
Abstract
Chronic anxiety is a common and debilitating result of traumatic brain injury (TBI) in humans. While little is known about the neural mechanisms of this disorder, inflammation resulting from activation of the brain's immune response to insult has been implicated in both human post-traumatic anxiety and in recently developed animal models. In this study, we used a lateral fluid percussion injury (LFPI) model of TBI in the rat and examined freezing behavior as a measure of post-traumatic anxiety. We found that LFPI produced anxiety-like freezing behavior accompanied by increased reactive gliosis (reflecting neuroimmune inflammatory responses) in key brain structures associated with anxiety: the amygdala, insula, and hippocampus. Acute peri-injury administration of ibudilast (MN166), a glial cell activation inhibitor, suppressed both reactive gliosis and freezing behavior, and continued neuroprotective effects were apparent several months post-injury. These results support the conclusion that inflammation produced by neuroimmune responses to TBI play a role in post-traumatic anxiety, and that acute suppression of injury-induced glial cell activation may have promise for the prevention of post-traumatic anxiety in humans.
Introduction
T
There is increasing evidence that excessive inflammatory actions of the neuroimmune system may contribute to the development of anxiety disorders following TBI (Gasque et al., 2000; Hoge et al., 2009; Shiozaki et al., 2005; Spivak et al., 1997; Tucker et al., 2004; von Känel et al., 2007). Microglial cells are the first line of defense and primary immune effector cells in the central nervous system (CNS), and respond immediately to even small pathological changes from damaged cells, producing proinflammatory cytokines and toxic molecules that compromise neuronal survival (Aloisi, 2001; Gehrmann, 1996; Gonzalez-Scarano and Baltuch, 1999; Town et al., 2005). This rapid microglial response often precedes the more delayed, yet prolonged activation of astrocytes, and is thought to be involved with the onset and maintenance of astrogliosis (Graeber and Kreutzberg, 1988; Hanisch, 2002; Herber et al., 2006; Iravani et al., 2005; McCann et al., 1996; Zhang et al., 2010). It is well established that microglia and astrocytes are activated during the innate immune response to brain injury, leading to the expression of high levels of proinflammatory cytokines, most notably interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α). While glial activation is typically neuroprotective (Aloisi, 2001; Farina et al., 2007), the chronic inflammatory responses and exaggerated proinflammatory cytokine levels observed following injury initiate neurotoxic processes resulting in secondary tissue damage (Gasque et al., 2000; Hailer, 2008; Lehnardt, 2010; Simi et al., 2007), neuronal death (Beattie et al., 2010; Brown and Bal-Price, 2003; Schmidt et al., 2005; Sternberg, 1997), secondary injury cascades (Ansari et al., 2008a,2008b; Bains and Shaw, 1997; Cernak et al., 2001b,2001a), and neuronal hyperexcitability (Beattie et al., 2010; Hailer, 2008; Riazi et al., 2008; Rodgers et al., 2009), all of which may contribute to the dysfunction of brain regions associated with anxiety.
Recently developed animal models of post-traumatic anxiety (Baratz et al., 2010; Fromm et al., 2004; O'Connor et al., 2003; Jones et al., 2008; Liu et al., 2010; Sönmez et al., 2007; Vink et al., 2003; Wagner et al., 2007) permit examination of the possible contributions of brain inflammation. Tests of post-traumatic anxiety in these models have typically included standard measurements of exploratory preference in mildly stressful environments, such as an open-field or elevated-plus testing apparatus. However, it has been frequently noted that measures of exploratory preference may be confounded by a marked overall decrease in exploration in brain-injured animals (Fromm et al., 2004; O'Connor et al., 2003; Vink et al., 2003). Decreased exploration cannot be attributed to TBI-induced motor deficits since numerous studies report only transient (∼ 1 week) deficits following trauma (Baratz et al., 2010; Bouilleret et al., 2009; Cutler et al., 2005,2006a,2006b; Dixon et al., 1996; Fassbender et al., 2000; Frey et al., 2009; Goss et al., 2003; Kline et al., 2007; Liu et al., 2010; Taupin et al., 1993; Wagner et al., 2007; Yan et al., 1992). Rather, TBI-induced decreases in exploration have been attributed to the indirect effects of freezing (a primary component of the rodent's natural defensive behavior repertoire; Blanchard and Blanchard, 1988), suggesting an abnormally heightened response to stress in brain-injured rats (O'Connor et al., 2003; Fromm et al., 2004; Vink et al., 2003).
Based on these results, we tested the hypothesis that trauma-induced innate immune responses contribute to the development of anxiety-like behaviors in rats by directly examining freezing responses to a minor (novel environment), and major (foot-shock) stressor, following lateral fluid percussion injury (LFPI; a clinically-relevant animal model of human closed-head injury). We also tested the effectiveness of a glial cell activation inhibitor, ibudilast (MN166), in attenuating post-injury freezing behavior and reducing reactive gliosis in brain regions associated with hyperexcitability in anxiety disorders.
Methods
Sixty adult virus-free male Sprague-Dawley rats (275–325 g; Harlan Laboratories, Madison, WI) were housed in pairs in temperature-controlled (23±3°C) and light-controlled (12-h:12-h light:dark cycle) rooms with ad libitum access to food and water. All procedures were performed in accordance with the University of Colorado Institutional Animal Care and Use Committee guidelines for the humane use of laboratory rats in biological research. The rats were randomly assigned to 1 of 10 groups (n=6/group). Six groups (surgically-naïve, sham-operated, sham-operated + vehicle, sham-operated + MN166, LFPI + vehicle, and LFPI + MN166) were shocked immediately after behavioral testing at 1 month post-surgery (sham surgery or LFPI in the experimental rats). Surgically-naïve rats received no injections or surgery, whereas sham-operated rats received surgery but were not injected. The final four groups received sham or LFPI surgery and either vehicle injections or MN166 treatment. Another four groups (sham-operated + vehicle, sham-operated + MN166, LFPI + vehicle, and LFPI + MN166) were run separately in a sucrose preference test to assess anhedonia (the inability to experience pleasure, a core symptom of human depression), without exposure to stressors (anxiety tests and foot shock).
Lateral fluid percussion injury
LFPI rats were anesthetized with halothane (4% induction, 2.0–2.5% maintenance) and mounted in a stereotaxic frame. The LFPI used in this study has been described previously (Frey et al., 2009; McIntosh et al., 1989; Thompson et al., 2005). A PV820 Pneumatic PicoPump (World Precision Instruments, Inc., Sarasota, FL) was used to deliver standardized pressure pulses of air to a standing column of fluid. A 3.0-mm-diameter craniotomy was centered 3 mm caudal to the bregma and 4.0 mm lateral to the sagittal suture, with the exposed dura remaining intact. A female Luer-Lok hub (inside diameter 3.5 mm) was secured over the craniotomy with cyanoacrylate adhesive. Following hub placement, the animal was removed from the stereotaxic frame and connected to the LFPI apparatus. The LFPI apparatus delivered a moderate impact force (2.0 atmospheres; 10 msec). The injury cap was then removed, the scalp was sutured, and the rats were returned to their home cages for recovery. Sham-operated rats underwent identical surgical preparation, but did not receive the brain injury.
Ibudilast (MN166) administration
MN166 (MediciNova Inc., San Diego, CA) is a relatively non-selective phosphodiesterase inhibitor with anti-inflammatory actions via glial cell attenuation, which has been found to reduce glia-induced neuronal death through the suppression of nitric oxide, reactive oxygen species, and proinflammatory mediators (Mizuno et al., 2004; Rolan et al., 2009). Treated rats received a 5-day dosing regimen of once-daily MN166 injections (10 mg/kg, 1 mL/kg subcutaneously in corn oil) 24 h prior to LFPI, the day of surgery and LFPI, and 3 days following LFPI. Weight was recorded prior to each dosing, and treatment was administered at the same time each day to maintain constant levels across a 24-h period. Dose selection was based on prior animal pharmacology results (Ledeboer et al., 2007), demonstrating MN166 to be safe and well tolerated, yielding plasma concentration-time profiles commensurate with high-dose regimens in clinical development. MN166 administered using this regimen yields plasma and CNS concentrations that are linked to molecular target actions, including most potently, macrophage migration inhibitory factor (MIF) inhibition (Cho et al., 2010), and secondarily, phosphodiesterase (PDE)-4 and PDE-10 inhibition (Gibson et al., 2006). The relevance of MIF inhibition in disorders of neuroimmune function such as neuropathic pain has recently been well demonstrated (Wang et al., 2011). Such dosing regimens have clearly been linked to glial attenuation in other animal models (Ledeboer et al., 2007), and the anti-inflammatory actions of MN166 have recently been shown to suppress cerebral aneurysms in a dose-dependent manner (Yagi et al., 2010).
Tests of motor, vestibular, and locomotive performance
Baseline testing of motor, vestibular, and locomotive performance in all groups was conducted immediately prior to surgery, and again following a 1-week recovery period. These tests included ipsilateral and contralateral assessment of forelimb and hindlimb use to assess motor function, locomotion, limb use, and limb preference (Bland et al., 2000,2001), toe spread to assess gross motor response (Nitz et al., 1986), placing to assess visual and vestibular function (Schallert et al., 2000; Woodlee et al., 2005), a catalepsy rod test to assess postural support and mobility (Sanberg et al., 1988), bracing to assess postural stability and catalepsy (Morrissey et al., 1989;Schallert et al., 1979), and air righting to assess dynamic vestibular function (Pellis et al., 1991a,1991b). Scoring ranged from 0 (severely impaired) to 5 (normal strength and function). The individual test scores were summed and a composite neuromotor score (0–45) was then generated for each animal. In addition to the composite neuromotor score, limb-use asymmetry was assessed during spontaneous exploration in the cylinder task, a common measure of motor forelimb function following CNS injury in rats (Schallert et al., 2000,2006), and post-injury locomotor activity was assessed through distance traveled on a running wheel. Both tasks were scored for 5 min under red light (∼ 90 lux).
Behavioral measures
A novel environment was used to assess freezing behavior in response to a minor stressor (Dellu et al., 1996). The environment consisted of a standard rat cage with one vertically-striped and one horizontally-striped wall. No aversive stimuli were introduced in this context and no conditioning occurred. The rats were tested (5 min), and the percent of freezing behavior was assessed. Freezing was defined as the absence of movement except for heartbeat/respiration, and was recorded in 10-sec intervals.
Freezing behavior in the novel environment was measured before and after administration of a foot shock in a separate shock apparatus. The shock apparatus consisted of two chambers placed inside sound-attenuating chests. The floor of each chamber consisted of 18 stainless steel rods (4 mm diameter), spaced 1.5 cm center-to-center and wired to a shock generator and scrambler (Colbourn Instruments, Allentown, PA). An automated program delivered a 2-sec/1.5-mA electric shock. The rats were transported in black buckets and shocked immediately upon entry to the chambers. Following shock, the rats were returned to their home cages.
A sucrose preference test was also performed in separate groups of rats that did not receive foot shock or testing in the novel environment. This task is commonly used to measure anhedonia in rodent models of depression (Monleon et al., 1995; Willner, 1997). The sucrose preference task was included because anxiety and depression share high rates of co-morbidity in humans (Moore et al., 2006), and was assessed as a possible confound to freezing behavior, due to possible co-occurrence of depression-like behavior. The rats were first habituated to sucrose solution, and were tested during the dark phase of the light/dark cycle to avoid the food and water deprivation necessary when testing during the light phase. Day 1 and day 2 consisted of habituation, day 3 and day 4 were baseline (averaged), and day 5 was the first test day. The rats were presented with two pre-weighed bottles containing 2% sucrose solution or tap water for a period of 4 h. Thirty minutes into the task the bottles were swapped to force preference and counter for placement effects. Total sucrose intake and sucrose preference, calculate by: (sucrose intake/(sucrose intake + water intake * 100), were measured.
Timeline for behavioral testing
Following a 2-week recovery period from sham operation or LFPI in experimental animals, all groups except those to be evaluated for sucrose preference were tested in the novel context. Testing was performed at 2 weeks, and 1, 2, and 3 months post-surgery. Shock was delivered after behavioral testing was completed at the 1-month time point. Tests for sucrose preference were performed at 2 weeks, 1 month, and 3 months post-surgery, with no intervening foot shock.
Immunohistochemistry
Immunoreactivity for OX-42 (targets CD11b/c, a marker of microglial activation), and glial fibrillary acidic protein (GFAP; a marker of astrocyte activation) were measured using an avidin-biotin-horseradish peroxidase (ABC) reaction (Loram et al., 2009). Brain sections (12 μm) were cut on a cryostat and mounted onto poly-L-lysine-coated slides and stored at −80°C. Sections were post-fixed with 4% PFA for 15 min at room temperature, then treated with 0.03% H2O2 for 30 min at room temperature. The sections were incubated at 4°C overnight in either mouse anti-rat OX-42 (1:100; BD Biosciences Pharmingen, San Jose, CA), or mouse anti-pig GFAP (1:100; MP Biomedicals, Aurora, OH). The next day, the sections were incubated at room temperature for 2 h with biotinylated goat anti-mouse IgG antibody (1:200; Jackson ImmunoResearch, West Grove, PA). The sections were washed and incubated for 2 h at room temperature in ABC (1:400; Vector Laboratories, Burlingame, CA), and reacted with 3′,3-diaminobenzidine (DAB; Sigma-Aldrich, St. Louis, MO). Glucose oxidase and β-D-glucose were used to generate hydrogen peroxide. Nickelous ammonium sulfate was added to the DAB solution to optimize the reaction product. The sections were air-dried overnight and then dehydrated with graded alcohols, cleared in Histo-Clear, and cover-slipped with Permount (Fisher Scientific, Fairlawn, NJ). Densitometric analysis was performed using Scion Image software.
Image analysis
The slides were viewed with an Olympus BX-61 microscope, using Olympus Microsuite software (Olympus America, Melville, NY), with bright-field illumination at 10×magnification. The images were opened in ImageJ, converted into gray scale, and rescaled from inches to pixels. Background areas were chosen in the white matter or in cell-poor areas close to the region of interest (ROI). The number of pixels and the average pixel values above the set background were then computed and multiplied, giving an integrated densitometric measure (integrated gray level). Four measurements were made for each ROI; the measurements were then averaged to obtain a single integrated density value per rat, per region. All measurements were taken while blind to treatment group.
Statistical analyses
Results are expressed as mean±standard error of the mean (SEM). Analyses for all behavioral variables used analysis of variance (ANOVA) with repeated measures (time after injury), and treatment as the independent variable. The integrated density from the histology was only conducted at one time point, and utilized one-way ANOVAs to compare regions between groups. Data were analyzed using SPSS software, and in all cases statistical significance was set at p<0.05.
Results
Neuromotor composite scores of the brain-injured groups (LFPI + MN166 and LFPI + vehicle) did not significantly differ from controls [F(3,20)=0.803, p=0.508]. Rats in all groups consistently received normal scores on forelimb and hindlimb use, toe spread, placing, catalepsy rod, bracing, and air righting tests, indicating no impairments in motor, vestibular, or locomotive functioning due to TBI. There were also no significant between-group differences in limb-use asymmetry observed for contralateral [F(5,29)=0.544, p=0.741] and ipsilateral [F(5,29)=0.428, p=0.826] forelimb use during vertical exploratory behavior in the cylinder task, indicating no limb-use bias due to injury (Fig. 1A). No significant between-group differences were found in locomotor performance as evidenced by distance traveled during the running wheel activity [F(5,29)=0.069, p=0.996], revealing no post-injury impairments in locomotion (Fig. 1B). There were no significant between-group differences in the sucrose preference task [F(3,21)=0.338, p=0.798], indicating no impairments in hedonic states post-injury.
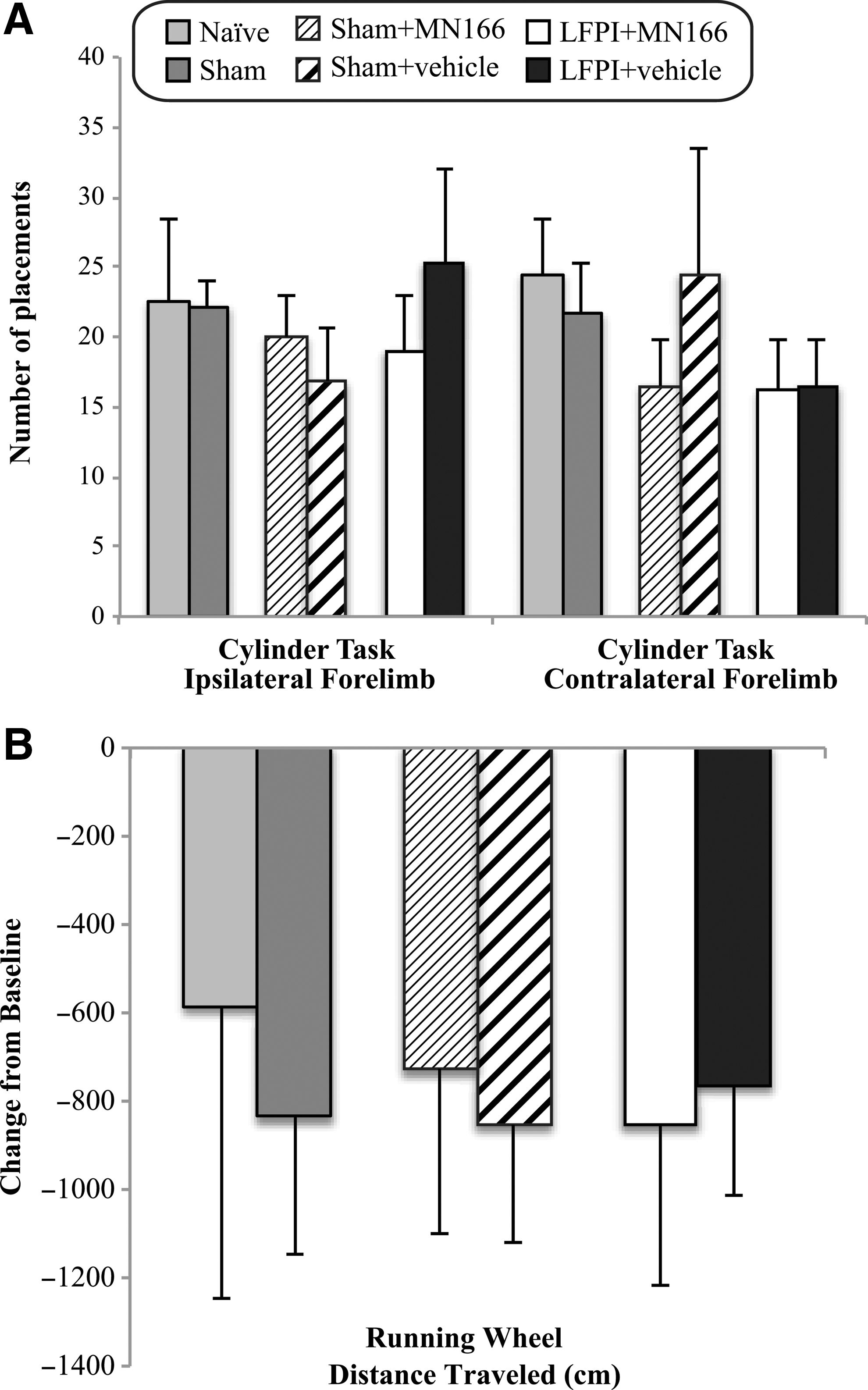
Cylinder task and running wheel activity at 1 week post-injury. (
Despite normal motor, vestibular, and locomotive function, LFPI produced large increases in freezing behavior when rats were placed in a novel context [Fig. 2; F(5,30)=9.539, p<0.0001]. Exposed only to this minor stressor (i.e., at 2-week and 1-month post-injury measurements conducted prior to shock), LFPI rats injected with either MN166 or vehicle (Fig. 2, white and black bars, respectively) froze approximately twice as long as naïve or sham-operated rats (Fig. 2; light and dark grey bars, respectively; p<0.01). At the 2- and 3-month measurement time points, following the additional major stressor of shock (arrow in Fig. 2), freezing in both naïve and sham-operated rats remained constant at approximately 10%. Freezing in LFPI rats treated with MN166 remained consistently higher than their controls (p<0.001) but, while appearing higher compared to earlier post-injury measurements in the same animals, this increased freezing compared to naïve and sham-operated rats before (1 month) and following (2 months) shock did not reach significance (p=0.316). By contrast, LFPI + vehicle rats nearly doubled their freezing time, to approximately 50% (Fig. 2, black bars) compared to pre-shock values (p<0.001), freezing approximately twice as long as LFPI + MN166 rats (p<0.001), and five times as long as naïve and sham-operated controls (p<0.001), at the 2- and 3-month post-injury time points.
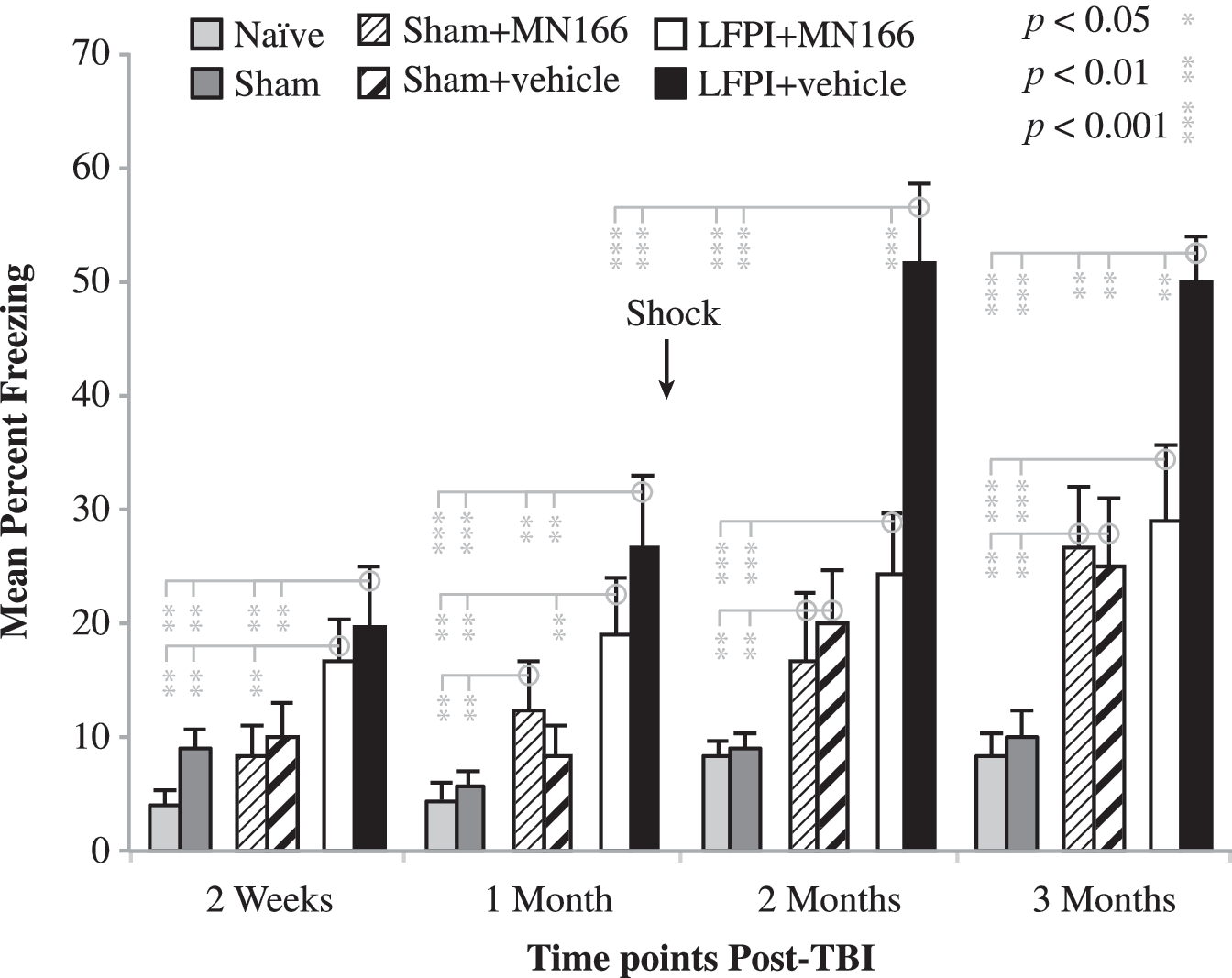
Freezing behavior in a novel context. Both surgically-naïve and sham-operated rats froze approximately 5–10% at post-surgical measurement points before (2 weeks and 1 month) after (2 and 3 months) foot shock (arrow). In contrast, LFPI rats froze significantly longer (∼ 20%) than controls before shock. After shock, untreated LFPI rats (LFPI + vehicle) nearly doubled their freezing time (∼ 50%), whereas treated LFPI rats (LFPI + MN166) showed only a slight increase (∼25%), that did not reach significance (p=0.316). The effects of injection alone (sham + MN166 and sham + vehicle) were to increase freezing behavior compared to un-injected naïve and sham-operated rats, particularly at the 2- and 3-month post-shock measurement time points, when freezing in these rats could not be distinguished from LFPI rats treated with MN166. Data represent mean±standard error of the mean (LFPI, lateral fluid percussion injury; TBI, traumatic brain injury).
The behavioral effects of injections alone, independent of LFPI, are reflected in the sham-surgery groups with injections of either MN166 or vehicle (Fig. 2, narrow and broad diagonal lines, respectively). Sham-operated rats tended to freeze more than un-injected naïve and sham-operated controls, reaching significance for both groups at the 2- and 3-month measurement time points (p<0.01), and suggesting that injections alone are aversive and can contribute to subsequent freezing. However, even at pre-shock measurement points, LFPI animals that received the same injections of MN166 or vehicle froze significantly more than injected controls (p<0.01), indicating substantial enhancement of freezing produced by LFPI. This effect became more apparent following shock, when LFPI + vehicle rats froze twice as long as the injected controls (p<0.001). By contrast, LFPI + MN166 rats were not distinguishable from either injected control group following shock, suggesting that their elevated freezing compared to naïve and sham-operated animals was the result of injections alone, and that MN166 eliminated the exaggerated freezing response to shock characterizing LFPI + vehicle rats.
OX-42 and GFAP immunoreactivity (reflecting microglia and astrocytic activation) was assessed in the insula, amygdala, and hippocampus in brain-injured rats for comparison to sham-operated and surgically-naïve rats. Representative images (40×), showing GFAP immunoreactivity in several of these regions, are shown in Figure 3, revealing normal astrocyte morphology in surgically-naïve and sham-operated rats. LFPI + vehicle rats showed clear signs of reactive astrocytes (Fig. 3, bottom row). LFPI rats treated with MN166 (Fig. 3, third row) were difficult to differentiate from sham-operated or surgically-naïve control groups.
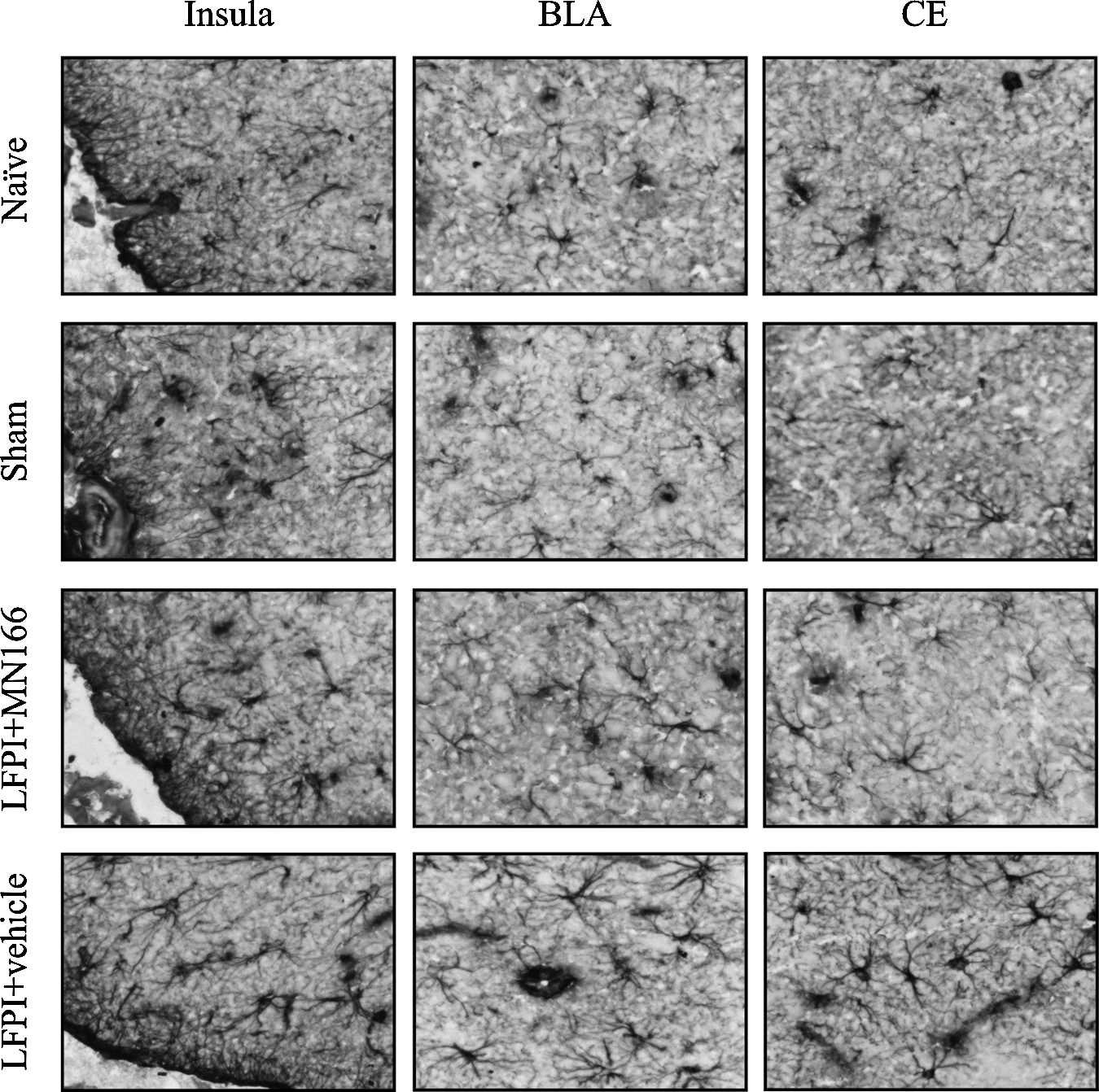
Representative images depicting GFAP immunoreactivity (reflecting astrocytic activation) as assessed in the hippocampus, amygdala, and insula at 3 months post-injury. LFPI rats injected with vehicle showed clear signs of reactive astrocytes (bottom row), while naïve and sham-operated rats appeared to have normal astrocyte morphology. LFPI rats treated with MN166 (third row from top) were difficult to differentiate from surgically-naïve and sham-operated animals (LFPI, lateral fluid percussion injury; GFAP, glial fibrillary acidic protein; BLA, basolateral amygdala; CE, central amygdala).
Densitometry of GFAP labeling in all areas examined confirmed that activation of astrocytes was significantly greater in LFPI compared to all other groups in the insula [Fig. 4A, left bars; F(3,19)=13.17, p<0.0001], amygdala [Fig. 4B, left bars; F(3,18)=7.54, p<0.002], and hippocampus [Fig. 4C, left bars; F(3,15)=8.47, p<0.002]. In contrast, no differences in GFAP labeling were observed between the surgically-naïve, sham-operated, and LFPI + MN166 groups, in any of the regions examined. While MN166-treated LFPI rats were not distinguishable from surgically-naïve or sham-operated controls, post-hoc analyses revealed that LFPI + vehicle rats had significantly greater astrocyte activation in all three brain regions compared to controls (Fig. 4A–C): insula (p<0.002 versus the surgically-naive, sham-operated, and LFPI + MN166 groups), amygdala (p<0.02 versus the surgically-naive, sham-operated, and LFPI+MN166 groups). and hippocampus (p<0.03 versus the surgically-naive, sham-operated and LFPI + MN166 groups).
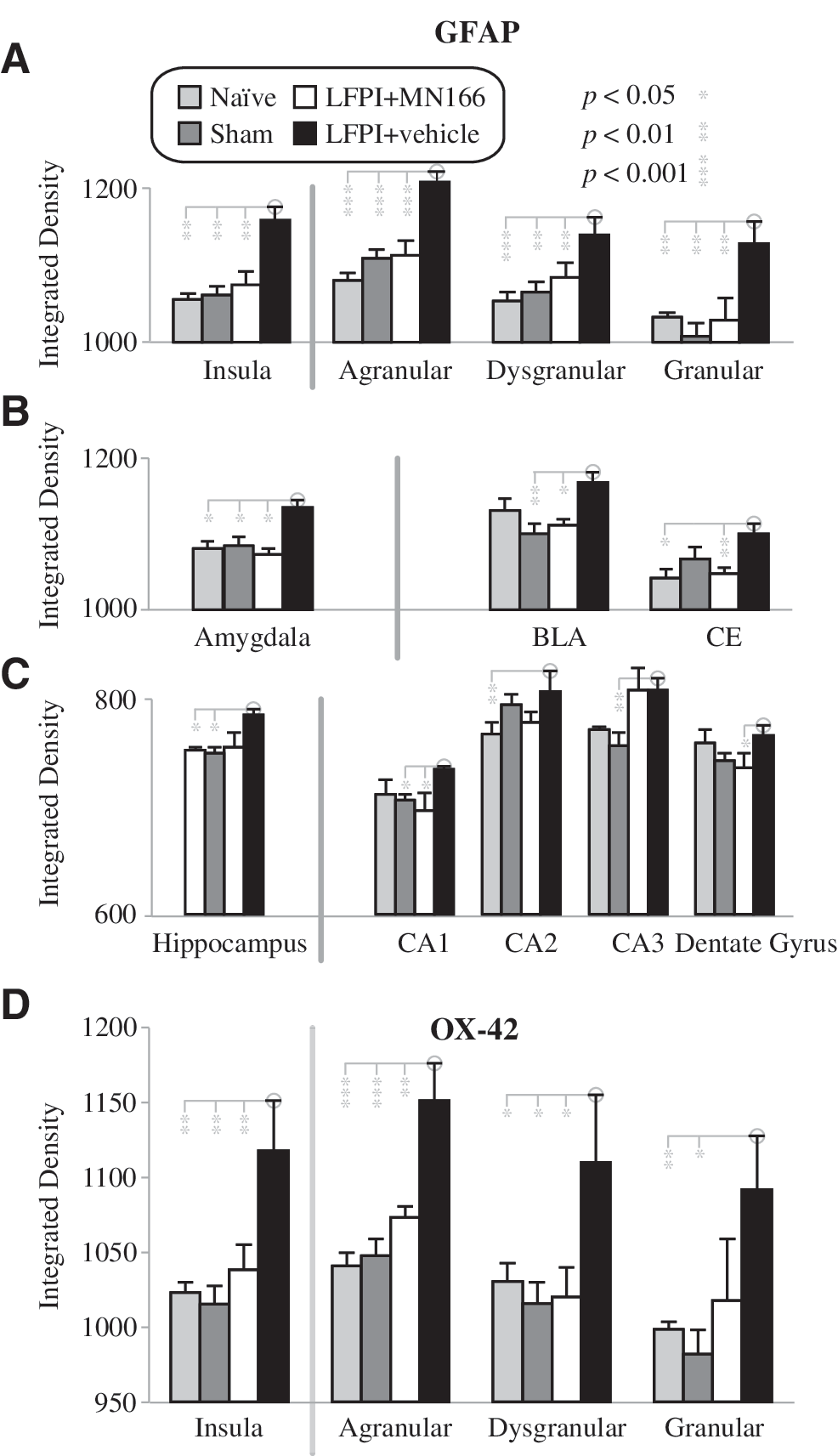
Regional and sub-regional analyses of microglial and astroglial activation in the hippocampus, amygdale, and insula at 3 months post-injury. (
Analysis of GFAP immunoreactivity in sub-regions of the insula (Fig. 4A, right bars), amygdala (Fig. 4B, right bars), and hippocampus (Fig. 4C, right bars), also revealed no differences between the surgically-naïve, sham-operated, and LFPI + MN166 groups. As in the regional analysis, LFPI + vehicle rats showed increased astrocyte activation over controls in most sub-regions examined. In the insula, LFPI + vehicle rats showed significantly increased GFAP labeling in agranular [F(3,19)=16.778, p<0.0001], dysgranular [F(3,19)=6.042, p<0.005], and granular [F(3,19)=5.277, p<0.008] regions, compared to control groups. In the amygdala, GFAP labeling in LFPI + vehicle rats was significantly increased in the basolateral amygdala (BLA) [F(3,18)=4.050, p<0.023] and central amygdala (CE) [F(3,18)=5.012, p<0.011] nuclei, compared to controls. LFPI + vehicle rats also showed increased GFAP expression in the hippocampus, but this was only significant in CA3 [F(3,18)=3.810, p<0.03], and approached significance in CA1 [F(3,17)=3.234, p=0.055].
LFPI + vehicle rats also showed significantly increased microglia activation compared to control groups as measured by OX-42 labeling, but this was restricted to the insula [Fig. 4D; F(3,19)=5.59, p<0.007]. Analysis of sub-regions of the insula also revealed increases in microglial activation for LFPI + vehicle rats, and post-hoc comparisons showed that LFPI alone significantly increased OX-42 labeling in agranular [F(3,19)=11.186, p<0.0001] and granular [F(3,18)=3.740, p<0.03] areas, and that it approached significance [F(3,19)=2.742, p<0.072] in dysgranular areas. No differences in OX-42 labeling were observed between the surgically-naïve, sham-operated, and LFPI + MN166 groups, in any insular regions examined. No significant between-group differences were found in OX-42 expression for the amygdala or hippocampus.
Discussion
These data suggest a link between injury-induced brain inflammation and post-traumatic anxiety. Rats with LFPI display freezing responses to the minor stress of a novel environment that is 2–3 times normal, and which unlike controls, is nearly doubled by the delivery of a major foot-shock stressor. LFPI also results in marked reactive gliosis in brain regions associated with anxiety. The possibility that post-traumatic brain inflammation and gliosis may contribute to the anxiety-like behavior observed here is supported by the effects of the glial-cell activation inhibitor MN166. MN166 reduces reactive gliosis and TBI-induced freezing behavior, rendering these animals histologically and behaviorally indistinguishable from naïve and sham-operated controls. To our knowledge, this is the first study to report pharmacological immunosuppression resulting in the reduction of anxiety-like behaviors following TBI.
A possible mechanism for neuroimmune-induced post-traumatic anxiety
Our finding of prolonged reactive gliosis in brain structures including, but likely not confined to, the hippocampus, amygdala, and insular cortex, suggests that these structures may contribute to the persistent enhanced freezing of our brain-injured animals in reaction to a novel environment. All three structures have been implicated in rodent research investigating the pathogenesis of anxiety (Canteras et al., 2010; Davidson, 2002; Davis, 1992; Davis et al., 1994; Paulus and Stein, 2006; Rauch et al., 2006; Vyas et al., 2004) and fear behavior in the rat (Liu et al., 2010; Milad et al., 2009; Rosen and Donley, 2006; Sullivan, 2004).
The mechanisms by which immune responses may contribute to dysfunction of these structures remain to be determined. It is well established that LFPI in the rat results in activation of microglia and astrocytes as part of the innate immune response to insult. A number of studies indicate that LFPI-induced reactive gliosis follows a distinct time course, beginning with predominant microglia activation that peaks within a week (Clausen et al., 2009; Grady et al., 2003; Gueorguieva et al., 2008; Hill et al., 1996; Nonaka et al., 1999; Yu et al., 2010), but continues for several weeks and overlaps later with persistent astrocytic activation (D'Ambrosio et al., 2004; Yu et al., 2010). Microglia are resident macrophages and first responders to pathogens and neuronal insults in the CNS. They react rapidly, leading to activation of astrocytes and prolonged disruption of neuronal function (Herber et al., 2006; Iravani et al., 2005; Zhang et al., 2009,2010). Several lesion paradigms have also shown a rapid microglial response, followed by delayed astrocyte reaction (Dusart and Schwab, 1994; Frank and Wolburg, 1996; Gehrmann et al., 1991; Liberatore et al., 1999; McCann et al., 1996).
Our results support this well-documented temporal relationship, suggesting that microglial activation precedes astrocytic activation and plays a role in the onset and maintenance of astrogliosis (Graeber and Kreutzberg, 1988; Hanisch, 2002; Herber et al., 2006; Iravani et al., 2005; McCann et al., 1996; Zhang et al., 2010). This time course is consistent with behavioral freezing responses in the present study, appearing rapidly within 2 weeks, but persisting unabated for the 3-month post-injury measurement period. It is also consistent with our immunohistochemistry results, indicating injury-induced astrocytic activation in all three regions of interest, the insula, amygdala, and hippocampus, at 3 months post-injury, but less activation of microglia, which was only significant in the insula. The lower levels of microglia expression are likely due to assessment at 3 months post-injury.
Trauma-related reactive gliosis is well known to result in the release of high levels of proinflammatory cytokines, specifically tumor necrosis factor-α (TNF-α; Fan et al., 1996; Lloyd et al., 2008; Taupin et al., 1993), interleukin-1β (IL-1β; Fan et al., 1995; Fassbender et al., 2000; Lloyd et al., 2008; Taupin et al., 1993; Yan et al., 2002), and interleukin-6 (IL-6; Lloyd et al., 2008; Taupin et al., 1993; Yan et al., 2002), which are central mediators of neuroinflammation following head injury (Fan et al., 1995,1996; Rothwell and Hopkins, 1995; Rothwell and Strijbos, 1995; Simi et al., 2007). Release of these proinflammatory cytokines, particularly IL-1β and TNF-α, pathologically increases neuronal excitability in all brain regions where it has been measured (Beattie et al., 2010; Maroso et al., 2010; Riazi et al., 2008; Rodgers et al., 2009; Schafers and Sorkin, 2008). While neuronal excitability and proinflammatory cytokine levels were not measured in the present study, neuroinflammation has been implicated in neuronal excitability of the amygdala and insular cortex and anxiety-like behavior by others using c-Fos labeling (Abrous et al., 1999; Ikeda et al., 2003; Kung et al., 2010). These same regions have also consistently been reported to be hyperexcitable in human imaging data across a variety of anxiety disorders (Carlson et al., 2011; Rauch et al., 1997; Shin and Liberzon, 2010; Shin et al., 2006; Simmons et al., 2006; Stein et al., 2007).
Attenuation of post-traumatic anxiety with MN166
Meta-analysis of the impact of pharmacological treatments on behavioral, cognitive, and motor outcomes after TBI in rodent models (Wheaton et al., 2011) indicates that of 16 treatment strategies evaluated to date, improved cognition and motor function have been reported, but few treatments have improved behaviors related to psychiatric dysfunction in general, and anxiety in particular. Exceptions to this are recent promising reports of treatments such as magnesium sulfate to limit excitotoxic damage (Fromm et al., 2004; O'Connor, 2003; Vink et al., 2003), and resveratrol to limit excitotoxicity, ischemia, and hypoxia (Sönmez et al., 2007), both increasing open-field exploration (resulting from decreased freezing), and therefore presumably decreasing post-injury anxiety.
Glial-targeted immunosuppression has also been found to be neuroprotective following TBI in rodents, resulting in increased structural preservation and improved functional outcomes (Hailer, 2008), including recent reports that MN166 significantly attenuated brain edema formation, cerebral atrophy, and apoptosis in neuronal cells following ischemic brain injury in rats, increasing neuronal survival rates (Lee et al., 2011). MN166 may reduce neuronal damage in regions involved in anxiety, mitigating the role of glial activation, neurotoxicity, and hyperexcitability in the subsequent development of anxiety-like behaviors. While not focused on post-traumatic anxiety, MN166 has been found to reduce intracellular calcium accumulation (Yanase et al., 1996), apoptosis, functional damage, and passive avoidance behaviors, following a transient ischemia model in rats (Yoshioka et al., 2002). Increasing evidence supports neuroinflammation, chronic inflammatory responses, proinflammatory cytokines, neuronal hyperexcitability, and secondary injury cascades in the pathophysiology of post-traumatic anxiety. The mechanisms of the effect of MN166 on TBI-induced anxiety-like behavior are not fully known. However, the results of this study provide evidence of a neuroprotective role for MN166 in attenuating and perhaps preventing development of post-traumatic anxiety.
Further exploration of the relationship between TBI, neuroimmune responses, neurocircuitry, and anxiety disorders, is important to better understand the sequelae of TBI, and will aid in the development of effective treatment strategies. The development of anxiety disorders following TBI is a complex and multifaceted problem, and finding treatments that work will require multifaceted approaches. The injury itself initiates many complex biological events, including glial activation, breakdown of the blood–brain barrier, excitotoxicity, and chronic neuroinflammation. While the primary injury often cannot be prevented, it may be possible to reduce the sequelae of secondary injury, leading to better functional and behavioral recovery following TBI. The present results, using peri-injury treatment with MN166 to prevent post-traumatic freezing behavior, not only suggest a role for neuroimmune inflammation in anxiety physiology, but similarly successful results with post-injury treatment could result in clinically-useful new agents to prevent post-traumatic anxiety in humans.
Footnotes
Acknowledgments
This work was supported by U.S. Army Medical Research and Material Command grant PR100040, the Craig Hospital Gift Fund, a University of Colorado Innovative Seed Grant, Autism Speaks Pilot Study grant 7153, National Institutes of Health grant NS36981 to D.S.B., and National Institutes of Health grants DA024044, DA01767 to L.R.W.
Author Disclosure Statement
Kirk W. Johnson is chief science officer of MediciNova Inc., the pharmaceutical firm providing MN166 for this research. No other competing financial interests exist.