
Abstract
Pituitary adenylate cyclase-activating polypeptide (PACAP) is effective in reducing axonal damage associated with traumatic brain injury (TBI), and has immunomodulatory properties. Toll-like receptor 4 (TLR4) is an important mediator of the innate immune response. It significantly contributes to neuroinflammation induced by brain injury. However, it remains unknown whether exogenous PACAP can modulate TBI through the TLR4/adapter protein myeloid differentiation factor 88 (MyD88)/nuclear factor-κB (NF-κB) signaling pathway. In this study, we investigated the potential neuroprotective mechanisms of PACAP pretreatment in a weight-drop model of TBI. PACAP38 was microinjected intracerebroventricularly before TBI. Brain samples were extracted from the pericontusional area in the cortex and hippocampus. We found that TBI induced significant upregulation of TLR4, with peak expression occurring 24 h post-trauma, and that pretreatment with PACAP significantly improved motor and cognitive dysfunction, attenuated neuronal apoptosis, and decreased brain edema. Pretreatment with PACAP inhibited upregulation of TLR4 and its downstream signaling molecules MyD88, p-IκB, and NF-κB, and suppressed increases in the levels of the downstream inflammatory agents interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), in the brain tissue around the injured cortex and in the hippocampus. Administration of PACAP both in vitro and in vivo attenuated the ability of the TLR4 agonist lipopolysaccharide (LPS) to increase TLR4 protein levels. Therefore, PACAP exerts a neuroprotective effect in this rat model of TBI, by inhibiting a secondary inflammatory response mediated by the TLR4/MyD88/NF-κB signaling pathway in microglia and neurons, thereby reducing neuronal death and improving the outcome following TBI.
Introduction
T
The pituitary adenylate cyclase-activating polypeptide (PACAP), which was initially isolated from the ovine hypothalamus, belongs to the secretin/glucagon/vasoactive intestinal peptide (VIP) superfamily (Miyata et al., 1989; Watanabe et al., 2007), and has two amidated forms, PACAP27 and PACAP38 (Vaudry et al., 2000). Based on its ability to activate cAMP in the pituitary gland, it acts as a neurotransmitter, neuromodulator, neurotrophic, or neuroprotective factor, via multiple signaling pathways (Das et al., 2007; Vaudry et al., 2009). PACAP has a protective role in several nervous system diseases/conditions, such as focal cerebral ischemia (Allais et al., 2010; Nakamachi et al., 2010; Stetler et al., 2010), Alzheimer's disease (Kojro et al., 2006), schizophrenia (Matsuzaki and Tohyama, 2008; Tanaka et al., 2006), anxiety disorders (Choi et al., 2008; Norrholm et al., 2005), Parkinson's disease (Reglodi et al., 2004,2006), and TBI (Johanson et al., 2011; Kovesdi et al., 2008; van Landeghem et al., 2007). Several reports have demonstrated that PACAP can attenuate axonal injury induced by trauma (Kovesdi et al., 2008; Tamas et al., 2006), and promote neural restoration through enhanced neurogenesis, angiogenesis, and neuroprotective effects (Johanson et al., 2011). Furthermore, PACAP has immunomodulatory properties and can inhibit production of inflammatory agents, such as TNF-α, from microglia activated by lipopolysaccharide (LPS) in vitro (Fang et al., 2010; Kim et al., 2010). However, whether PACAP can alleviate brain injury by modulating the secondary inflammatory response to TBI, as well as the precise mechanisms involved, are still under investigation.
The innate immune system recognizes different pathogens via highly conserved microbial motifs, namely pathogen-associated molecular patterns (PAMPs), through pathogen-recognition receptors (PRRs; Takeuchi and Akira, 2010; Mogensen, 2009). Toll-like receptors (TLRs) are a family of PRRs that recognize conserved microbial motifs in molecules such as bacterial LPS, peptidoglycan, flagellin, and double- and single-stranded RNA (Akira et al., 2006; Miyake, 2007). Microglia are macrophage-like glial cells that reside within the central nervous system. Injury or infection activates ramified microglia, which function as effectors of the inflammatory response. Rat microglia can express TLR1–9 (Arumugam et al., 2009). Among them, TLR4 is widely expressed in the brain and can detect endogenous agonists, such as the degradation products of macromolecules, heat shock proteins 60 and 70, and intracellular components of ruptured cells (Johnson et al., 2003). TLR4 has been shown to participate in the cerebral inflammatory response related to ischemia reperfusion injury (Arslan et al., 2010; Brea et al., 2010), TBI (Chen et al., 2009), Alzheimer's disease (Ding et al., 2011; Vollmar et al., 2010), and Parkinson's disease (Panaro et al., 2008; Waak et al., 2009). The TLR4 signaling pathway is mediated by interaction with the adapter protein myeloid differentiation factor 88 (MyD88), which leads to activation of nuclear factor-kappa B (NF-κB), and subsequent production of proinflammatory cytokines (Pulskens et al., 2008; Stetler et al., 2010). Recent studies have reported that TLR4-mediated NF-κB signaling contributes to cerebral ischemia reperfusion injury (Hyakkoku et al., 2010).
There are a few reports describing the ability of PACAP to regulate TLR responses. For instance, PACAP38 inhibits TLR4/MyD88/tumor necrosis factor receptor-associated factor adaptor protein (TRAF6) expression in mouse kidney and proximal tubule epithelial cells, and protected mice from ischemia- and hypoxia-induced acute renal tubule damage (Li et al., 2010). Furthermore, vasointestinal peptide (VIP) participates in the regulation of TLRs by inhibiting the expression of the leukocyte differentiation antigen CD14, and reducing the inflammatory response induced by LPS (Gomariz et al., 2007). In mammalian tissue, PACAP shares 68% identity with VIP, and PACAP38 constitutes about 90% of the endogenous PACAP pool. Therefore, we hypothesized that exogenous PACAP38 may inhibit the TLR4/MyD88/NF-κB signaling pathway in the brain and thus decrease inflammatory mediators, attenuate the secondary inflammatory response, and promote motor and cognitive recovery following TBI.
Methods
Animals
Adult male Sprague-Dawley rats (220–250 g) were provided by the Animal Center of Xuzhou Medical College. All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animals were housed in temperature- and humidity-controlled animal quarters with a 12-h light/dark cycle and water and food provided ad libitum.
Reagents
PACAP38 peptide (A1439) and LPS (L4391) were obtained from Sigma-Aldrich (St. Louis, MO). BV2 microglial cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). Goat polyclonal anti-TLR4 (sc-16240), rabbit polyclonal anti-MyD88 (sc-11356), mouse monoclonal anti-NF-κB p65 (sc-8008), rabbit polyclonal anti-IκB-α (sc-371), mouse monoclonal anti-p-IκB-α (sc-8404), rabbit polyclonal anti-Bcl-2 (sc-492), rabbit polyclonal anti-Bax (sc-526), and mouse monoclonal anti-Iba-1 antibody (SC-32725), were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-neuronal nuclei Alexa Fluor-conjugated monoclonal antibody (MAB377X), and FITC-conjugated anti-mouse antibody (AP160F) were obtained from Millipore (Billerica, MA). Glial fibrillary acidic protein (GFAP) mouse monoclonal antibody (#3655) was obtained from Cell Signaling (Boston, MA). Mouse anti-β-actin monoclonal antibody (TA-09), alkaline phosphatase goat anti-rabbit IgG (ZB-2308), alkaline phosphatase horse anti-mouse IgG (ZB-2310), alkaline phosphatase rabbit anti-goat IgG (ZB-2311), and rhodamine (TRITC)-conjugated affinipure rabbit anti-goat IgG (H+L; ZF-0317) were purchased from ZSGB-Bio (Beijing, China). PageRuler™ Plus Prestained Protein Ladder (SM1811) was purchased from Fermentas (Glen Burnie, MD). Primary antibody dilution buffer (P0023A), secondary antibody dilution buffer (P0023D), phenylmethanesulfonyl fluoride (ST506), BCA protein assay kit (P0012), SDS-PAGE sample loading buffer (P0015), and BCIP/NBT alkaline phosphatase color development kit (C3206) were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). TNF-α and IL-1β ELISA kits were purchased from R&D Systems (Minneapolis, MN).
Intracerebroventricular treatment with PACAP38 or LPS
Rats were anesthetized with chloral hydrate (400 mg/kg) and mounted onto a stereotaxic apparatus. The location of the right lateral cerebral ventricle was obtained according to The Rat Brain in Stereotaxic Coordinates (Paxinos and Watson, 1986); that is: AP 1.0 mm, R 1.5 mm, H 4.5 mm. The incisor bar was positioned 3.3 mm below the center of the aural bars. PACAP38 (1 μg in a volume of 5 μL saline for the lateral cerebral ventricle; Gressens et al., 2000; Reglodi et al., 2004), or LPS (0.5 μg in 5 μL saline; Xia et al., 2006) were injected via a cannula connected to a microsyringe with a polyethylene tube (Reglodi et al., 2002; Somogyvari-Vigh and Reglodi, 2004). The injection lasted for 2 min, and the injection cannula was left in place for an additional 10 min to prevent backflow. PACAP was injected into the lateral cerebral ventricle before TBI or 24 h following injection of LPS. In the sham control group, animals received the same surgical procedures, including a burr hole for microinjection into the lateral ventricle with vehicle (saline), followed by the craniotomy (see below), but without the cortical impact.
Experimental traumatic brain injury model
Immediately after bolus injections of PACAP (after the last bolus injection), the rats underwent TBI using a Feeney weight-drop contusion model with slight modifications (Feeney et al., 1981; Yu et al., 2010). Rats underwent craniotomy in a circular region of skull over the right parietal cortex between the lambda and the bregma (4.0 mm in diameter, centered 4 mm caudal and 2.5 mm lateral to the bregma), leaving the dura intact. A weight-drop device was placed over the dura. An impact transducer (foot plate) was adjusted to stop at a depth of 2.5 mm below the dura. Then a 20-g weight was dropped from 30 cm above the dura through a guide tube onto the foot plate. This model is generally associated with 12.5% mortality within the first 12 h after injury, and no delayed mortality was observed thereafter. Body temperature was maintained with an overhead heating lamp during the experiments. Dural tears were not repaired and the bone flap was not re-inserted. If the animals demonstrated dural tears or excessive bleeding, they were excluded. After injury, the skin was closed tightly. To maintain normal body temperature during surgery and recovery, the rats were maintained with isothermic (37°C) heating. Rats in the sham-operated group were subjected to the same surgical procedure, including craniotomy, but received no cortical impact. The surrounding brain tissue of the injured cortex demonstrated obvious hyperemia and edema, and was depressed (Fig. 1).
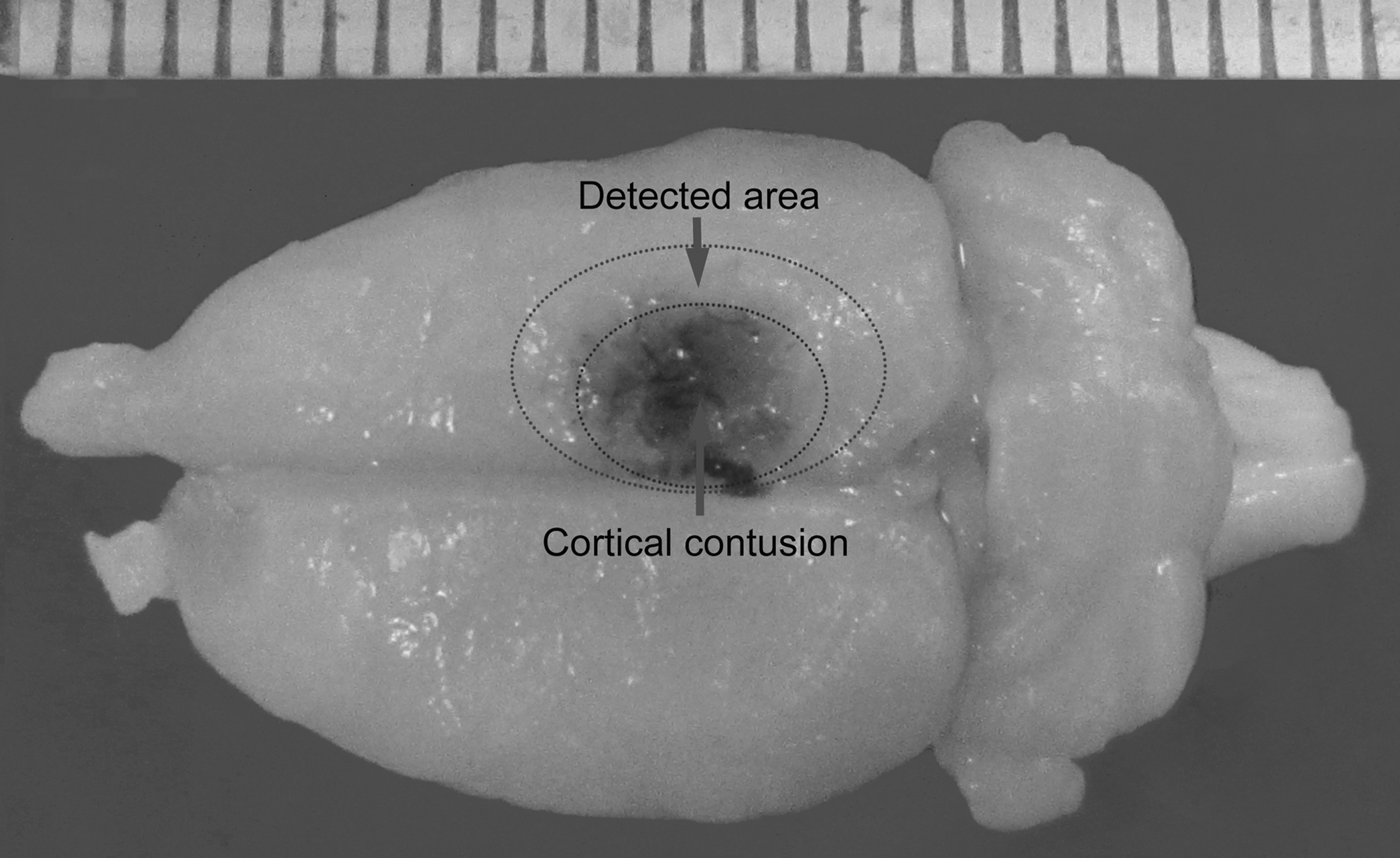
Image of a representative brain showing the cortical contusion area produced by weight-drop trauma in macroscopic appearance, and the area surrounding the injured brain cortex.
Behavioral analysis
Neurobehavioral, motor, and cognitive functional assessments in the normal (n=8), sham (n=8), vehicle+TBI (n=8), and PACAP+TBI groups (n=8) were performed 1, 3, 7, 10, 14, and 21 days following TBI, by a blinded observer unaware of the experimental treatments. The battery of tests included the mNSS (modified Neurological Severity Score) system, the inclined plane task, and the Morris water maze (MWM).
Modified Neurological Severity Score
The mNSS assesses functional neurological status based on the presence of some reflexes, and the ability to perform motor and behavioral tasks such as beam walking, beam balance, and spontaneous locomotion (Chen et al., 2001; Tsenter et al., 2008). Neurological function was graded on a scale from 0–10. One point was scored for the inability to perform the test, increasing with the severity of dysfunction. The rat training consisted of 3 trials per day.
The inclined plane task
The inclined plane task evaluates the rats' ability to maintain a horizontal body position on an inclined board, followed by assessment for injured-side hindlimb foot faults. The rats were placed at the top of a 50-cm-long, 30-cm-wide wooden board set at 10° above horizontal. The animals were placed in a right side (the injured side) position on the inclined plane task. The angle of the board was incrementally raised until the animal was no longer able to maintain the horizontal position. The maximum angle at which the animal was able to maintain its position on the board for 10 sec was recorded 3 times per day (Beaumont et al., 1999).
Morris water maze acquisition
The cognitive test was performed in a Morris water maze (MWM). A black pool (1.8 m diameter) was subdivided into four equal quadrants formed by imaging lines. At the beginning, the rats were randomly placed at one of four fixed starting points (north, east, south, and west), and then trained in the other three fixed starting points per day, starting 1, 3, 7, 10, 14, and 21 days following TBI. Each trial lasted until the rat found the platform, or for a maximum of 2 min. If the rat failed to find the platform in the allocated time, it was gently placed on the platform for 20 sec. At the end of each trial, the rats remained on the platform for 20 sec to make associations. The escape latencies to find the platform were recorded (Aiguo et al., 2010; Marklund et al., 2009; Molteni et al., 2004; Thompson et al., 2006).
Brain water content
Twenty-four hours post-injury, brain edema was determined using the wet/dry method: percent brain water=[(wet weight–dry weight)/wet weight]×100% (Roof et al., 1996). The brains from mice in each treatment group (n=6) were rapidly removed from the skull, and the brains were separated bilaterally, weighed, and then placed in an oven for 72 h at 100°C. The brains were then reweighed to obtain dry weight content.
H&E staining and immunostaining
At 24 h and 21 days post-injury, the animals were deeply anesthetized and perfused via the right ventricle with 0.9% saline, followed by 4% paraformaldehyde. The entire brain was removed and fixed in 4% paraformaldehyde for 48 h at 4°C. A series of groups of rats were used for hematoxylin and eosin (H&E) staining in order to observe the brain injury. After the brains were fixed, the forebrains were paraffin-embedded and sectioned on a microtome. Sections (5-μm thick) were cut every 500 μm throughout the injured cortex and deparaffinized, serially hydrated, stained with H&E, and then cover-slipped for tissue analysis. Five sections were selected for each animal, and five nonoverlapping adjacent fields of each section were selected that surrounded the edge of the cortical lesion.
Another group of rats was used for double immunofluorescence labeling in order to identify the cell types of TLR4 localization. After the brains were fixed, they were transferred to 30% sucrose diluted in 0.01 M phosphate-buffered saline (PBS) at 4°C for 2 days. The blocks were then rapidly frozen with dry ice and sectioned with a cryostat. A series of 30-μm-thick sections of the brain were collected in six vials to obtain sets of sections at regularly spaced intervals (150 μm) for each vial. The position of the cannula track protruding into the lateral ventricle was histologically verified for each brain. Animals with incorrectly placed injection sites were excluded from further data analysis. Selected sections of the brain were washed with PBS for 3×5 min, and then incubated in 1% BSA containing 0.3% Triton-X-100 in PBS for 2 h at room temperature. The sections were then incubated with goat anti-TLR4 antibody (1:50), and cell type-specific antibodies recognizing neuronal nuclear antigen (anti-NeuN for neurons; dilution 1:50), the astrocytic marker anti-GFAP (dilution 1:50), and the microglia marker mouse anti-ionized calcium binding adaptor molecule 1 (Iba-1; dilution 1:50) at 4°C for 24 h. TRITC-conjugated anti-goat antibody (1:50), and FITC-conjugated anti-mouse antibody (1:50) were added to the sections at 4°C overnight. Each step was followed by 3×5-min rinses in PBS. Tissue sections were mounted with 50% glycerol mounting medium. Negative control sections received identical preparation for immunohistochemical staining, except that the primary antibodies were omitted. Images were viewed by fluorescence microscopy. Immunostained sections were quantitatively characterized by digital image analysis using Image Pro-Plus 5.0 software (MediaCybernetics, Silver Spring, MD). TLR4 was quantified as the average number of positive cells per section. Six serial sections containing a cross-section of injured cortex, and the hippocampus with centralized TLR4 immuno-positive cells, were chosen from the same aspect of brain parenchyma in each rat. Each cross-section was analyzed at a 400×magnification under the microscope to count the number of TLR4-positive cells per section (Du et al., 2009).
Western blot analysis
The peripheral cortex surrounding the contusion site (Fig. 1) and ipsilateral hippocampus were collected on ice and then mechanically lysed in RIPA Lysis Buffer (P0013B; Beyotime Institute of Biotechnology) containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride (PMSF). The tissue was homogenized for 30 sec, and then kept on ice for 20 min. BV2 cells were similarly lysed in RIPA buffer after treatments. The homogenates were centrifuged at 12,000g for 30 min at 4°C, and the supernatants containing the cytoplasmic components were retained and stored at −80°C, and were thawed once. The nuclear pellets were again dissolved in RIPA Lysis Buffer and PMSF, and centrifuged at 12,000g for 20 min at 4°C. The supernatants were then collected. Nuclear extracts were aliquotted and stored at −80°C for Western blot analysis of p65 protein activity. Protein concentrations were determined by the bicinchoninic acid (BCA) protein assay. Equal amounts of protein were resolved on SDS-PAGE gels, and transferred electrophoretically to a nitrocellulose membrane. The membrane was blocked with 5% skim milk for 2 h at room temperature, incubated overnight at 4°C with primary antibodies directed against TLR4, MyD88, NF-κB p65, IκB-α, phosphorylated-IκB-α (p-IκB-α; all diluted 1:500 in primary antibody dilution buffer), and β-actin (loading control diluted 1:1000 in primary antibody dilution buffer). After the membrane was washed 3×5 min in wash buffer, it was incubated in the appropriate AP-conjugated secondary antibody (diluted 1:1000 in secondary antibody dilution buffer) for 2 h at room temperature. Protein bands were visualized by the BCIP/NBT Alkaline Phosphatase Color Development Kit (S3771). Developed films were digitized using an Epson Perfection 2480 scanner (Seiko Corp., Nagano, Japan). Optical densities of detected proteins were obtained using Glyko Bandscan software (Glyko, Novato, CA).
Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR)
Twenty-four hours following TBI, TLR4 mRNA levels in the cerebral cortex around the injury area (Fig. 1) and hippocampus were determined by qRT-PCR. Total cellular RNA was isolated from tissue samples using Trizol reagent (15596-026) per the manufacturer's instructions. RNA quantity was measured by spectrophotometric analysis (OD260/280). RNA was transcribed to cDNA using M-MLV Reverse Transcriptase (D263915) and dT primers. PCR amplification was performed with Taq polymerase for 30 cycles at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min. PCR primers for TLR4 were 5’′-CAGCAGGTCGAATTGTATCGCC-3′ (sense), and 5′-CCCTGTGAGGTCGTTGAGGTTAG-3′ (antisense), which were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). qRT-PCR was performed using the Rotor-Gene™ 3000 real-time DNA analysis system (Corbett Research, Sydney, Australia), applying real-time SYBR Green PCR technology. The reaction mixtures contained diluted cDNA, SYBR Premix Ex Taq II (2×) (DRR081), 10 μM of each gene-specific primer, and nuclease-free water, in a final volume of 10 μL. cDNA results were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) measured on the same plate. All samples were analyzed in triplicate.
Enzyme-linked immunosorbent assay (ELISA)
Levels of inflammatory mediators were quantified using specific ELISA kits for rats. Brain tissue in the cerebral cortex around the injured area (Fig. 1) and in the hippocampus were collected and homogenized in a buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, sodium orthovanadate, sodium fluoride, EDTA, and leupeptin. The homogenates were centrifuged at 4°C and 12,000g for 15 min. Supernatants were removed and assayed in duplicate using TNF-α and IL-1β assay kits, according to the manufacturer's guidelines. Tissue cytokine concentrations were expressed as nanograms per milliliter protein.
BV2 microglial cells culture
The cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. They were maintained at 37°C in a 5% CO2/air environment. The BV2 microglial cells were unstimulated (controls) or treated with LPS (100 ng/mL) for 24 h (Aravalli et al., 2008), to increase TLR4 expression. Alternatively, the cells were treated with PACAP (100 nM) to attenuate TLR4 expression.
Statistical analysis
Data are reported as mean±standard deviation (SD). SPSS version 13.0 was used for statistical analysis. Data for behavioral evaluation were analyzed with repeated-measures one-way analysis of variance (ANOVA). Other measurements were subjected to one-way ANOVAs. Differences between experimental groups were determined by the Fisher's Student Neuman Keuls (SNK) test. p Values<0.05 were considered statistically significant.
Results
Pretreatment with PACAP improved behavioral outcomes following TBI
Trauma induced neurologic, motor, and cognitive deficits in TBI rats, as determined by mNSS scores, the inclined plane task, and the MWM in each group. To identify the consistency of injury severity, we observed histologic changes of the injured brain after 24 h and 21 days post-injury in the vehicle+TBI group at low levels. The brain tissue around the injured cortex at 24 h post-injury showed edema, hemorrhage, and hyperemia, widened perivascular spaces, an increased gap between nerve cells, neuronal swelling, lightly-stained cytoplasm, and nuclear vacuolization. The molecular layer of the hippocampus demonstrated neuron swelling and agglutinated chromatin. The brain tissue around the injured cortex at 21 days post-injury also showed edema, widened perivascular spaces, increased gaps between nerve cells, and some neuronal swelling, lightly-stained cytoplasm, and nuclear vacuolization, but the hemorrhage and hyperemia were alleviated. The molecular layer of the hippocampus demonstrated neuron swelling and agglutinated chromatin (Fig. 2A). There were no differences between the normal and sham groups in the three tests. Injury in the right hemispheric cortex resulted in functional deficits in the evaluated rats using the mNSS scoring system. The vehicle+TBI group exhibited severe neurological deficits, and mNSS scores were significantly higher on days 1–21 (all p<0.05). However, the PACAP-pretreated rats exhibited a significant reduction in neurological deficits on days 1–21 post-trauma (all p<0.05; Fig. 2B).

Pretreatment with PACAP improved motor performance and neurological and cognitive recovery in rats following TBI. Histologic damage at 24 h and 21 days post-injury were assessed to evaluate injury consistency (
Motor deficits resulting from injury in the right cortical hemisphere were measured by the inclined plane task, which strongly depends on the integrity of sensorimotor cortical processing. As expected, the vehicle+TBI group demonstrated significant sensorimotor functional deficits. The critical angle at which rats did not fall was significantly decreased on days 1–21 (all p<0.05). PACAP-pretreated rats showed less severe sensorimotor deficits than the vehicle+TBI group 1–21 days post-trauma (all p<0.05). At the end of the test, the vehicle+TBI group still had motor function deficits. These data imply that pretreatment with PACAP improves sensorimotor functional deficits post-TBI in rats (Fig. 2C).
All rats, regardless of age and injury, were able to locate and climb onto the platform during the training period in the MWM test, indicating that none of the rats had significantly impaired visual or motor deficits. However, injury in the right hemispheric cortex resulted in cognitive deficits as measured by the MWM. The mean latency (in seconds) to reach the goal platform for each group is presented in Figure 2E. All injured rats spent most of their time swimming around the perimeter of the maze (Fig. 2D). They all demonstrated longer latency to find the hidden platform on days 1–21 (all p<0.05; Fig. 2D). However, PACAP-pretreated rats demonstrated improved target searching compared to the vehicle+TBI group, and had significantly greater reductions in cognitive deficits on days 1–21 post-trauma (all p<0.05). Moreover, at the end of the MWM test, there were still some cognitive deficits in the vehicle+TBI group, whereas the PACAP-pretreated rats had noticeably recovered, suggesting that PACAP-pretreated rats exhibited better performance and faster learning in the MWM (Fig. 2E).
Neuroprotective effects against TBI following PACAP pretreatment
To observe the effect of PACAP on histopathology, brain sections were stained with H&E, and assessed for morphological changes under a light microscope. Normal and sham-operated control rats showed normal-appearing neural structures. The brain tissue around the injured cortex of TBI rats showed edema, hemorrhage, and hyperemia, widened perivascular spaces, increased gaps between nerve cells, neuronal swelling, lightly-stained cytoplasm, and nuclear vacuolization. The molecular layer of the hippocampus demonstrated edema, neuron swelling, and agglutinated chromatin. Pretreatment with PACAP alleviated the histologic presentation of edema, hemorrhage, and neuronal necrosis, in both the injured cortex and hippocampus (Fig. 3A).
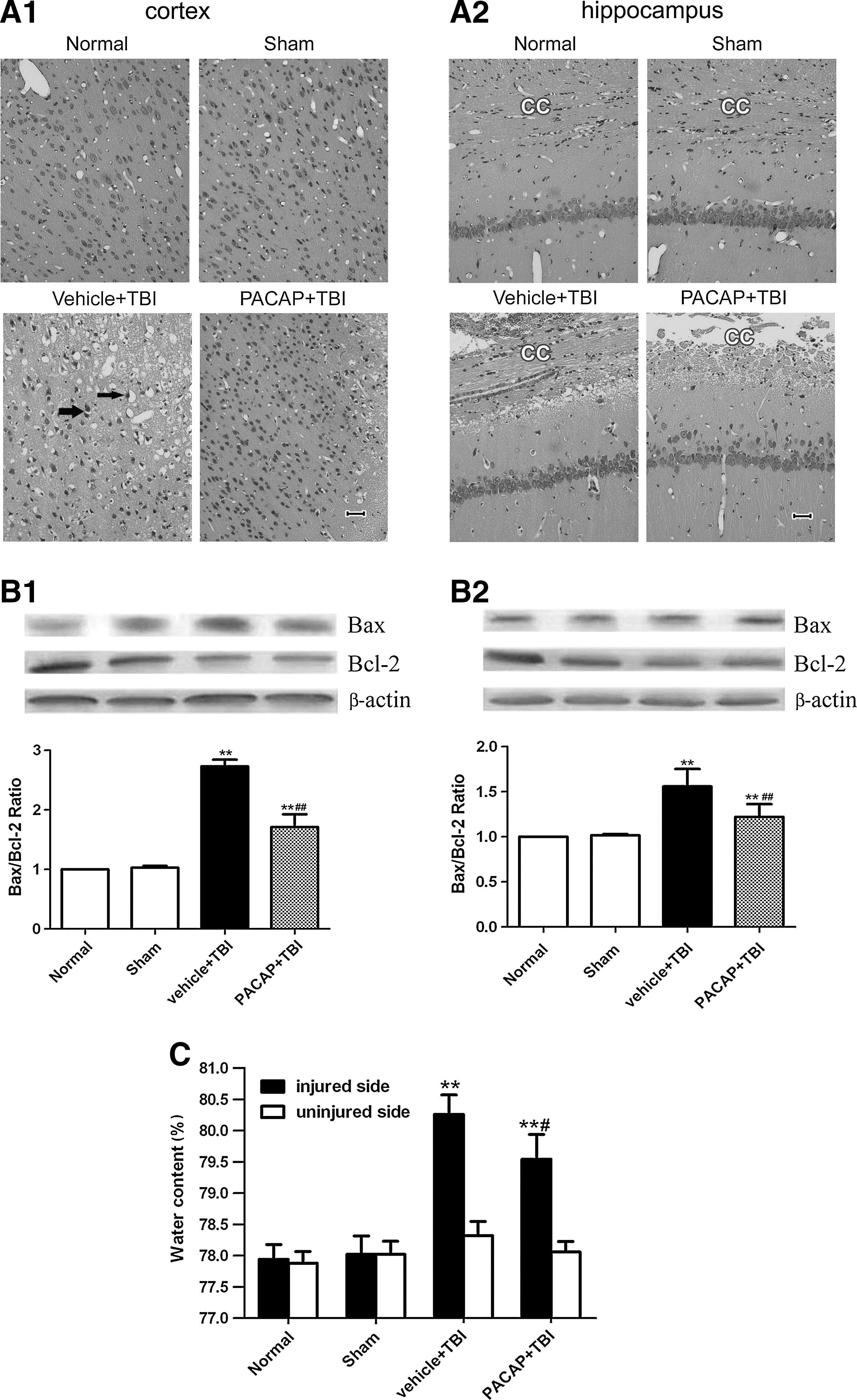
Pretreatment with PACAP reduced trauma-induced brain injury. (
The expression of Bax and Bcl-2 in the cerebral cortex around the injury site in both normal and sham-treated rats was low, with no significant differences between them (p>0.05). However, protein levels of Bax increased (p<0.01), and Bcl-2 levels significantly decreased, in the vehicle+TBI group (p<0.01). PACAP pretreatment inhibited increases in Bax expression, enhanced Bcl-2 expression, and depressed the ratio of Bax/Bcl-2 compared to the vehicle+TBI group (p<0.01). In the hippocampus, PACAP pretreatment significantly upregulated Bcl-2 protein expression (p<0.05), but did not change Bax expression compared to the vehicle+TBI group (Fig. 3B).
Brain edema was next determined using the wet/dry method. The water content in brain samples in the injured side was clearly increased at 24 h post-TBI compared to that seen in the normal and sham groups (p<0.01). Pretreatment with PACAP markedly decreased brain water content (p<0.05) following TBI. On the uninjured side, there were no significant differences in brain water content in any group (p>0.05). These results suggest that PACAP pretreatment can attenuate brain edema following TBI in rats (Fig. 3C).
Time-dependent expression of TLR4 in the cerebral cortex around the injured brain area and in the hippocampus post-TBI
We used Western blots to analyze TLR4 protein expression at different time points in the cerebral cortex around the injured brain area and in the hippocampus. In the sham group, TLR4 protein levels were very low in the cerebral cortex around the injured brain area and in the hippocampus. In the vehicle+TBI group, TLR4 protein levels were significantly increased 12, 18, 24, 48, and 72 h post-TBI (Fig. 4; p<0.05). There were no significant differences 0 and 6 h post-TBI between the sham and vehicle+TBI groups (p>0.05). TLR4 protein expression peaked at 24 h in both the cortex and hippocampus (p<0.01), and declined 48 and 72 h post-TBI (p<0.05; Fig. 4). Thus, the 24-hour time point was used for analysis in subsequent experiments.
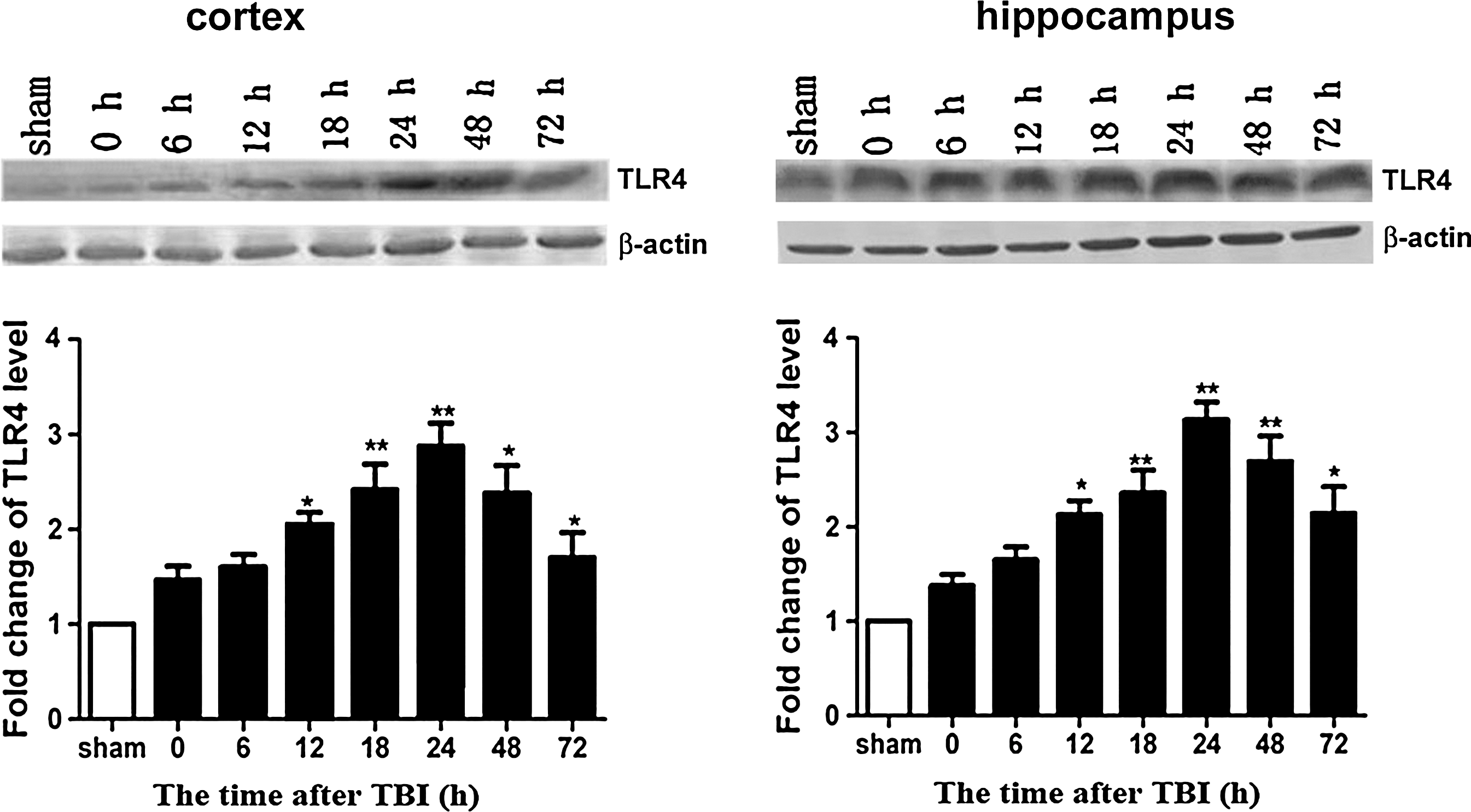
Time course of Toll-like receptor 4 (TLR4) expression in the rat brain following traumatic brain injury (TBI). (Top) Shown here are representative Western blots showing protein levels of TLR4 and β-actin (loading control) in the cerebral cortex around the injury site and the hippocampus 0, 6, 12, 18, 24, 48, 72 h post-trauma (n=6). (Bottom) Densitometric analysis of TLR4 protein levels in the cerebral cortex around the injured brain area and hippocampus 0, 6, 12, 18, 24, 48, 72 h post-trauma was normalized to sham levels, averaged, and graphed (*p<0.05, **p<0.01 compared with the sham group).
Pretreatment with PACAP inhibited TLR4 expression around the injured cerebral cortex site and in the hippocampus post-TBI
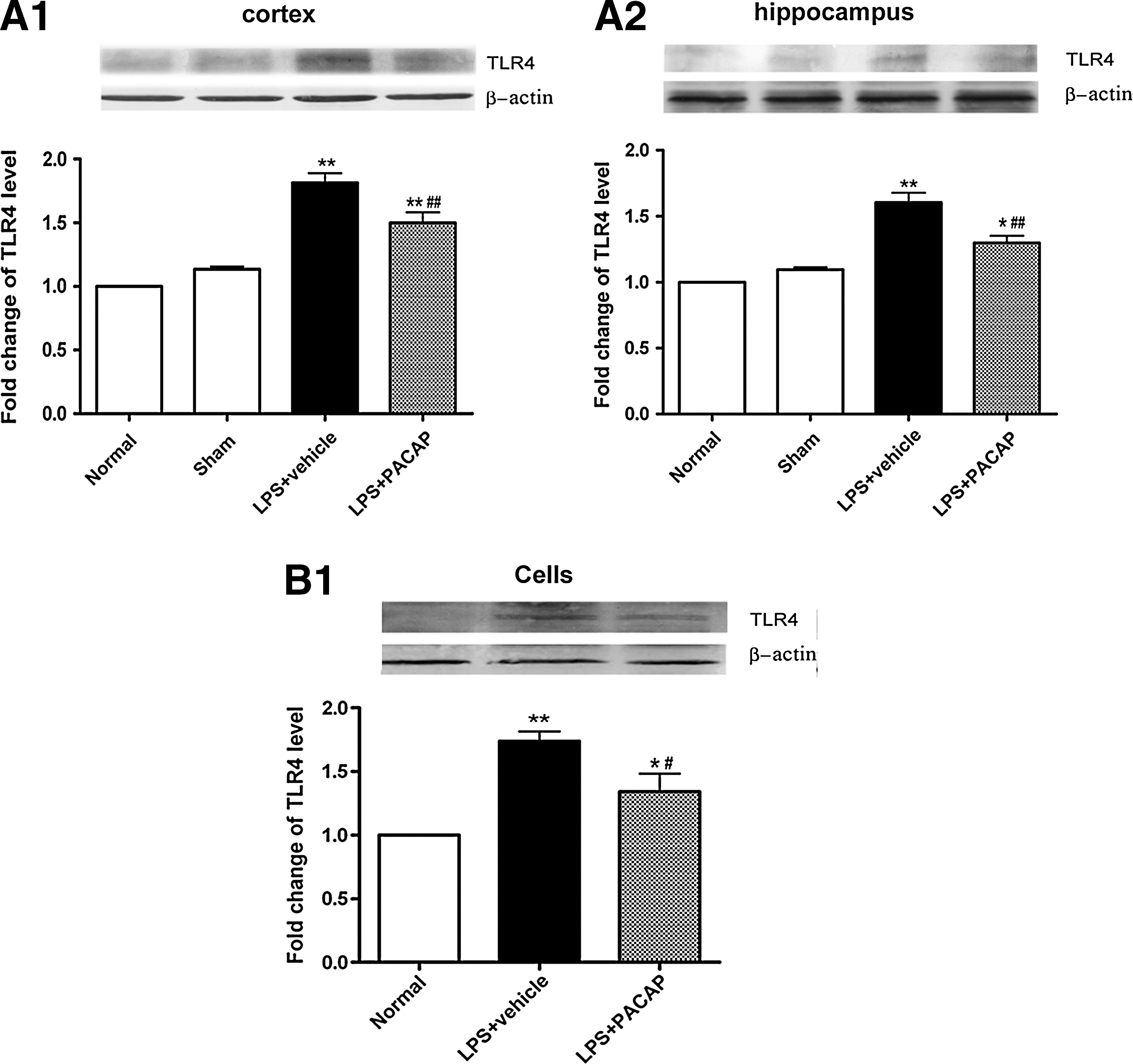
To determine whether PACAP attenuated brain injury by modulating TLR4, real-time qRT-PCR was performed to detect changes in TLR4 mRNA. Western blotting and immunohistochemistry were used to detect changes in TLR4 protein levels. In the cerebral cortex around the injured brain area and in the hippocampus, TLR4 mRNA levels were low in the normal and sham groups, with no significant differences between them (p>0.05). As expected, at 24 h post-TBI, TLR4 mRNA levels in the cerebral cortex in the area around the injured brain and in the hippocampus were significantly increased (p<0.01). Pretreatment with PACAP markedly suppressed increases in TLR4 mRNA levels post-TBI (p<0.05; Fig. 5A). The protein levels of TLR4 and the number of TLR4-positive cells were also significantly increased in vehicle+TBI rats. Changes in TLR4 protein levels were similar to those observed for TLR4 mRNA. As with mRNA, pretreatment with PACAP significantly inhibited TBI-induced increases in TLR4 protein levels, and decreased the number of TLR4-positive cells in the injured cortex and hippocampus (p<0.05; Figs. 5B and 5C).
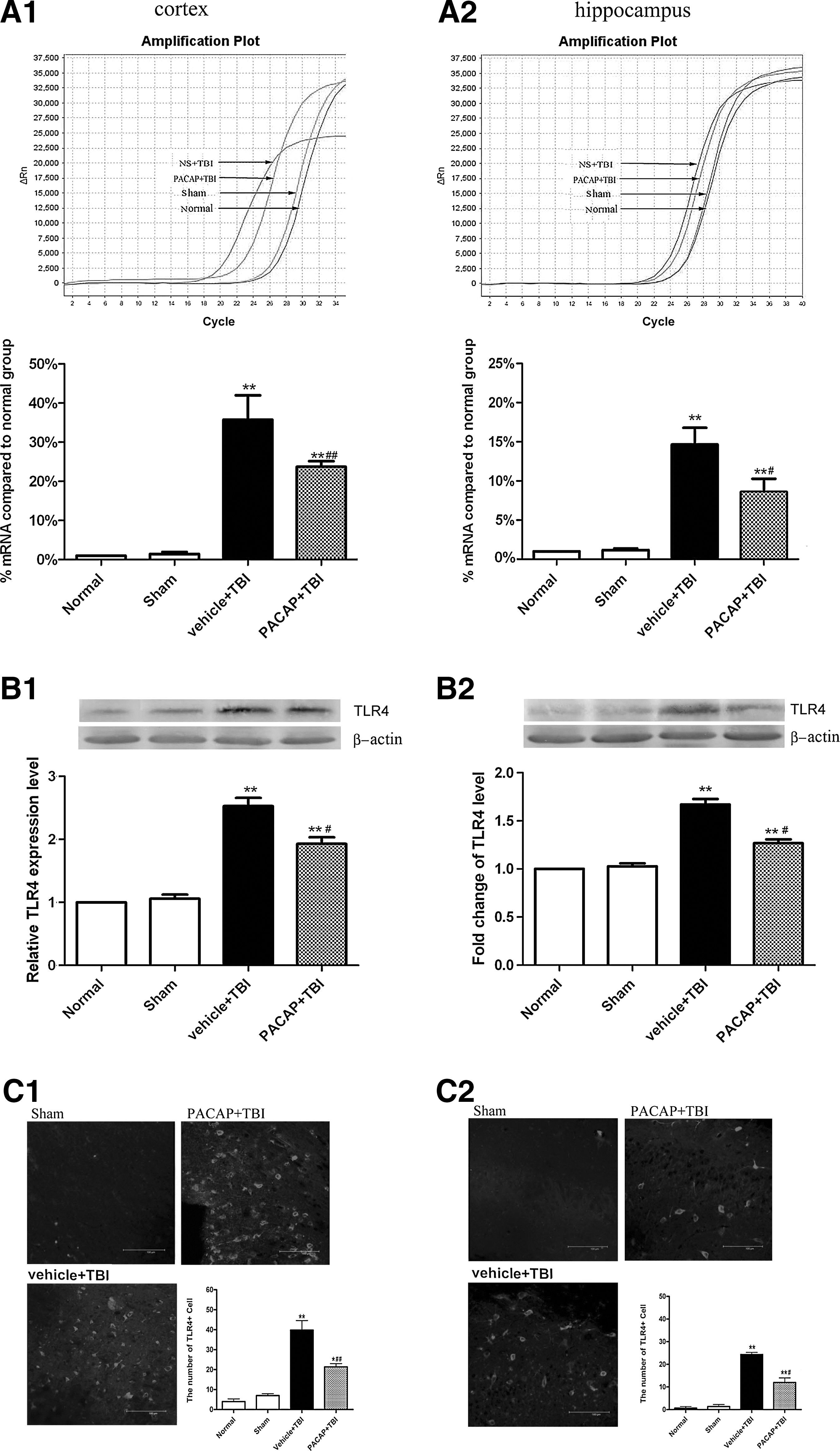
Pretreatment with PACAP downregulated TLR4 mRNA and protein levels in the cerebral cortex around the injured brain area and in the hippocampus of rats 24 h post-trauma (n=6). (
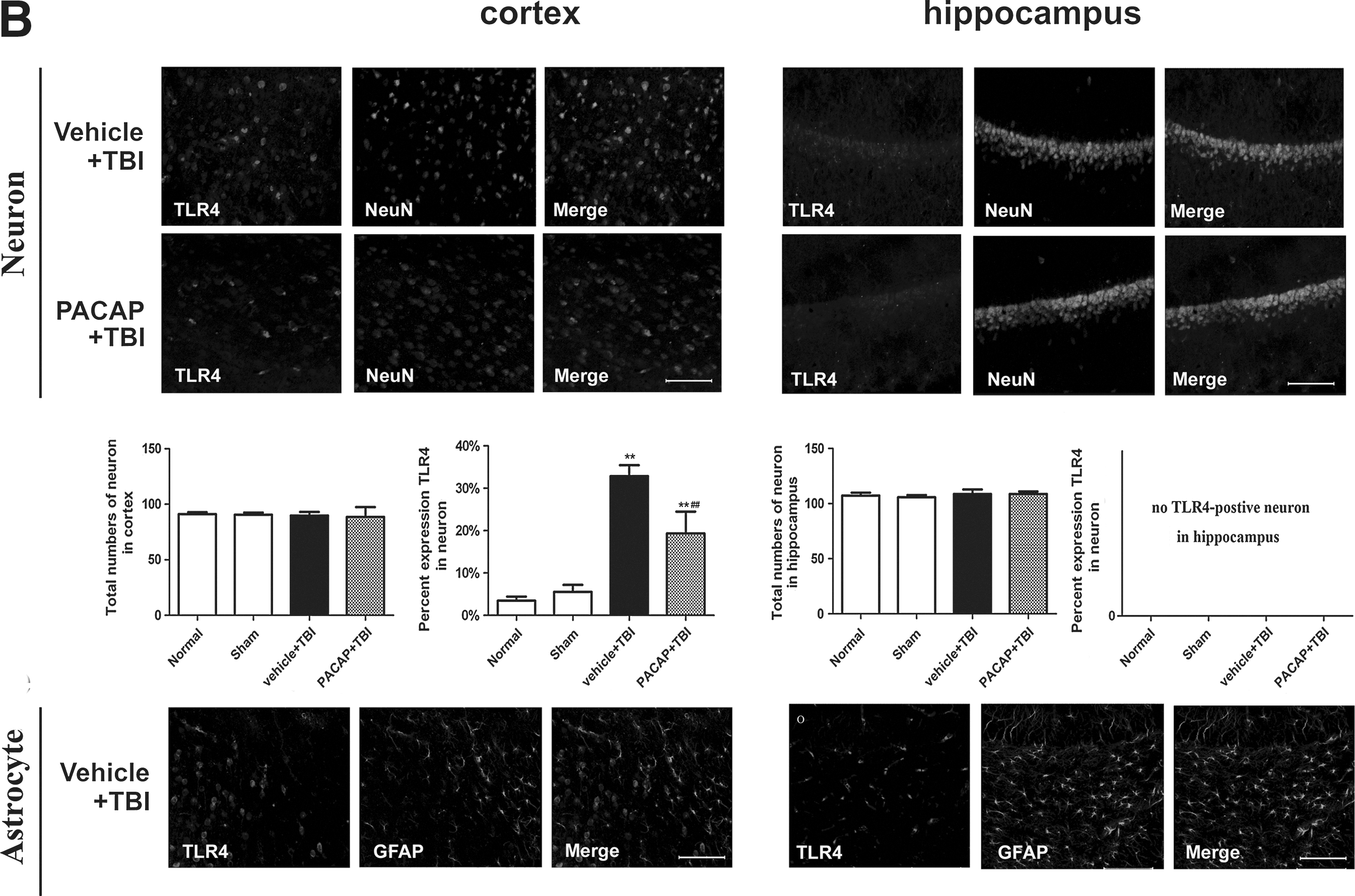
Immunofluorescence was used to elucidate the cell types with increased TLR4 protein levels in the injured cortex and hippocampus. In general, we observed that TLR4 immunoreactivity was localized to microglia and neurons, but not astrocytes. In the cortex and hippocampus of normal and sham control rats, a few quiescent microglia were observed, with small cell bodies and fine, ramified processes. Few or no TLR4-positive microglia were detected (Fig. 6A). However, at 24 h post-injury the density of Iba-1+microglia was increased, along with altered cellular morphology in the vehicle+TBI group. Trauma triggered microglia to acquire a reactive state with a larger cell body, and thickened, slightly shorter processes. Activated microglia exhibited robust TLR4 immunoreactivity in the traumatized cerebral cortex and hippocampus. Pretreatment with PACAP inhibited increases in TLR4-positive microglia post-TBI, although microglia still exhibited reactive morphology (Fig. 6A). We also observed immunoreactivity of TLR4 in neurons. In the cerebral cortex of normal and sham control rats, few or no TLR4-positive neurons were observed. At 24 h post-trauma, neurons in the traumatized cortex exhibited robust TLR4 immunoreactivity. Pretreatment with PACAP inhibited increases in TLR4-positive neurons. TLR4-positive neurons were not detected in the hippocampus of any group (Fig. 6B). Furthermore, we did not observe TLR4-positive astrocytes in any group (Fig. 6B). PACAP treatment attenuated the decrease in total neuron numbers and the increase in total microglia numbers induced by the injury. In addition, the percentages of neurons and microglia immunoreactive for TLR4 increased with injury, but were attenuated by PACAP treatment (Fig. 6B).
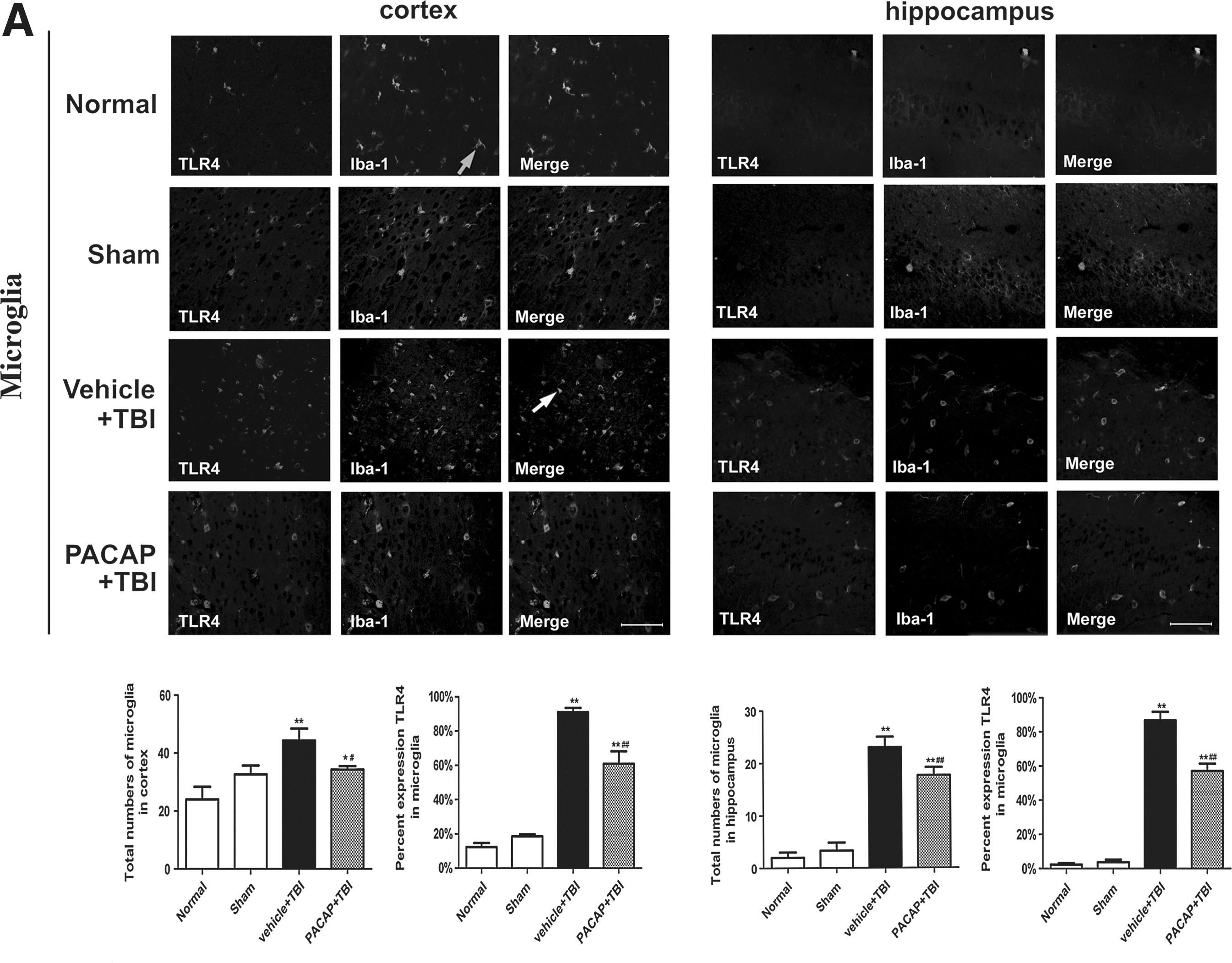
Localization of Toll-like receptor 4 (TLR4) in the cerebral cortex around the injured brain area and in the hippocampus following traumatic brain injury (TBI) in rats. Brain tissue sections were double-immunofluorescence stained with anti-TLR4 and NeuN, Iba-1, or glial fibrillary acidic protein (GFAP) antibodies. Representative images of cortical immunoreactivity around the injured brain area and from the hippocampus are shown (n=6). (
Pretreatment with PACAP suppressed expression of proteins in the MyD88/NF-κB pathway in the injured cortex and hippocampus post-TBI
To further understand the effect of PACAP pretreatment on TLR4 downstream signaling pathways, Western blotting was performed to examine the expression of the adapter protein MyD88, as well as NF-κB p65, p-IκB-α, and IκB-α, in the cerebral cortex around the injured brain area and in the hippocampus 24 h post-TBI. In the normal and sham groups, MyD88 (Fig. 7A), NF-κB p65 in nuclei, and p-IκB-α (Fig. 7B) were detected at low levels, with no significant differences between them (p>0.05). In the TBI group, MyD88 protein levels, NF-κB p65 in the nucleus, and p-IκB-α in the injured cortex and hippocampus were notably increased (p<0.01). Pretreatment with PACAP significantly decreased MyD88 protein levels, NF-κB p65 in nuclei, and p-IκB-α (p<0.01). IκB-α demonstrated an inverse profile to both p-IκB-α and NF-κB p65 in the nucleus, with PACAP causing increases in IκB-α levels (Fig. 7).
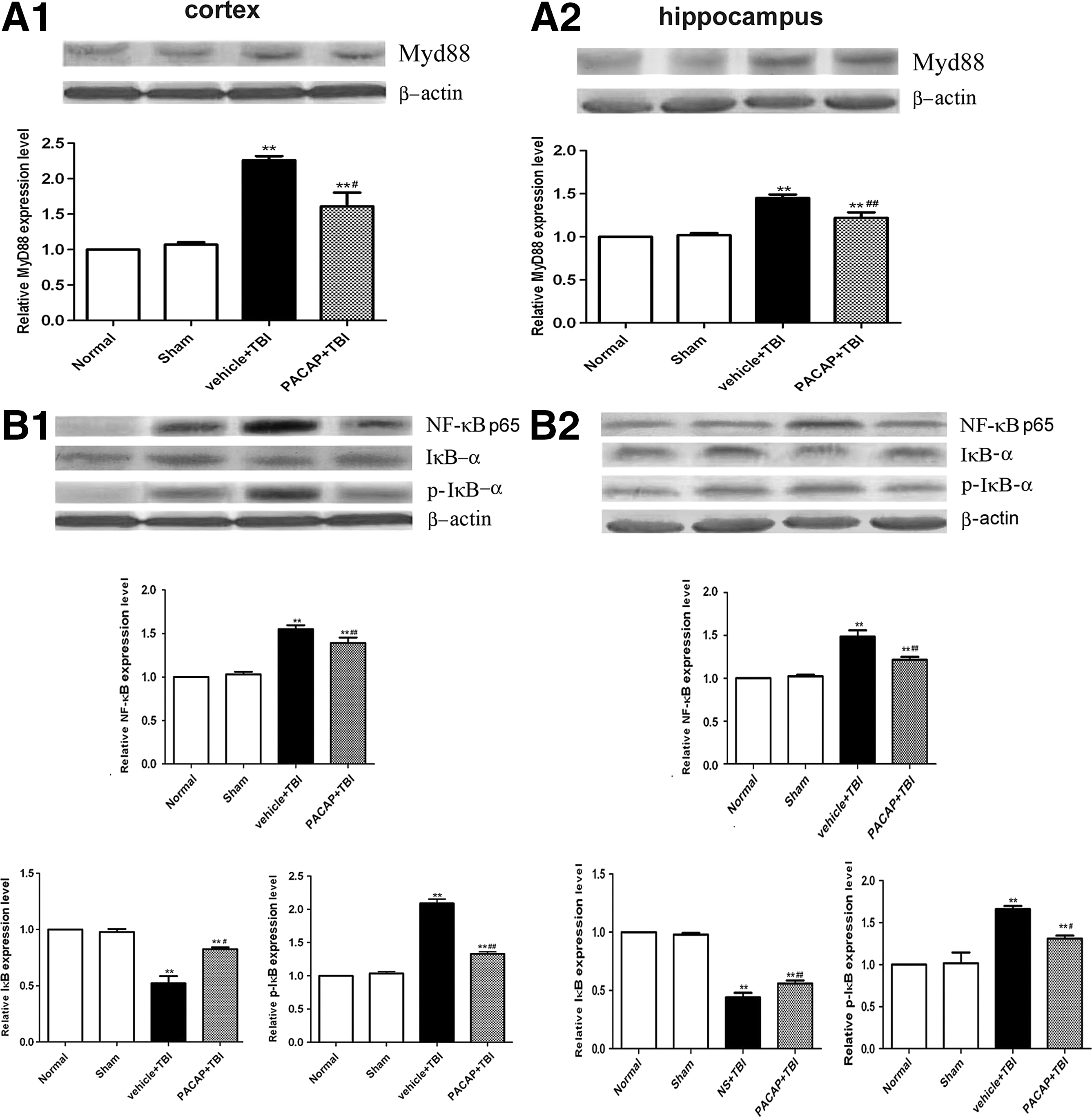
Pretreatment with PACAP inhibited the increase in MyD88 and NF-κB protein levels in the cortex area around the injury site and in the hippocampus of rats at 24 h post-trauma (n=6). (
Pretreatment with PACAP decreased levels of proinflammatory cytokines in the injured cortex and hippocampus post-TBI
Since brain injury caused by PACAP correlated with decreased protein levels of components of the TLR4/MyD88/NF-κB pathway, we hypothesized that a reduction in NF-κB-mediated transcription and expression also occurred. To address this, we measured levels of proinflammatory cytokines in the cerebral cortex around the injured brain area and in hippocampal tissue. To examine the effects of PACAP on TNF-α and IL-1β levels, ELISA was used to confirm their concentrations 24 h post-injury. TNF-α and IL-1β were detected at low levels in the cortex around the injury area and in the hippocampus of the normal and sham groups. There was no significant difference between groups (p>0.05). The levels of TNF-α and IL-1β in the cortex around the injured area and in the hippocampus increased significantly post-injury (p<0.01). As expected, PACAP inhibited increases of the two cytokines (p<0.01) in the injured cortex and hippocampus (Fig. 8).
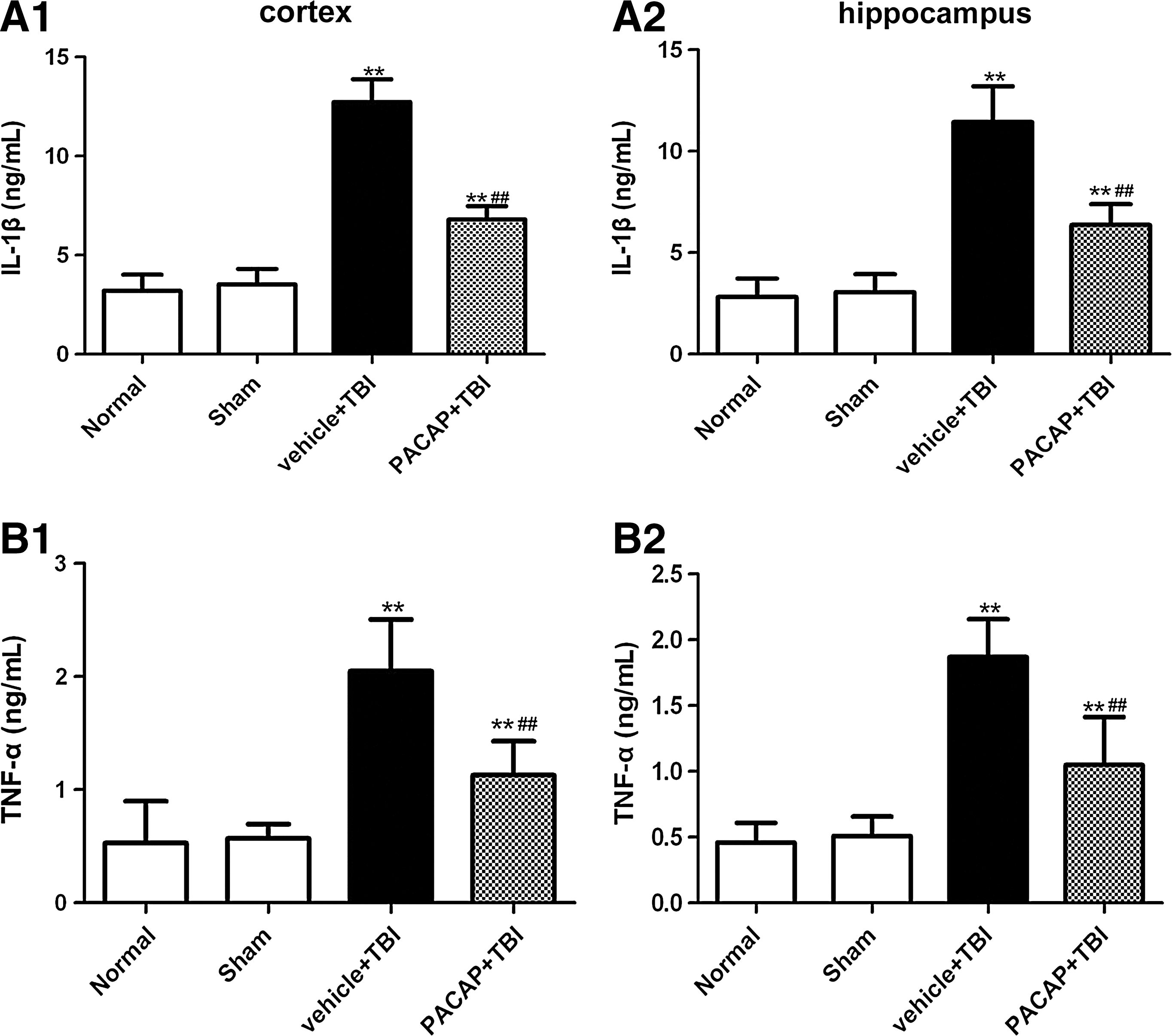
Pretreatment with PACAP decreased inflammatory cytokine IL-1β and TNF-α levels in the cerebral cortex around the injured brain area and in the hippocampus in rats at 24 h post-trauma (n=6). ELISA analyses were used to quantify protein levels of IL-1β (
PACAP inhibited the increase in TLR4 protein levels induced by LPS
Although we had confirmed that PACAP exerted a neuroprotective effect in this rat model of TBI, these findings did not directly demonstrate attenuated TLR4 signaling, but instead correlated with attenuated protein levels of TLR4, MyD88, and NF-κB. In order to provide additional evidence of a direct ability of PACAP to attenuate TLR4 function, we performed in vitro and in vivo experiments using a well-described TLR4 agonist, LPS. Intracerebroventricular administration of LPS induced a significant increase in TLR4 protein levels that was attenuated by a 24-h post-treatment with PACAP (Fig. 9A; p<0.01). We observed similar findings in vitro, demonstrating that LPS significantly increased TLR4 in BV2 microglia cells, but this increase was also attenuated by 24-h post-treatment with PACAP (Fig. 9B; p<0.05). Therefore, PACAP appears able to directly regulate TLR4-mediated signaling by attenuating protein levels of key molecules in the signaling response, including TLR4 itself.
Post-treatment with PACAP directly reduces TLR4 protein levels. In vivo (
Discussion
The present study used a rat model to provide further evidence of the neuroprotective effects of PACAP in TBI. We found that pretreatment with PACAP significantly attenuated TBI-induced motor and cognitive dysfunction, and protected the brain against apoptosis, inflammation, and edema. One possible molecular mechanism for PACAP's protection may be attenuation of the TLR4/MyD88/NF-κB signaling pathway in microglia and neurons, based on our immunohistochemical and Western blot analyses of PACAP-treated rats. This suggests that attenuating the secondary inflammatory response induced by TBI with PACAP protects against brain injury. Although pretreatment with PACAP may not be the ideal paradigm for clinical application, a primary purpose of this study was to determine whether the neuroprotective effect of PACAP is mediated by the TLR4/MyD88/NF-κB pathway. These mechanisms were explored and may provide insight into the clinical actions of PACAP. It is likely that no single drug will maximally offer neuroprotection, suggesting that combination therapy will be necessary. Understanding the mechanism of PACAP-mediated protection may help direct future combinatorial strategies.
Our first goal was to confirm that PACAP could protect against TBI in rats. Clinically, although numerous pharmacologic agents such as corticosteroids (Roberts et al., 2004), albumins (Myburgh et al., 2007), calcium channel blockers (Clausen and Bullock, 2001), and excitatory amino acid inhibitors (Narayan et al., 2002), have all been evaluated in TBI patients, none has demonstrated a significantly convincing benefit in the overall TBI population. Pharmacological compounds that have produced worsened outcome measures post-TBI include haloperidol, which acts on D2-like dopamine receptors (Kline et al., 2004), bicuculline, the ROS scavenger PBN, the iNOS inhibitors aminoguanidine and L-N-iminoethyl-lysine, and tetrahydroaminoacridine, a cholinesterase inhibitor (Marklund et al., 2006). Therefore the efficacy of existing neuroprotective treatments for TBI has remained uncertain. Thus it is necessary to identify novel approaches to improve motor, sensory, and cognitive outcomes in TBI patients (Leker and Shohami, 2002; Xiong et al., 2009). Our rationale for this study was that the neuropeptide PACAP could improve outcomes and offer one such new direction, based on prior work demonstrating that PACAP may be a promising therapeutic agent in stroke, global ischemia, and other central nervous system disorders (Somogyvari-Vigh and Reglodi, 2004; Vaudry et al., 2009). However, there are only a few studies that indicate the potentially protective effect of PACAP in TBI. In a rat model of TBI induced by central fluid percussion, post-injury PACAP treatment significantly reduced diffuse axonal injury and protected the corticospinal tract (Johanson et al., 2011; Kovesdi et al., 2008). In the present study we determined that pretreatment with 1 μg PACAP could significantly improve motor and cognitive outcomes following TBI, as evaluated by the beam balance walk, inclined plane task, and the MWM test. Since the hippocampus is critically involved in learning behavior, the MWM results indicated that the hippocampus may be vulnerable to TBI, although there was no direct injury to it. Therefore, we assessed morphological changes in the brain tissue around the injured cortex and in the ipsilateral hippocampus. Pretreatment with PACAP decreased the appearance of swollen and necrotic neurons in the brain tissue around the injured cortex and in the molecular layer of the hippocampus post-TBI. Moreover, pretreatment with PACAP depressed increases in the Bax/Bcl-2 ratio following TBI, indicating that pretreatment with PACAP may also inhibit cell apoptosis. The Bax/Bcl-2 ratio has been proposed to be an important factor in apoptosis because Bax is a critical initiator of the intrinsic apoptotic pathway, and Bcl-2 is an anti-apoptotic protein (Autret and Martin, 2009; Fletcher and Huang, 2008).
Based upon the significant behavioral benefits afforded by PACAP pretreatment, we attempted to identify the mechanism by which it offered neuroprotection. Prior work to understand its neuroprotective mechanisms has largely focused on direct neuronal effects. However, according to some studies, indirect mechanisms mediated by astrocytes might also play a role, since PACAP can regulate glial glutamate uptake and metabolism (Reglodi et al., 2002; Somogyvari-Vigh and Reglodi, 2004). Whether PACAP also regulates microglial function and the molecular pathways activated by PACAP to exert its neuroprotective roles in the brain is incompletely understood. Microglia activation and the release of mediators have been suggested to play a role in local inflammatory reactions following TBI (Rock and Peterson, 2006). Emerging evidence demonstrates a central role for TLRs expressed on microglia as a pivotal factor in generating these neuroimmune responses (Aravalli et al., 2007). However, despite the demonstrated role of PACAP in neuroprotection, none of the previous studies have focused on the TLR signaling pathway in relation to cerebral inflammation after TBI. The family of TLRs is part of the innate immune system and is critical in brain injury (Armant and Fenton, 2002; Jang and Rabb, 2009). TLR4 is well positioned to be the initiator of inflammation post-TBI. In a model of thrombotic focal cerebral ischemia, TLR4 expression noticeably increased, peaking at 24 h in the cortex and 4 h in the hippocampus (Dong et al., 2011). In the present study, levels of TLR4 mRNA and protein in the contused brain increased and peaked at 24 h, and decreased at 48 and 72 h post-TBI. The increased TLR4 expression may be related to cellular stress and upregulated protein synthesis induced by the brain trauma. The subsequent decrease of TLR4 expression at 48 and 72 h may be related to an anti-injury downregulatory response during the recovery process. The fact that increases in TLR4 mRNA and protein expression at 24 h post-TBI were suppressed by PACAP pretreatment supports the notion that the mechanism underlying the neuroprotective effect of PACAP against TBI is related to inhibition of TLR4 activation. This suggests that administering PACAP at different time points post-injury may have dramatically different benefits, which may be more effective earlier than later.
Since a number of cells can express TLR4, it was necessary to assess which cells were upregulating TLR4 protein levels post-injury. We observed that TLR4 was not only expressed in microglia, but was also expressed also in neurons, although not astrocytes, after brain injury. Neurons have previously been shown to express molecules related to innate immunity, including TLR4, which is activated in response to focal cerebral ischemia/reperfusion injury, and may contribute to the neuronal death process (Arumugam et al., 2009; Mack et al., 2006). In this study, we observed increased immunoreactivity of TLR4 in neurons 24 h following TBI in the cerebral cortex. In response to potential pathogen invasion, microglia react by destroying infectious agents before potential pathogen damage to neural tissue can occur. Moreover, microglial activation is critical in the progression of multiple inflammatory diseases via release of inflammatory cytokines (Rock et al., 2006). Therefore, activation of TLR4 in microglia likely contributes to the inflammatory processes that occur in TBI. Although we have yet to determine the stimuli leading to increased TLR4 expression in neurons and microglia, we speculate that the loss of neurons is the source of microglial increases in TLR4. However, the stimuli underlying neuronal increases may involve some sort of activity change rather than the release of cellular debris like the microglia. Traumatic injury-activated microglia exhibited a larger cell body with thickened, shorter processes, and robust TLR4 immunoreactivity in the traumatically-injured cerebral cortex and hippocampus. Interestingly, pretreatment with PACAP attenuated increases in TLR4 protein levels in microglia following TBI, but not the morphologically reactive state. This suggests that PACAP treatment is not sufficient to completely attenuate microglial activation. It is well known that astrocytes also participate in innate immune responses and serve as a major source of chemokines (Farina et al., 2005). Several studies have demonstrated that astrocytes do not express TLR4 in vitro (Farina et al., 2005), or in vivo (Lehnardt et al., 2003). However, others have been able to detect low, constitutive expression of TLR4 in astrocytes that increases upon cell activation (Farina et al., 2005). In our study, we did not observe astrocytic TLR4 immunoreactivity, suggesting that although these cells may be involved in the inflammatory response post-injury, their activation is likely not dependent upon TLR4-dependent signaling. In our paradigm, we hypothesize that microglial TLR4 activation following TBI could result in microglial production of a number of immune mediators that influence the ability of neurons to process information (Aravalli et al., 2007). Simultaneously, neuronal TLR4 activation could stimulate neuronal production of immune mediators to alter function as well.
Presumably, the TLR4-activated downstream signaling pathway, including MyD88 and NF-κB activation, can promote the production of proinflammatory cytokines as part of the response to TBI. Based upon the ability of PACAP to attenuate increases in TLR4 protein levels, we expect that a component of its neuroprotective benefits is attenuation of TLR4-dependent signaling in microglia and neurons. MyD88 is a central adaptor protein for the majority of TLRs, acting as a link between the receptors and downstream signaling molecules (O'Neill, 2003). One such molecule is the transcription factor NF-κB, which requires phosphorylation of IκB as a prerequisite for its activation (Hoffmann et al., 2002). NF-κB has a well-characterized role in regulating cytokine production (Covert et al., 2005). Indeed, the TLR4/NF-κB signaling pathway has previously been described as mediating the cerebral inflammatory response, which aggravates brain injury following TBI (Chen et al., 2009; Dong et al., 2011). In this study we found that increased protein levels of MyD88 and NF-κB following TBI was accompanied by a reduction of IκB-α protein and an increase of p-IκB-α protein, offering support for the idea that increases in TLR4-mediated signaling events occur in injured brains. PACAP suppressed the increased protein levels of MyD88 and NF-κB in the cortex and hippocampus post-TBI. It is well known that NF-κB activation increases gene transcription and protein synthesis of proinflammatory cytokines such as TNF-α and IL-1β, which can then further activate NF-κB (Neurath et al., 1996). This positive feedback is considered a means of amplifying inflammatory signals. Prior work indicates that inflammatory mediators play an important role in the pathogenesis of experimental traumatic brain injury (Chen et al., 2008). For instance, TNF-α and IL-1β have been shown to affect the integrity of the blood–brain barrier, cerebral edema, and neuronal damage, and participate in the mechanisms of TBI (Barichello et al., 2011; Li et al., 2011; Lloyd et al., 2008). A number of studies have demonstrated that PACAP can modulate inflammatory factor production, including proinflammatory cytokines and ICAM-1, in vivo or in vitro (Armstrong et al., 2008; Arumugam et al., 2009; Reglodi et al., 2002). In the present study, PACAP treatment similarly reduced TBI-induced production of TNF-α and IL-1β. Based on our results, we speculate that microinjection of PACAP into the lateral cerebral ventricle suppressed TLR4/MyD88 activation and subsequent NF-κB activation. This downregulation of key signaling proteins attenuated transcription and production of TNF-α and IL-1β, thus decreasing the inflammatory response following TBI, and promoting behavioral benefits in rats.
We are aware that the proposed mechanism of PACAP protection is somewhat correlative at this point. However, we observed that the behavioral, neuroprotective, anti-inflammatory, and anti-apoptotic effects of PACAP did correlate with decreased protein levels of key components of the TLR4 pathway, including TLR4, MyD88, and NF-κB. This supports the notion that this pathway is attenuated in microglia and neurons following treatment. We have not yet resolved whether TLR4-dependent signaling in neurons versus microglia is more critically involved in the TBI injury response or PACAP-mediated protection at this time. However, it is quite likely that both cells exhibit a TLR4-mediated response, based on the observed increased immunoreactivity in both following TBI. It was also interesting that although cytokine levels were attenuated, and components of the TLR response pathway decreased, microglia still retained their morphologically reactive phenotype. This suggests that something other than TLR4 stimulation is responsible for the initial activation of these cells. Indeed, we do not know what ligand most likely activates microglia post-TBI, although we speculate that many cellular products from dying neurons are involved. Finally, this study employed a PACAP pretreatment paradigm to offer a maximal protective response as an initial means of assessing its efficacy in TBI. Future work using post-treatment administration of PACAP will resolve the effectiveness and the mechanisms of this neuroprotective peptide approach to attenuating TBI.
We demonstrated a neuroprotective effect of PACAP in this rat model of TBI, which appeared to involve attenuation of the TLR4/MyD88/NF-κB signaling pathway in microglia and neurons, in correlation with attenuated levels of the proteins following PACAP administration. We are aware that we have not directly assessed attenuation of TLR4 signaling, and have provided only correlative evidence of attenuated signaling. It is possible that other effects of PACAP, including direct neuroprotection, may also be involved. However, our in vitro and in vivo studies using LPS also demonstrated an ability of post-treatment PACAP to attenuate TLR4 protein levels. Therefore, in this additional paradigm a characteristic of the neuroprotective effect of PACAP was once again downregulation of TLR4 levels, and presumably any TLR4-mediated responses. Microglia recognize the major gram-negative bacterial cell wall component LPS through TLR4 (Jung et al., 2005). TLR4 serves as a specific receptor for LPS and is localized on the surface of microglia cells. In support of our conclusions, there are a few reports also describing the ability of PACAP to regulate TLR responses. PACAP38 inhibits TLR4/MyD88/tumor necrosis factor receptor-associated factor adaptor protein (TRAF6) expression in mouse kidney and proximal tubule epithelial cells (Fang et al., 2010). Furthermore, vasointestinal peptide (VIP) participates in the regulation of TLRs by inhibiting the expression of the leukocyte differentiation antigen CD14, and reducing the inflammatory response induced by LPS (Gomariz et al., 2007), which shares 68% identity with PACAP38.
In conclusion, PACAP treatment reduced brain edema, neuronal death and apoptosis, and inflammatory cytokine production, and improved motor function and cognition, demonstrating that administration of the neurotrophic factor is protective in this rat model of TBI. In addition, correlative evidence supports a mechanism by which PACAP inhibits TLR4 activation and the downstream MyD88/NF-κB signaling pathway in reactive microglia and neurons 24 h following TBI. This includes diminished levels of inflammation-related cytokines in the brain, reinforcing the importance of inflammatory reactions in the pathogenesis of secondary neuronal injury following TBI (Fig. 10). Moreover, these data lend support to activation of the TLR4/MyD88/NF-κB signaling pathway as a potential therapeutic target in TBI, and suggest that PACAP should be considered a candidate for clinical trials in TBI.

Schematic diagram depicting proposed mechanisms by which pretreatment with PACAP alleviated traumatic brain injury (TBI) through attenuating the TLR4/MyD88/NF-κB pathway in microglia and neurons. (
Footnotes
Acknowledgments
This project was supported by grants from the National Natural Science of China (81171041 and 81171013), the Natural Science Foundation of Jiangsu Province (BK2011197), the Educational Department Science Research Foundation of Jiangsu Province (09KJD310009), a project from the Jiangsu Key Laboratory of Brain Disease Bioinformation (JSBL0804), the Innovation Project of Nanjing Military Region (09MA036), the Research Foundation of President of Xuzhou Medical College (no. 09KJZ17), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions of Jiangsu Province.
Author Disclosure Statement
No conflicting financial interests exist.