methanol/2% H2O2/PBS (BrdU). For BrdU immunohistochemistry, DNA was denatured in 2 M HCl (37°C for 45 min), then all sections were blocked in skim milk, normal serum (goat or horse), and Triton X-100/Tween-20 for 1 h. The sections were incubated overnight at 4°C with primary antibodies to detect activated microglia/macrophages (monoclonal rat α-mouse F4/80, 1:500 in 2% milk/PBS; Serotec, Kidlington, Oxford, U.K.), newly differentiated immature neurons (polyclonal goat α-human Dcx, 1:500 in 5% NHS/PBS/0.3% Triton; Santa Cruz Biotechnology, Santa Cruz, CA), and newly proliferated cells that incorporated BrdU (monoclonal rat α-BrdU, 1:1000 in 1% BSA/PBS; Abcam, Cambridge, U.K.). The following biotinylated secondary antibodies were applied for 1 h: goat α-rat IgG, 1:500 in 1% NGS/2% milk/PBS; horse α-goat IgG, 1:500 in 2% NHS/PBS; and goat α-rat IgG 1:600 in 2% NGS/PBS/0.1% Tween 20 (Vector Laboratories, Burlingame, CA). Positive staining was detected using the Vectastain ABC kit (Vector Laboratories), and 3-3′-diaminobenzidine tetrahydrochloride (DAB; Sigma-Aldrich), with (BrdU) or without (F4/80, Dcx) nickel enhancement. Each experiment included negative control sections on which primary antibodies were omitted. The brain sections were visualized under an Olympus BX50 microscope, and the images were captured using an Olympus DP71 digital camera. Cell counts were performed using AnalySIS LisProfessional software. The investigator was blinded to treatment while performing the analyses.
Cell counts
Activated microglia/macrophages
As we have described previously, F4/80-labeled cells surrounding the injured cortex exhibit distinct morphologies of resting microglia, with small elongated cell bodies and several thin processes. Reactive microglia have thickened processes and more intense F4/80 immunoreactivity. Activated microglia have enlarged ameboid cell bodies and fewer, shrunken processes. Highly-activated microglia are indistinguishable from macrophages, with dark F4/80 immunoreactivity and no processes (Bye et al., 2007). In this study, all F4/80-immunopositive cells displaying a reactive or activated microglial phenotype, or an activated microglial/macrophage phenotype, were counted. Cell counts in the pericontusional cortex were performed at 7 days post-CHI, which is the peak of macrophage infiltration in this model, as reported previously (Semple et al., 2010). Five frames per section were captured (1730×1730 mm images at 10×magnification) of four sections per brain spanning the lesioned hemisphere. A 400×400-mm grid was overlaid on each frame and F4/80-positive cells were counted within four squares systematically selected out of a possible 12 squares. Cell counts were calculated as total cells per mm2.
In the hippocampus, numerous F4/80-positive cells were present and were often tightly clustered. Therefore, rather than counting individual labeled cells, F4/80 immunolabeling in this region was quantified by densitometric analysis using ImageJ software (National Institutes of Health, v1.40). F4/80-staining intensity was examined within a selected region of interest (ROI) on each brain section, which included the hippocampal tissue surrounding the DG and hilus. Analysis was performed on grey scale images adjusted to a set threshold to ensure consistency between sections. Data were expressed as “% ROI F4/80-positive,” corresponding to F4/80-staining intensity averaged across four sections spanning the anterior hippocampus.
New cells
BrdU-immunolabeled sections were imaged at 20×magnification, and BrdU-labeled cells were identified as those displaying intensely stained nuclei. Cell counts were performed for mice in the sham and trauma groups at 1 and 6 weeks, in approximately 10 sections per brain, separated by 400 μm. BrdU-positive cells were counted in both the ipsilateral and contralateral brain hemisphere relative to the side of injury, in the DG SGZ and granule cell layer (GCL) in 4–6 sections spanning bregma −1.7 to −2.9 mm, and in the SVZ of the lateral ventricle in 8–10 sections spanning bregma +1.2 to −1.7 mm. The total number of BrdU-positive cells per region per section were counted and averaged across all sections for each brain.
Immature neurons
To assess the effect of minocycline treatment on neuronal differentiation in the hippocampus at 1 week following trauma, Dcx-positive immature neurons were counted in the SGZ and GCL as described for BrdU-labeled cells. To assess the apparent migration of neuroblasts from the SVZ to the injured cortex, 20×images were taken of three brain sections spanning the cortical lesion and containing the SVZ, and all Dcx-positive cells from the lateral corner of the SVZ, through the corpus callosum, and into the pericontusional cortex, were counted and averaged per section.
Immunofluorescence triple labeling of BrdU, NeuN, and glial fibrillary acidic protein (GFAP)
Tissue sections
To identify and quantify the phenotype of the new cells surviving to 6 weeks post-injury, 5–6 sections per brain separated by 200 μm throughout the lesioned cortex and hippocampus were processed for triple-labeling immunofluorescence to detect co-labeling of BrdU with markers for mature neurons and astrocytes. After DNA denaturation as described above, the sections were blocked in 5% NGS/2% BSA/0.1% Triton/4% milk/PBS before simultaneous incubation with primary antibodies to detect new cells (monoclonal rat α-BrdU, 1:1000; Abcam), mature neurons (monoclonal anti-mouse NeuN, 1:500; Chemicon, Billerica, MA), and astrocytes (polyclonal rabbit α-GFAP, 1:1000; Santa Cruz Biotechnology) in blocking solution overnight at 4°C. The following Alexa-Fluor-conjugated secondary antibodies were applied for 2 h (all at 1:200 in blocking solution from Molecular Probes, Eugene, OR): goat anti-rat Alexa 488 (green BrdU-positive cells), goat anti-mouse Alexa 647 (blue NeuN-positive cells), and goat anti-rabbit Alexa 568 (red GFAP-positive cells). The sections were then cover-slipped with Vectashield Hardset mounting medium (Vector Laboratories) and stored at 4°C in the dark until analysis.
Cell counts
In order to estimate the percentages of new neurons and astrocytes among the newly generated cells, confocal analysis of triple-immunofluorescence staining was used to quantify co-labeling of BrdU with NeuN or GFAP in the hippocampus and pericontusional cortex. Five sections at 480-μm intervals spanning the hippocampus and containing the cortical lesion site were selected for fluorescence immunohistochemistry. Using a Zeiss LSM Meta 510 or Nikon AR1 confocal laser scanning microscope with excitation/emission wavelengths of 488 nm (green), 543 nm (red), and 633 nm (far red), by argon and He-Ne lasers, confocal z sectioning was performed at 1-μm intervals using a Plan-Apochromat X 63 (NA=1.40) water-immersion objective, for 5–6 randomly selected cell fields per DG (ipsilateral and contralateral to the injury) on each of 3 sections, and on 3 randomly selected cell fields per section within the pericontusional cortex, on 3 sections containing the lesion site (a total of 9–15 cell fields in the DG and 9 in the pericontusional cortex per brain). Analyses were performed in sequential scanning mode to rule out cross-bleeding between detection channels. Images were acquired and a z-stack was reconstructed using Zeiss LSM Image software or the NIS viewer. BrdU-positive nuclei were analyzed in their entire z-axis, and were also viewed in orthogonal planes (x–y) to verify double-labeling and exclude false double-labeling caused by overlay of signals from different cells. The total number of BrdU-positive cells and the number of those double-labeled with NeuN or GFAP were counted in each z-stack and summed per region per brain, and the percentage of total BrdU-positive cells that was co-labeled for each marker was then calculated per brain. All cell counts were performed by an investigator blinded to the experimental groups.
Statistical analysis
Statistical analysis of data was performed using Sigma Stat 2.03 (SPSS Inc., Chicago, IL). Behavioral data from the NSS and ledged beam walk test were analyzed by two-way repeated-measure (RM) analysis of variance (ANOVA) using the factors of time point post-surgery and treatment group. Cell count data of F4/80-positive cells in the cortex was assessed by t-test, while intensity of F4/80 immunoreactivity in the hippocampus was assessed by two-way ANOVA using the factors of treatment group and side of brain relative to injury. Cell count data following BrdU or Dcx immunolabeling was analyzed by one-way ANOVA to assess differences between treatment groups within a specific brain hemisphere and single time point, and by two-way ANOVA to assess whether the effect of minocycline treatment may be dependent upon the time post-surgery, or on brain hemisphere relative to the site of injury. Tukey's test was used for all post-hoc analyses following ANOVAs. Differences were considered statistically significant at the 5% level. Stated p values are those from Tukey's post-hoc analysis unless otherwise indicated. All data are expressed as mean±standard error of the mean (SEM).
Image manipulation
For figure preparation, images were adjusted for size, brightness, and contrast only, using Adobe Photoshop version 8.0.
Results
Improved neurological recovery in minocycline-treated mice after CHI
Earlier work by us and others has shown that treatment with minocycline is often, but not always, associated with improved behavioral outcomes (Bye et al., 2007; Fox et al., 2005). We therefore investigated whether our current minocycline treatment strategy would affect neurological recovery of CHI mice (Fig. 1). Using the NSS (Fig. 1A) to assess neurological deficits in all mice within the first week post-trauma or sham (n=12–14) showed that the vehicle-treated and minocycline-treated CHI groups had similar initial scores at 1 h post-injury that were significantly higher than those of sham-operated mice (p<0.001 by two-way RM ANOVA for effect of treatment, time, and interaction between treatment and time), indicating comparable trauma severity. While vehicle-treated CHI mice displayed some spontaneous recovery at 7 days (p<0.05 for 7 days versus 1 h), minocycline-treated mice had an early and sustained improvement in NSS from 1 day post injury (p<0.05 for 1 h versus all other time points, and 1 day versus 3, 4, and 7 days). Moreover, on days 3–7 after injury, CHI mice treated with minocycline displayed significant reductions in NSS compared to the vehicle group (p<0.05). Assessment of the cohort of mice surviving to 6 weeks (n=6–7) showed that the beneficial effect of minocycline persisted. While the overall elevation of the NSS in the vehicle-treated mice was sustained throughout the 6-week duration (p<0.05 versus sham controls, two-way RM ANOVA effect of treatment p<0.001, time p<0.001, and the interaction between treatment and time p=0.081), the scores of minocycline-treated mice were significantly lower than the vehicle group (p<0.05), returning to sham levels (p>0.05).
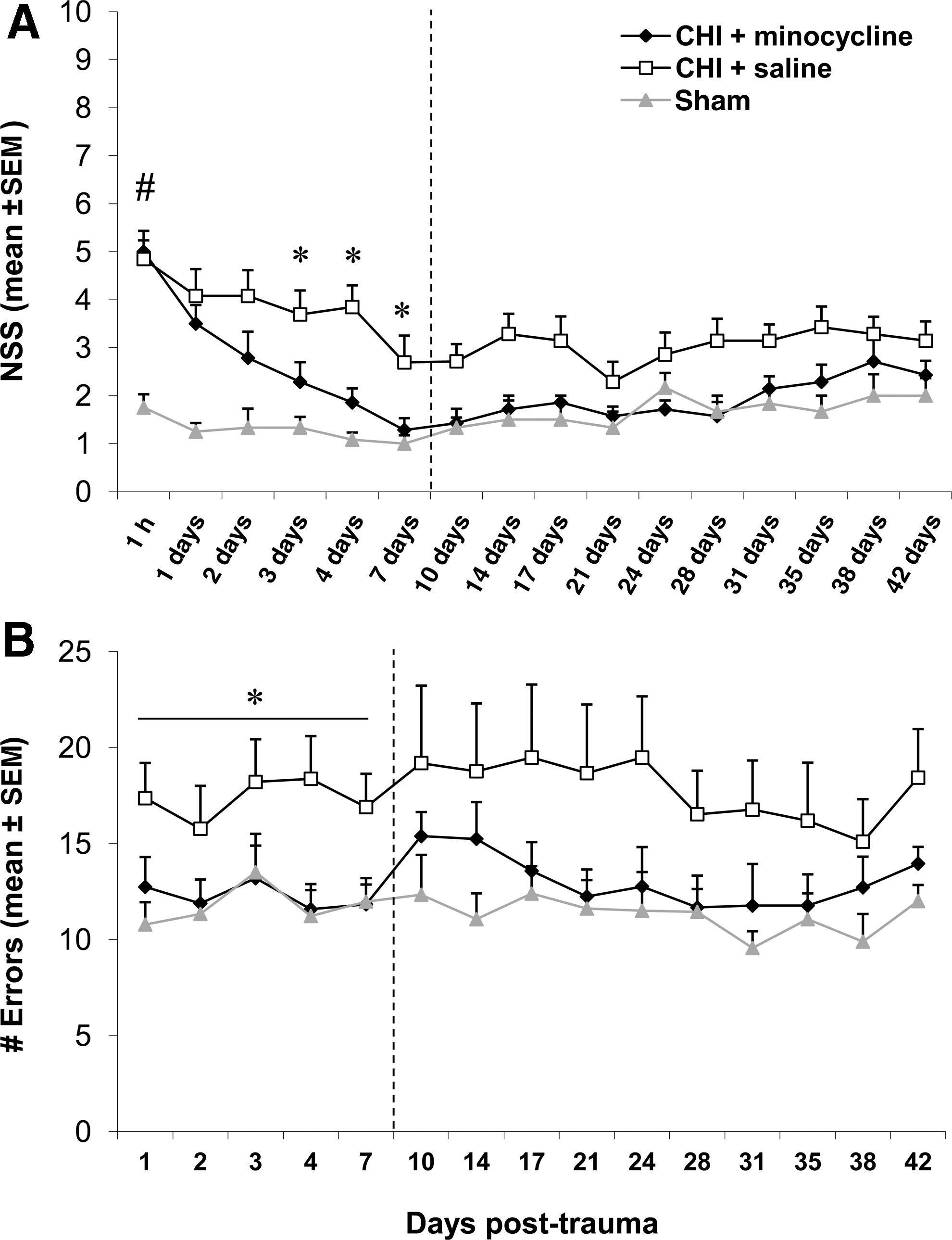
Minocycline improves neurological recovery after closed head injury (CHI). (
Similarly, minocycline-treated mice showed improved motor recovery after trauma, as assessed by the ledged beam test (Fig. 1B). Within the first week post-CHI (n=12–14), the minocycline-treated mice performed the task with similar efficiency to sham-operated controls, making significantly fewer errors than the vehicle-treated group (p>0.05 for minocycline versus sham, and p<0.05 for minocycline versus vehicle; two-way RM ANOVA effect of treatment p=0.021, time p=0.116, and interaction between treatment and time p=0.691). Conversely, the vehicle-treated CHI mice had significantly more errors than shams within the first week (p<0.05). When the smaller cohort of mice surviving to 6 weeks (n=6–7) was assessed to evaluate the long-term effect of minocycline treatment, no significant differences were detected between the three experimental groups (two-way RM ANOVA effect of treatment p=0.222), although the motor deficits in the vehicle-treated group appeared to persist compared to the minocycline-treated and sham-operated groups across the time course. Interestingly, statistical analysis detected a significant effect of time, regardless of treatment group, with scores on days 28 to 38 being lower than those of day 3 (p<0.05; two-way RM ANOVA effect of time p<0.001, and interaction between treatment and time p=0.926). This appears to be most likely due to some spontaneous recovery of motor function in the vehicle-treated CHI mice by these late time points.
Reduced activation of microglia/macrophages in minocycline-treated mice at 1 week after CHI
Based on the anti-inflammatory properties of minocycline, we investigated whether treatment with minocycline reduces the activation of resident microglia and/or recruitment of peripheral macrophages following CHI. Abundant F4/80-positive cells were evident surrounding the lesioned cortex and within the ipsilateral hippocampus at 1 week following CHI (Fig. 2A), and the accumulation of these cells was attenuated with minocycline treatment (Fig. 2B). Consistent with this observation, counts of F4/80-positive cells revealed that the density of activated microglia/macrophages in the pericontusional cortex was significantly reduced, by 40%, following minocycline treatment, compared with vehicle controls (p=0.019; Fig. 2E). Similarly, when F4/80 immunoreactivity was evaluated by densitometry in the hippocampi ipsilateral and contralateral to the lesion site, we found an overall significant reduction in minocycline-treated mice, regardless of side, by greater than 50% compared to the vehicle-treated CHI group (p<0.05 saline versus minocycline and sham; p>0.05 minocycline versus sham; two-way ANOVA effect of treatment p=0.002, side p=0.011, and interaction between treatment and side p=0.065; Fig. 2F).

Minocycline reduced the accumulation of activated microglia/macrophages at 1 week post-CHI. (
Hippocampal neurogenesis is not affected by minocycline treatment after CHI
Cell proliferation and survival in the dentate gyrus
Following the injection of BrdU for 4 days after either CHI or sham surgery, BrdU-positive cells were quantified in the hippocampal DG at 1 and 6 weeks post-CHI/sham in order to assess the effects of minocycline treatment on the proliferation and survival of new cells, respectively. As shown in Figure 3, in sham-operated mice at 1 week (Fig. 3A), only a few BrdU-positive cells were evident in the DG, predominantly in the SGZ. Following trauma at this time point, BrdU labeling increased dramatically in the DG ipsilateral to injury (Fig. 3B), with abundant labeled cells detected within the GCL as well as the SGZ. The distribution and extent of BrdU-positive cells within the DG appeared similar in the minocycline-treated CHI mice (Fig. 3C). It is interesting to note that the spread of BrdU-positive cells throughout the hilus and molecular layer of the hippocampus in the CHI mice appears to be attenuated in the minocycline group; we speculate that this could potentially reflect reduced proliferation of microglia in these regions with minocycline, especially in light of the decreased F4/80 staining seen after minocycline treatment in the same region, as described above. However, this observation needs to be confirmed with double-labeling to identify the cell type. The post-traumatic increase in BrdU-positive cells in the DG contralateral to injury was apparently lower than that in the ipsilateral hemisphere in both vehicle- and minocycline-treated mice. A similar pattern of BrdU labeling was evident 6 weeks in all treatment groups; however, the overall number of positive cells was markedly reduced compared to 1 week in the corresponding regions (not shown).
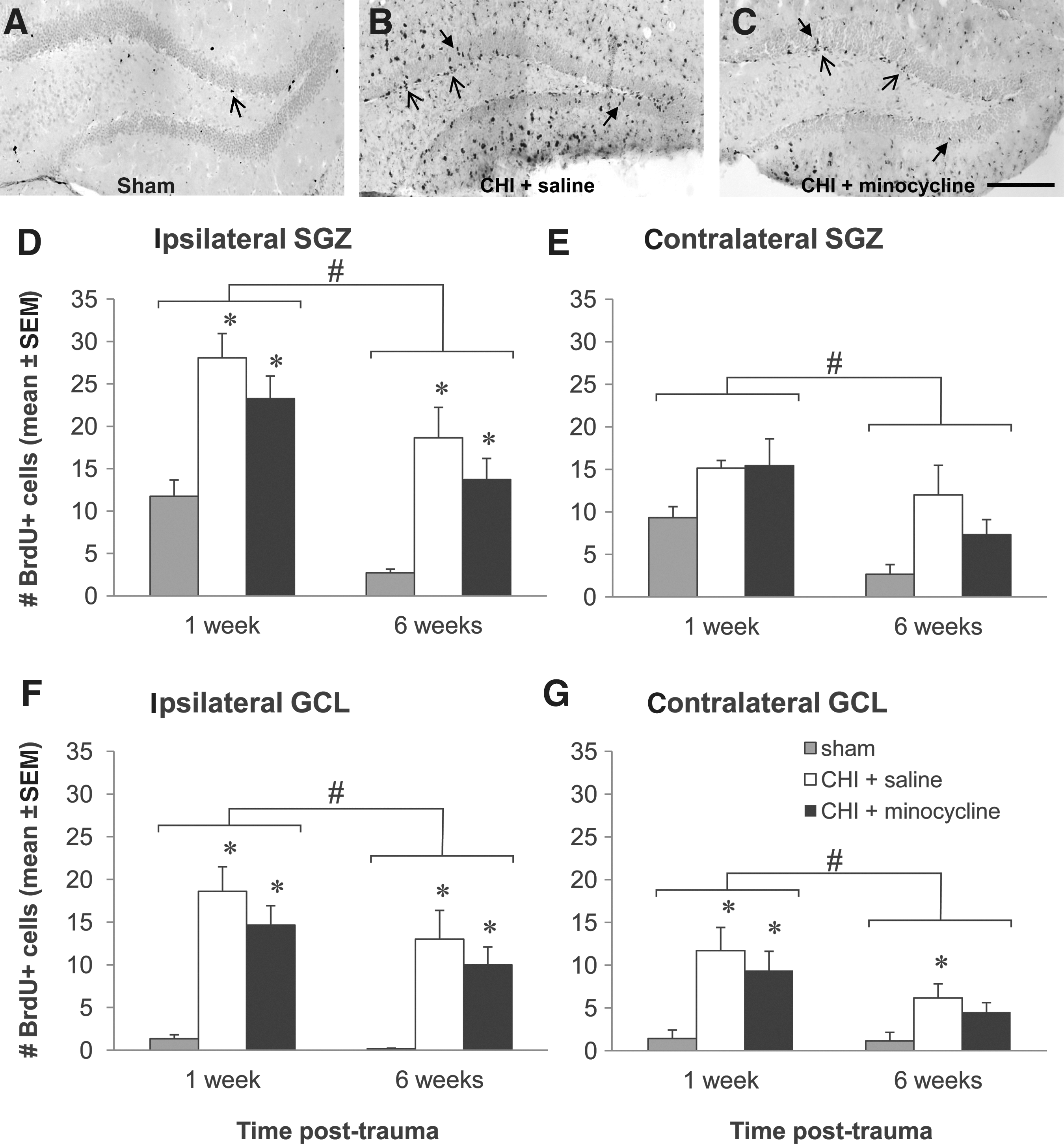
Minocycline does not affect proliferation or survival of new cells in the dentate gyrus (DG) after trauma. (
These observations were supported by quantitative assessment of BrdU-positive cells at 1 and 6 weeks in the SGZ (Fig. 3D and E) and GCL (Fig. 3F and G) of the hippocampi ipsilateral and contralateral to the cortical lesion. In the DG ipsilateral to injury, CHI induced significant twofold and 14-fold increases in the number of BrdU-positive cells at 1 week in the SGZ (Fig. 3D) and GCL (Fig. 3F), respectively, compared to shams, and these levels were not significantly affected by minocycline treatment (p<0.05 sham versus vehicle and minocycline; SGZ: one-way ANOVA, p=0.001; GCL: one-way ANOVA, p<0.001).
At 6 weeks post-injury in the same DG regions, the trauma groups maintained sevenfold and 20-fold more BrdU-positive cells compared to shams in the SGZ (Fig. 3D) and GCL (Fig. 3F), respectively, with no differences between the vehicle- and minocycline-treated groups (p<0.05 sham versus vehicle and minocycline; SGZ: one-way ANOVA, p=0.002; GCL: one-way ANOVA, p=0.005). Moreover, in both DG regions, regardless of the treatment group, the number of BrdU-positive cells was significantly decreased at 6 weeks compared to 1 week (two-way ANOVA for the effect of time and the effect of interaction between time and treatment, respectively, for the SGZ: p<0.001 and 0.995, and GCL: p=0.002 and 0.378). Interestingly, in both the SGZ and GCL, BrdU-positive cell numbers decreased over time, by approximately 80% in shams, but only by 30–40% in the CHI groups, which may suggest that trauma is not only affecting the rate of precursor proliferation, but also the ultimate survival of the new cells, with no additional effect of minocycline observed.
In the DG contralateral to injury (Fig. 3E and G), trauma did not induce cell proliferation to the same extent as in the ipsilateral side (Fig. 3D and F); two-way ANOVA revealed that there were significantly fewer BrdU-labeled cells, regardless of treatment, in the SGZ and the GCL at both 1 and 6 weeks, compared to the ipsilateral DG regions at each time point (for each DG region at each time point, p<0.05 for ipsilateral versus contralateral). Moreover, in contrast to the ipsilateral data, in the contralateral SGZ (Fig. 3E), CHI BrdU-positive cell values were only 40% higher than shams at 1 week post-trauma, and this increase was not significant (one-way ANOVA, p=0.096). Conversely, in the GCL (Fig. 3G), BrdU-positive cells did increase at 1 week post-trauma, by eightfold (p<0.05 sham versus vehicle and minocycline; one-way ANOVA, p=0.006). However, there was no effect of minocycline treatment on the number of BrdU-positive cells in either contralateral DG region at 1 week.
A similar pattern of BrdU labeling was observed in the contralateral SGZ and GCL at 6 weeks as seen with the 1-week data, with no effect of trauma on the number of BrdU-positive cells in the SGZ (Fig. 3E; one-way ANOVA, p=0.052), but a significant fivefold increase in the GCL of the CHI-vehicle group compared to shams (Fig. 3G; p<0.05; one-way ANOVA, p=0.034). Also, similar to the effect seen in the ipsilateral hemisphere, in both contralateral DG regions, regardless of the treatment group, the number of BrdU-positive cells was significantly decreased at 6 weeks compared to 1 week (two-way ANOVA effect of time and effect of interaction between time and treatment, respectively, for SGZ: p<0.001 and 0.202, and GCL: p=0.010 and 0.213).
Neuronal differentiation in the dentate gyrus
To investigate whether early and ongoing neuronal differentiation in the DG following CHI is affected by minocycline treatment, Dcx immunolabeling was used to quantify immature neurons in the SGZ and GCL of the DG ipsilateral and contralateral to injury, at 1 and 6 weeks. As shown in the representative images of the ipsilateral DG in Figure 4, Dcx-positive cells were predominantly located in the SGZ of sham-operated mice of both the 1- and 6-week groups (Fig. 4A and D), while in the vehicle- (Fig. 4B and E) and minocycline-treated (Fig. 4C and F) CHI mice, there appeared to be more Dcx-positive cells deeper within the GCL at 1 week (Fig. 4B and C), but not at 6 weeks (Fig. 4E and F), suggesting that an early enhanced production, and possibly migration, of new neurons deeper into the GCL may occur in the DG after trauma.
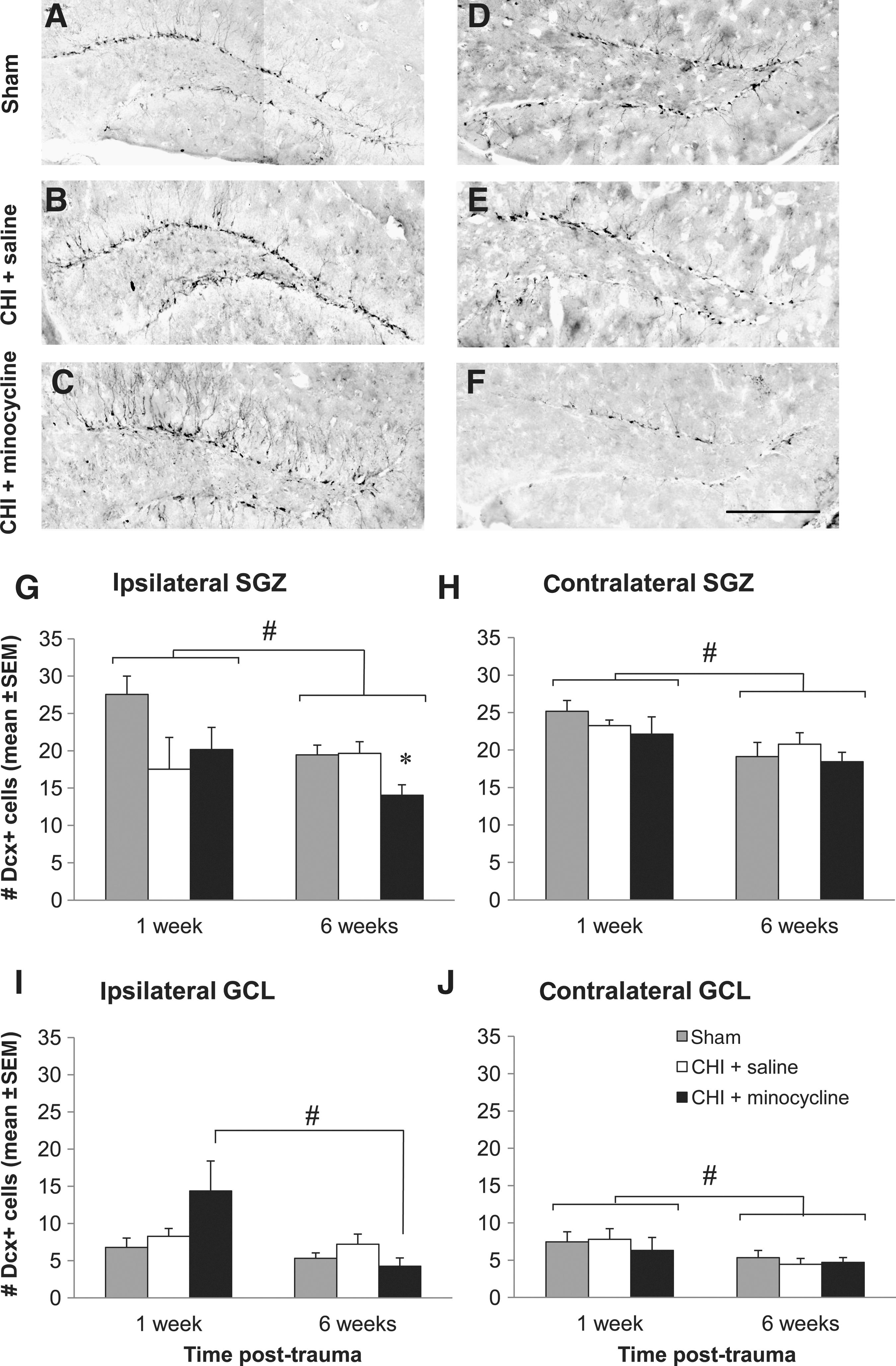
Minocycline does not influence the number of immature neurons in the dentate gyrus (DG) after trauma. (
However, quantitative analysis of Dcx-positive cells showed no significant differences in the number of immature neurons in the ipsilateral DG at 1 week, in either the SGZ (Fig. 4G; one-way ANOVA, p=0.105), or GCL (Fig. 4I; one-way ANOVA, p=0.127). Despite the lack of statistical significance, it was interesting to note that within the SGZ of both CHI groups, the number of Dcx-positive cells was approximately 30% lower than in shams, while in the GCL, the number in the minocycline group was twofold higher, supporting our qualitative assessment. These apparent differences were not observed in the DG contralateral to injury, with numbers of Dcx-positive cells remaining very similar to those of shams 1 week after CHI and minocycline treatment in both the SGZ (Fig. 4H; one-way ANOVA, p=0.447), and GCL (Fig. 4J; one-way ANOVA, p=0.775), suggesting that trauma and/or minocycline treatment may have a lesser effect on immature neurons in the contralateral hemisphere. Overall, the abundance of Dcx-positive cells was similar between the ipsilateral and contralateral DG, with no significant differences detected in the 1-week groups (two-way ANOVA for the effect of side, p=0.408 and 0.151, for the SGZ and GCL, respectively).
By 6 weeks post-CHI or sham-surgery, the overall numbers of Dcx-positive cells were slightly, but significantly, lower in each DG region regardless of treatment, compared to 1-week values (two-way ANOVA for the effect of time, ipsilateral SGZ: p=0.043; contralateral SGZ: p=0.005; contralateral GCL: p=0.020; ipsilateral GCL: p=0.012, and effect of interaction between treatment and time p=0.039, with p<0.05 for 1 week versus 6 weeks in the minocycline-treated group only). Interestingly, in the ipsilateral SGZ at 6 weeks post-CHI (Fig. 4G), minocycline treatment reduced Dcx-positive cells by 28% of sham and vehicle values (one-way ANOVA, p=0.020), suggesting perhaps that minocycline may be inhibiting ongoing neuronal differentiation, or conversely, when considered with the 1-week GCL data, that minocycline may stimulate an early migration of new neurons from the SGZ into the GCL, resulting in a depletion in the SGZ population at later time points. Further investigation of these hypotheses is required before these data can be confidently interpreted. In all other DG regions, ipsilateral and contralateral to injury, there were no differences between the treatment groups at the 6-week time point (one-way ANOVA, p=0.187, 0.543, and 0.736, for the ipsilateral GCL, contralateral SGZ, and contralateral GCL, respectively).
Maturation and survival of new neurons in the dentate gyrus
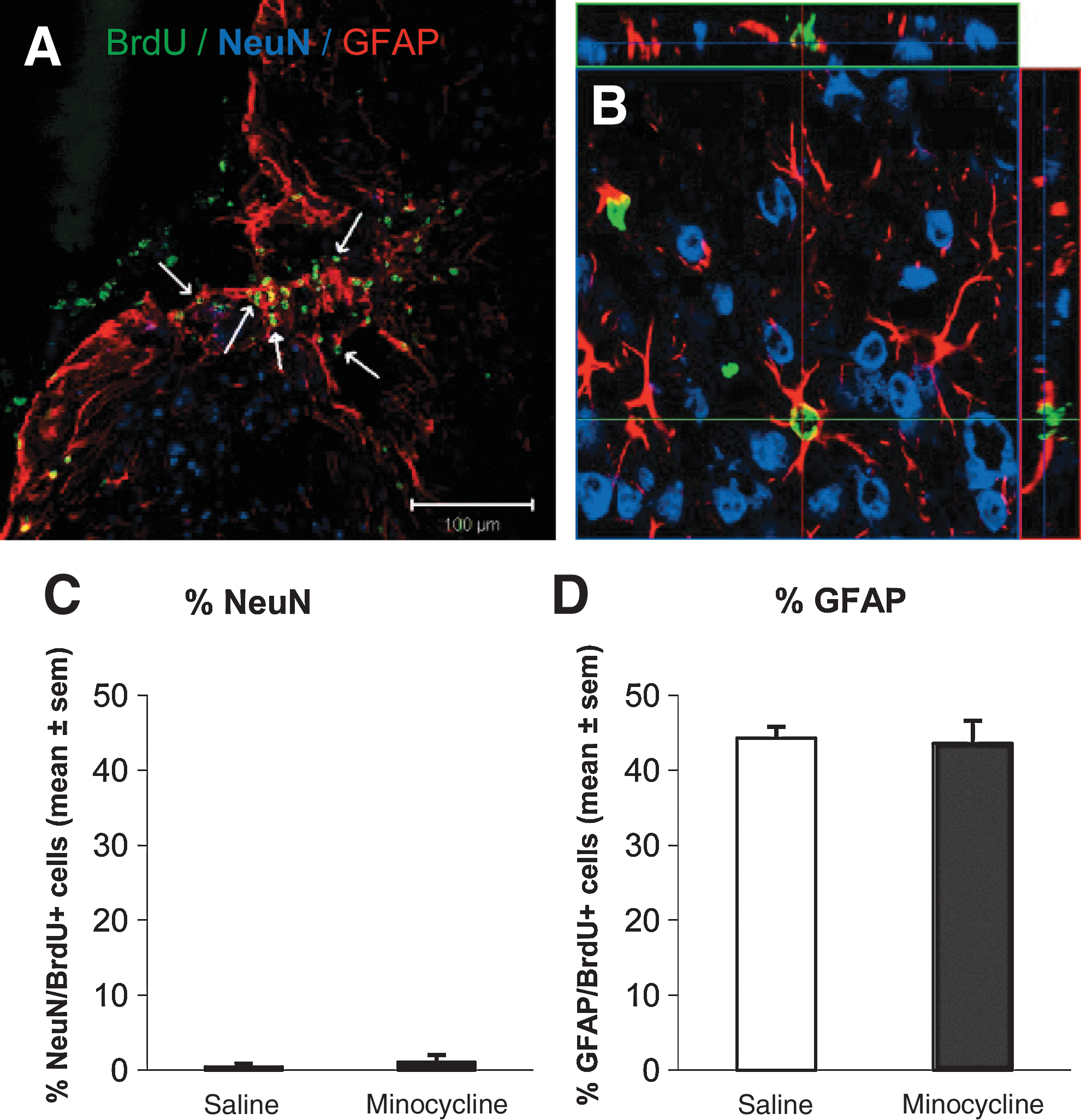
In order to investigate the ability of minocycline to enhance long-term survival of new mature neurons in the DG following CHI, immunofluorescence triple-labeling for BrdU with the mature neuron marker NeuN and the astrocyte marker GFAP was assessed at 6 weeks post-CHI or sham-surgery. As demonstrated in Figure 5A–D, BrdU immunolabeling was clearly co-localized with NeuN in a small number of cells, while a larger proportion were co-labeled with the astrocyte marker GFAP. These two different cell populations were quantified and expressed as the percentage of total BrdU-positive cells co-labeled with NeuN (NeuN-positive/BrdU-positive; Fig. 5E), or with GFAP (GFAP-positive/BrdU-positive; Fig. 5F).
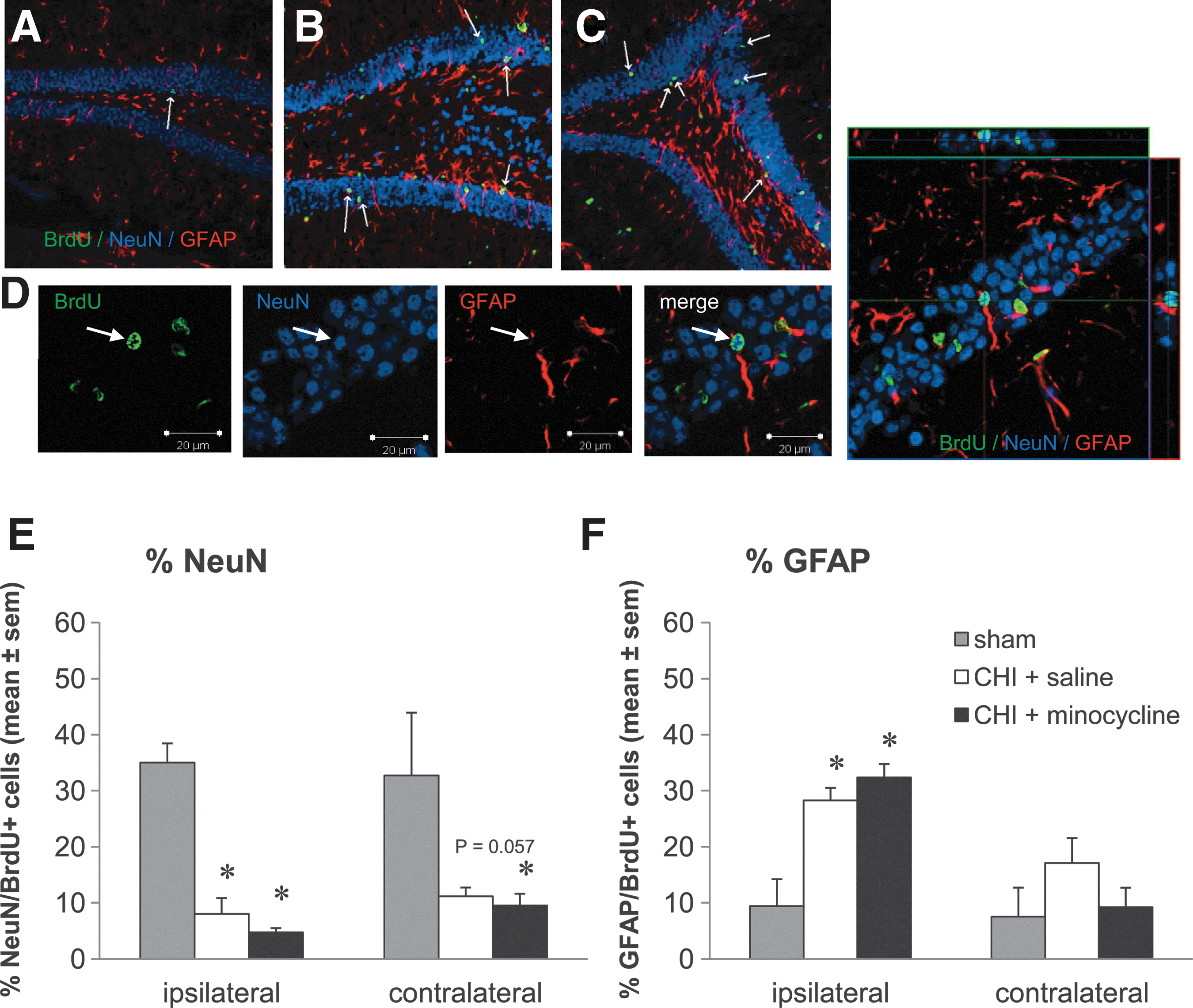
Minocycline does not affect the production of new mature neurons or astrocytes in the dentate gyrus (DG) after trauma. (
In sham-operated mice, approximately 35% of new BrdU-positive cells surviving in the DG at 6 weeks were mature NeuN-positive neurons (Fig. 5E). Interestingly, following trauma, this percentage dramatically decreased, with only approximately 8% of new cells expressing NeuN in the ipsilateral DG, and 11% in the contralateral side, with no detectable differences between the vehicle- and minocycline-treated groups (ipsilateral DG: p<0.05 sham versus vehicle and minocycline; one-way ANOVA, p<0.001; contralateral DG: p<0.05 sham versus minocycline, p=0.057 sham versus vehicle; one-way ANOVA, p=0.029), and no differences in any group between the ipsilateral and contralateral sides (two-way ANOVA for effect of side, p=0.623).
In order to determine whether the decreased percentage of new cells becoming neurons could be due to an increase in new astrocytes following trauma, we next assessed the percentage of BrdU-positive cells that co-labeled with GFAP (Fig. 5F). As expected, although in sham-operated mice fewer than 10% of new cells in the DG were astrocytes, in the DG ipsilateral to injury the proportion of new GFAP-positive cells was markedly increased, to approximately 30% after trauma, with no effect of minocycline treatment (p<0.05 sham versus vehicle and minocycline; one-way ANOVA, p<0.001). Conversely, in the contralateral DG, although the percentage of GFAP-positive/BrdU-positive cells was increased in the vehicle-CHI mice by twofold compared to shams, there was no significant effect of trauma or treatment with minocycline (one-way ANOVA, p=0.275). Furthermore, the levels of GFAP-positive/BrdU-positive cells were significantly higher in the ipsilateral DG of the CHI groups compared to the contralateral side (p<0.05; two-way ANOVA for the effect of side and of the interaction between treatment and side, p<0.001 and p=0.032, respectively).
Neurogenesis in the SVZ and pericontusional cortex is not affected by treatment with minocycline following CHI
Cell proliferation in the subventricular zone
We next assessed whether minocycline-treatment of CHI mice affected the neurogenic response of progenitors in the SVZ of the lateral ventricles. To assess proliferation in the SVZ, BrdU-positive cells were counted at 1 week post-CHI in the SVZ ipsilateral and contralateral to the site of injury. BrdU-positive cells were also counted at 6 weeks to determine whether CHI and/or minocycline influenced the population of newly proliferated cells remaining in the SVZ, from which the majority of cells normally emigrate soon after their generation.
At 1 week, BrdU-positive cells were lining the ipsilateral and contralateral SVZ in sham-operated mice, and a similar increase was observed in both trauma groups within the SVZ ipsilateral to the site of injury, but not on the contralateral side. By 6 weeks, very few BrdU-positive cells were detected in the SVZ of sham or CHI mice. As shown in Figure 6, these observations were supported by cell count data. The number of BrdU-positive cells was significantly increased, by 40%, in the ipsilateral SVZ at 1 week post-injury to a similar extent in both the vehicle- and minocycline-treated CHI mice compared to sham controls (p<0.05 sham versus vehicle and minocycline; one-way ANOVA, p=0.011; Fig. 6A). Conversely, there was no effect of CHI or minocycline treatment on the number of proliferating cells in the contralateral SVZ at 1 week (one-way ANOVA, p=0.742). Furthermore, there were significantly fewer BrdU-positive cells in the contralateral SVZ of the vehicle- and minocycline-treated CHI groups compared to the ipsilateral side (p<0.05 ipsilateral versus contralateral SVZ; two-way ANOVA for the effect of side and the effect of interaction between side and treatment, respectively, p=0.018 and 0.029).
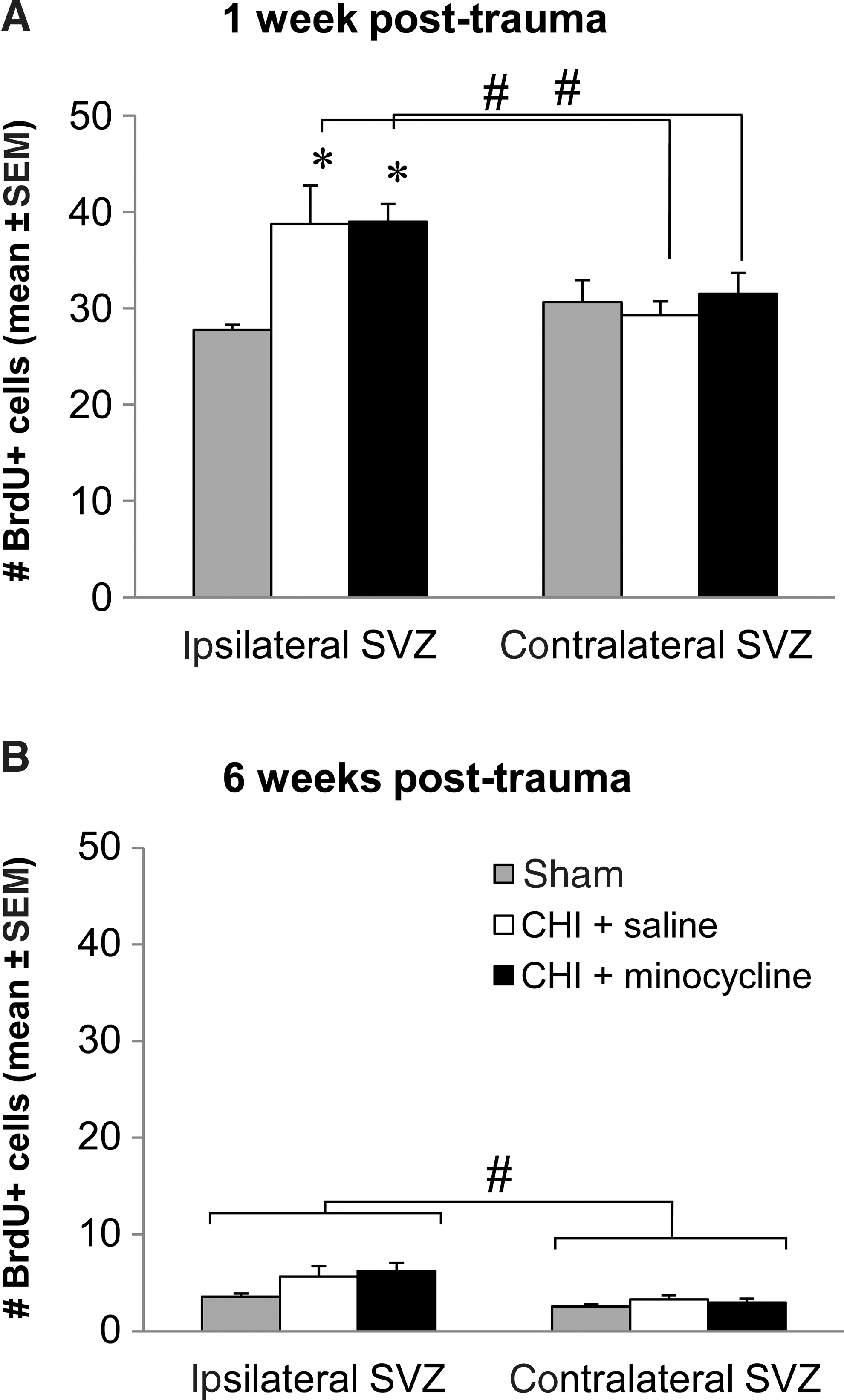
Minocycline does not affect cell proliferation or survival of new cells in the subventricular zone (SVZ) after trauma. (
By 6 weeks post-surgery (Fig. 6B), there was a dramatic reduction in BrdU-positive cells remaining in the SVZ compared to the number of newly proliferated cells present at 1 week, for all treatment groups in both the ipsilateral and contralateral SVZ (p<0.05 1 week versus 6 weeks; two-way ANOVA for the effect of time and the effect of the interaction between time and treatment, respectively, ipsilateral SVZ: p<0.001 and 0.690, contralateral SVZ: p<0.001 and 0.397). At 6 weeks, the number of BrdU-positive cells was similar between shams, vehicle-treated, and minocycline-treated CHI mice within each hemisphere (one-way ANOVA, ipsilateral SVZ: p=0.107, contralateral SVZ: p=0.355; Fig. 6B), but overall, there were significantly more cells in the ipsilateral SVZ compared to the contralateral side, regardless of treatment (two-way ANOVA for the effect of side and the effect of the interaction between side and treatment, respectively, p<0.001 and 0.275). Together these data suggest that early proliferation stimulated by CHI may contribute to a slight increase in the pool of resident stem/progenitor cells that remain in the ipsilateral SVZ, and that this effect is not impacted by treatment with minocycline.
Minocycline did not affect neuronal differentiation or migration of immature neurons
To investigate the potential effect of minocycline on the migration of immature neurons from the SVZ to the site of injury, Dcx-positive cells were quantified in the lateral corner of the SVZ, and extending into the corpus callosum and pericontusional cortex, of the injured brain hemisphere from vehicle- and minocycline-treated mice at 1 and 6 weeks post-CHI. While in sham-operated mice, Dcx-positive cells were restricted to the SVZ, at both 1 and 6 weeks after CHI, Dcx-positive neuroblasts were observed distributed throughout the tissue adjacent to the SVZ, along the overlying corpus callosum, and within the pericontusional cortex (Fig. 7A), with a greater abundance of cells detected at 1 week. Most ectopic Dcx-positive cells were grouped in clusters near the SVZ (Fig. 7A-ii), whereas chains of a few cells and more isolated Dcx-positive cells were observed within the corpus callosum and cortex (Fig. 7A-i). In the minocycline-treated CHI group, cell count data showed that there were approximately 20% more neuroblasts migrating to the lesion at 1 week post-CHI, and a 47% increase at 6 weeks, compared to the respective vehicle-treated groups. However, these differences were not statistically significant at either time point (t-test, 1 week: p=0.434 and 6 weeks: p=0.124; Fig. 7B). At 6 weeks post-CHI there was a significant reduction, by 60%, in the number of Dcx-positive neuroblasts compared to 1 week (p<0.05; two-way ANOVA for the effect of time and the effect of the interaction between time and treatment, respectively, p=<0.001 and 0.933), suggesting that CHI has a transient effect on recruiting neuroblasts to the injured cortex.
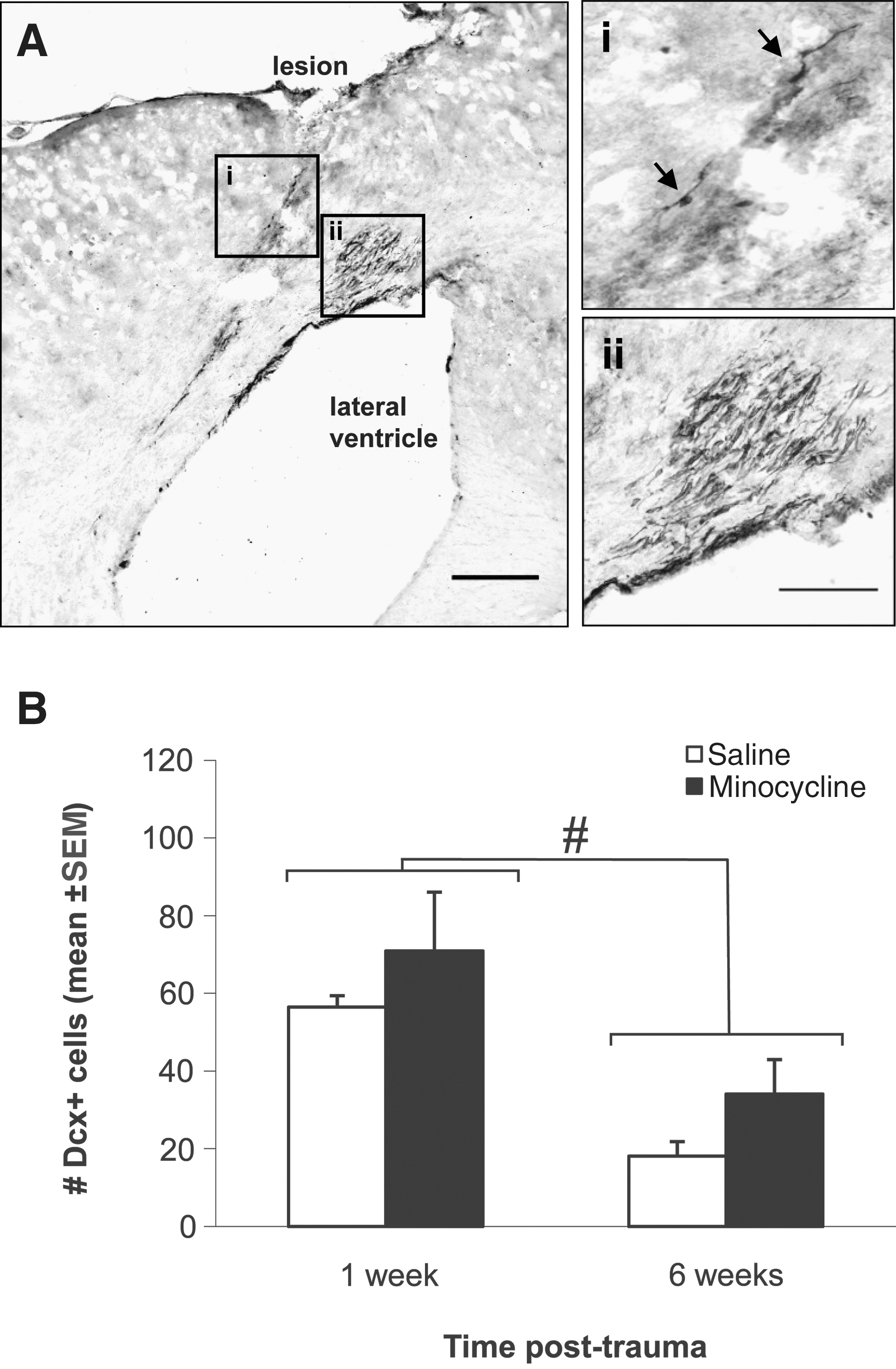
Minocycline does not affect the migration of neuroblasts into the pericontusional cortex. (
Minocycline did not influence cell fate/survival in the pericontusional cortex
Finally, we assessed whether minocycline treatment would affect the survival of new mature neurons and astrocytes in the pericontusional cortex at 6 weeks post-CHI. As shown in a representative image of the lesioned cortex at 6 weeks post-CHI in Figure 8A, numerous BrdU-positive cells remained in the core of the lesion and within the pericontusional region, and many were co-labeled with GFAP (Fig. 8B). When the percentage of BrdU-positive cells that co-labeled with either NeuN or GFAP was quantified, we found that fewer than 2% of new cells were mature neurons (Fig. 8C), while 44% were astrocytes (Fig. 8D), likely arising from post-traumatic gliosis that is known to occur around the lesion. Neither of these cell populations was affected by treatment with minocycline (t-test, p=0.576 and 0.846 for percent NeuN and percent GFAP, respectively).
Minocycline does not affect the production of new neurons or astrocytes in the pericontusional cortex after trauma. (
Discussion
In this study, we demonstrated that treatment of mice subjected to CHI with the anti-inflammatory drug minocycline significantly reduced the accumulation and activation of microglia/macrophages in the hippocampus and pericontusional cortex, and that this was associated with an early and sustained improvement in neurological outcome. However, minocycline treatment did not alter the extent of injury-induced neurogenesis in either brain region at any of the regulatory stages assessed, including precursor proliferation, neuronal differentiation, neuroblast migration, or survival of new neurons.
Minocycline-treatment reduces the activation of microglia/macrophages and improves neurological outcome
Neuroinflammation following TBI is characterized by the activation of resident astrocytes and microglia, and the infiltration of leukocytes into the injury site, which together release a multitude of pro- and anti-inflammatory cytokines and chemokines and neurotoxic molecules such as reactive oxygen species, which collectively contribute to progressive cell death and functional impairment (Morganti-Kossmann et al., 2007; Schmidt et al., 2004). Our present results show that minocycline treatment substantially decreased the accumulation of activated microglia and peripheral macrophages in the injured brain hemisphere of CHI mice, and that this anti-inflammatory action was associated with an early and sustained improvement in neurological outcome as assessed by motor and behavioral tasks. This finding supports the growing body of literature demonstrating that minocycline is beneficial to outcome following many types of experimental CNS injury, including intracerebral hemorrhage (Wasserman and Schlichter, 2007; Wu et al., 2009), ischemic injury (Arvin et al., 2002; Carty et al., 2008; Morimoto et al., 2005; Wang et al., 2003; Xu et al., 2004; Yrjanheikki et al., 1998), excitotoxicity (Tikka et al., 2001), spinal cord injury (Stirling et al., 2004; Teng et al., 2004; Wells et al., 2003), and TBI (Bye et al., 2007; Homsi et al., 2010; Sanchez Mejia et al., 2001), and that this effect may be largely mediated by its anti-inflammatory actions specifically targeting microglia (Bye et al., 2007; Das et al., 2011; He et al., 2001; Homsi et al., 2010; Krady et al., 2005; Wu et al., 2002; Yrjanheikki et al., 1998).
Minocycline-treatment does not increase neurogenesis after CHI
Similarly to other models of TBI (reviewed in Richardson et al., 2007), we have shown for the first time that CHI results in a robust 3.6-fold increase in cell proliferation in the hippocampal DG ipsilateral to the site of injury within the first week post-trauma, and a smaller 2.5-fold increase in the contralateral side. We have also established that approximately 70% of the new cells survive for at least 6 weeks following trauma; however, only a small proportion of these cells mature into a neuronal phenotype, which is consistent with our data showing no increased production of immature Dcx-positive neurons in the DG. Conversely, trauma increased the proportion of new cells expressing GFAP, which could indicate an increase in the population of hippocampal progenitor cells after injury, shown earlier to express GFAP (Liu et al., 2010). However, the increase in new GFAP-positive cells is likely to reflect increased proliferation of adult astrocytes after trauma, since abundant astrogliosis occurs in the DG following this model of injury (Semple et al., 2010). Likewise, cell proliferation in the SVZ was also stimulated by 40% within the first week following CHI, and differentially from the hippocampus, only in the hemisphere ipsilateral to injury and not in the contralateral region. One of the most striking effects we observed was the abundance of Dcx-positive migratory neuroblasts likely being recruited from the ipsilateral SVZ towards the site of injury, peaking at 1 week, but persisting at 6 weeks. However, at this time almost no new neurons were detected in the pericontusional cortex, with a large proportion of the new cells becoming mature astrocytes. This abortive attempt at neurogenesis in the pericontusional cortex has also been observed by others after experimental focal TBI, where despite the accumulation of SVZ neuroblasts in the cortex, few if any new neurons were ultimately detected in this region (Ramaswamy et al., 2005; Salman et al., 2004; Thau-Zuchman et al., 2010).
In light of these observations, we hypothesized that injury-induced neurogenesis could be supported by augmenting the survival of the new immature neurons, both in the cortex and the hippocampus, and proposed that this could be achieved by attenuating the accumulation and activation of microglia and macrophages. This reasoning was based on earlier studies showing that activated microglia negatively impact on neurogenesis, predominantly via inhibiting the survival of new neurons (Ekdahl et al., 2003, 2009; Monje et al., 2003). However, our results show that twice-daily intraperitoneal injections of minocycline for up to 2 weeks after trauma did not enhance the ultimate production of new neurons in the hippocampal DG or pericontusional cortex, despite its success in decreasing microglial activation in these regions.
While the extensive accumulation of activated microglia and macrophages was significantly reduced by treatment with minocycline in the pericontusional cortex and hippocampus by 40% and 50%, respectively, it is possible that this degree of attenuation was not sufficient to alleviate the inhibitory effect of microglia on new neuron survival. For example, in our earlier study using the same injury model, we found that that even though minocycline treatment reduced the levels of the cytokines IL-1ß and IL-6 in the injured cortex by 50% and 35%, respectively, their respective levels remained 4- and 11-fold higher than those of sham-operated controls (Bye et al., 2007). These are known anti-neurogenic cytokines (Koo et al., 2008; Monje et al., 2003; Vallieres et al., 2002), and therefore this finding demonstrates that even a substantial reduction in the number of activated microglia may not alleviate the restrictive inflammatory environment for neurogenesis.
However, our current findings are in contrast to an earlier study by Liu and associates (2007), in which chronic delivery of minocycline reduced the number of activated microglia in the hippocampus by approximately 40%, and concomitantly doubled the number of new mature BrdU-positive/NeuN-positive neurons in the DG at 4 weeks after middle cerebral artery occlusion. Similarly, in a model of epilepsy, when minocycline was administered over 5 weeks the number of newly-formed hippocampal neurons increased greater than twofold, while the microglial population decreased by half (Ekdahl et al., 2003). Kluska and colleagues (Kluska et al., 2005) also showed that treatment with the anti-inflammatory drug indomethacin for 2 weeks following focal ischemia resulted in a significant doubling of the number of newborn neurons in the ipsilateral DG; however, changes in inflammatory cells were not assessed. Indomethacin treatment also enhanced BrdU-positive cell density in the infarct penumbra regions in the cortex and striatum in adult rats subjected to focal cerebral ischemia from 1 to 4 weeks post-injury, and enhanced the percentage of BrdU-positive cells expressing markers of neuronal progenitors and immature glia and neuroblasts (Hoehn et al., 2005). However, despite the decrease of activated microglia by 50%, indomethacin treatment did not support the ultimate production of new BrdU-positive/NeuN-positive mature neurons (Hoehn et al., 2005), thus corroborating our results on the CHI model.
The diversity of outcomes in the above-mentioned studies is likely because a variety of injury models and experimental paradigms were employed, each one of them resulting in distinctive pathologies with varying degrees of damage, microglial activity, and injury-induced neurogenesis in the untreated groups. For example, in the study by Kluska and colleagues (2005), the size of the cortical infarct was much smaller than the contusion injury generated with our CHI model (Semple et al., 2010), and the number of new hippocampal cells detected at 4 weeks post-ischemia was only increased by approximately 50% compared to sham-operated animals, whereas we saw a much stronger 10-fold increase in new cells at 6 weeks post-trauma. Interestingly, this group found no difference in the number of BrdU-positive cells at 2 weeks post-injury relative to shams, whereas we detected a greater than threefold increase in proliferation at 1 week after CHI. These data suggest that the neurogenic response to injury is different between the two models, and it is likely that the inflammatory environment in the hippocampus is also quite distinct. Likewise, Liu and associates (2007), using a middle cerebral artery occlusion model, demonstrated that the number of surviving new cells in the hippocampus at 4 weeks was not increased by ischemia alone, and interestingly, despite the large infarct generated by this model, the authors reported only a small number of activated microglia within the hippocampus. Altogether, these studies by us and others demonstrate that the effectiveness of reduced microglial activation on supporting the production of new neurons in the injured brain is likely to be dependent upon the extent and location of tissue damage and the resulting degree of microglial activity, as well as on the profile of injury-induced neurogenesis that is elicited. This highlights the need for further studies to more fully understand the relationship between neurogenesis and neuroinflammation in different brain pathologies.
Indeed, the role of microglia in mediating injury-induced neurogenesis is complex, with emerging evidence demonstrating both supportive and detrimental actions. It appears that microglia can influence multiple stages of neurogenesis, and these effects may be dependent upon the microglial activation state, with acutely activated microglia being detrimental, and more mildly activated or T-cell-informed microglia being supportive (Das and Basu, 2008; Ekdahl et al., 2009). For example, the proinflammatory cytokines expressed by acutely-activated microglia following LPS stimulation or during status epilepticus, including IL-1ß, IL-6, IL-18, interferon-γ, and TNF-α, have been shown to specifically inhibit precursor proliferation, neuronal differentiation, and survival of new neurons (Ekdahl et al., 2003; Iosif et al., 2006; Liu et al., 2005; Monje et al., 2003), whereas microglia activated via an adaptive immunity response to the cytokine interferon-γ can instruct neuronal differentiation and survival (Butovsky et al., 2006). Furthermore, activated microglia in the injured brain also produce multiple chemokines, including SDF-1α and MCP-1, which may be necessary to recruit neuroblasts to the damaged region (Imitola et al., 2004; Yan et al., 2007), and also growth factors, such as brain-derived growth factor and epidermal growth factor, which in turn could promote precursor proliferation, neuronal differentiation, and survival (Craig et al., 1996; Pencea et al., 2001). Finally, it is interesting to note that progenitor proliferation is most strongly stimulated within the first week after CHI (N. Bye, unpublished data), which coincides with the peak period of microglial activity and cytokine production in this model (Bye et al., 2007; Semple et al., 2010; Ziebell et al., 2011). However, it is interesting to note that in our current study none of the stages of neurogenesis we investigated were affected following minocycline treatment, including proliferation in the DG and SVZ, or neuronal differentiation, migration, and survival, so it would appear that a partial reduction of microglial activation does not support or suppress neurogenesis in the present experimental paradigm. Consequently, future work should aim to distinguish sub-populations of activated microglia and infiltrated macrophages that are involved in mediating injury-induced neurogenesis, perhaps by examining cellular phenotypes and cytokine-expression profiles, in order to determine how and when these cells may be targeted therapeutically to best enhance regeneration in the injured brain.
In conclusion, we have characterized the neurogenic response for the first time in this CHI model of focal TBI, and have demonstrated that a robust increase in hippocampal and SVZ cell proliferation was followed by migration of neuroblasts to the pericontusional cortex, but ultimately, this did not result in increased production of new neurons. Secondly, our data support earlier work by showing that anti-inflammatory treatment with minocycline significantly reduced microglial activation, which was associated with an early neurological recovery that was maintained for over 6 weeks. However, these beneficial effects did not coincide with enhanced neurogenesis in either the hippocampus or pericontusional cortex, with none of the regulatory stages being affected by minocycline treatment, including proliferation in the DG and SVZ, neuronal differentiation, neuroblast migration, or survival of new mature neurons. These data suggest that further work is required to elucidate the relationship between microglial activation and neurogenesis after brain injury, which in turn could potentially guide the development of therapeutic strategies aimed at promoting endogenous repair following TBI.
Footnotes
Acknowledgments
This research was supported by the Victorian Neurotrauma Initiative (VNI), Transport Accident Commission (TAC), and Alfred Research Trust, through research grants and fellowships to C.M.K. and N.B., and by the Australian Government through an Australian Postgraduate Award to B.D.S. The authors would like to thank Dr. Iśka Carmichael of Monash Micro Imaging (AMREP), for training and assistance with confocal microscopy.
Author Disclosure Statement
No competing financial interests exist.