
Abstract
Traumatic brain injury (TBI) is a major cause of acquired cognitive disability in childhood. Such disability may be blunted by enhancing the brain's endogenous neuroprotective response. An important endogenous neuroprotective response is the insulin-like growth factor-1 (IGF-1) mRNA variant, IGF-1B. IGF-1B mRNA, characterized by exon 5 inclusion, encodes the IGF-1 and Eb peptides. IGF-1A mRNA excludes exon 5 and encodes the IGF-1 and Ea peptides. A region in the human IGF-1B homologue acts as an exon-splicing enhancer (ESE) to increase IGF-1B mRNA. It is not known if TBI is associated with increased brain IGF-1B mRNA. Epigenetic modifications may underlie altered gene expression in the brain after TBI. We hypothesized that TBI would increase hippocampal IGF-1B mRNA in 17-day-old rats, associated with DNA methylation and/or histone modifications at the promoter site 1 (P1) or exon 5/ESE region. Hippocampi from rat pups after controlled cortical impact (CCI) were used to measure IGF-1B mRNA, DNA methylation, and histone modifications at the P1, P2, and exon5/ESE regions. In CCI hippocampi, IGF-1B mRNA peaked at post-injury day (PID) 2 (1700±320% sham), but normalized by PID 14. IGF-1A peaked at PID 3 (280±52% sham), and remained elevated at PID 14. Increased IGF-1B mRNA was associated with increased methylation at P1, and increased histone modifications associated with gene activation at P2 and exon5/ESE, together with differential methylation in the exon 5/ESE regions. We report for the first time that hippocampal IGF-1B mRNA increased after developmental TBI. We speculate that epigenetic modifications at the P2 and exon 5/ESE regions are important in the regulation of IGF-1B mRNA expression. The exon 5/ESE region may present a means for future therapies to target IGF-1B transcription after TBI.
Introduction
T
While some therapeutic agents have improved cognitive outcome in the laboratory, translation to the clinical arena has been hampered by barriers such as ineffective or impractical administration. One way of overcoming such barriers may be to harness the endogenous production of neuroprotective factors produced by the injured brain. The insulin-like growth factor-1 mRNA variant, IGF-1B, is one such endogenously-produced neuroprotective factor.
IGF-1 plays an important role in the brain's endogenous response to injury (D'Ercole et al., 2002; Scheepens et al., 2000). IGF-1 promotes survival and proliferation of neurons and glia in the developing and adult brain (Aberg, 2010). In mature and immature rodents, either TBI (Li et al., 1998; Schober et al., 2010), or hypoxic ischemic injury (Beresewicz et al., 2010; Yan et al., 2006), increase brain IGF-1 expression. Indeed, blockade of endogenous IGF-1 activity in the adult brain worsened outcome after ischemia (Yan et al., 2006). IGF-1 administration improved histologic and functional outcomes in adult rodents after TBI (Kazanis et al., 2003; Lu et al., 2009; Rubovitch et al., 2010; Saatman et al., 1997), and in adult and neonatal rats after hypoxic-ischemic brain injury (Li et al., 2011; Lin et al., 2005,2009). Similarly, IGF-1 administration improved neurologic outcome in 21-day-old rat pups after lipopolysaccharide-induced brain injury (Cai et al., 2011). Thus, brain IGF-1 upregulation appears to be a neuroprotective response in the mature and immature brain.
A growing body of literature suggests that the IGF-1 mRNA variant IGF-1B is a particularly potent neuroprotective factor (Aperghis et al., 2004; Dai et al., 2010; Dluzniewska et al., 2005). Experimental brain ischemia increased IGF-1B mRNA levels in hippocampal regions resistant to ischemia, but not in CA1, several days before extensive CA1 neuronal death occurred. Further, administration of the Eb peptide (encoded by IGF-1B mRNA) increased neuronal survival after ischemic injury, from ∼ 12% to 74%, while administration of IGF-1 had little effect. Finally, in rat pup hippocampal cultures, the Eb peptide afforded greater protection against excitotoxic neuronal death than did IGF-1 (Dluzniewska et al., 2005). IGF-1B cDNA resulted in more than twice as much neuronal survival as IGF-1A (the predominant IGF-1 mRNA variant) gene transfer in rodent models of nerve avulsion (Aperghis et al., 2004) and neurodegenerative disease (Riddoch-Contreras et al., 2009). Little is known about the regulation of IGF-1B mRNA expression.
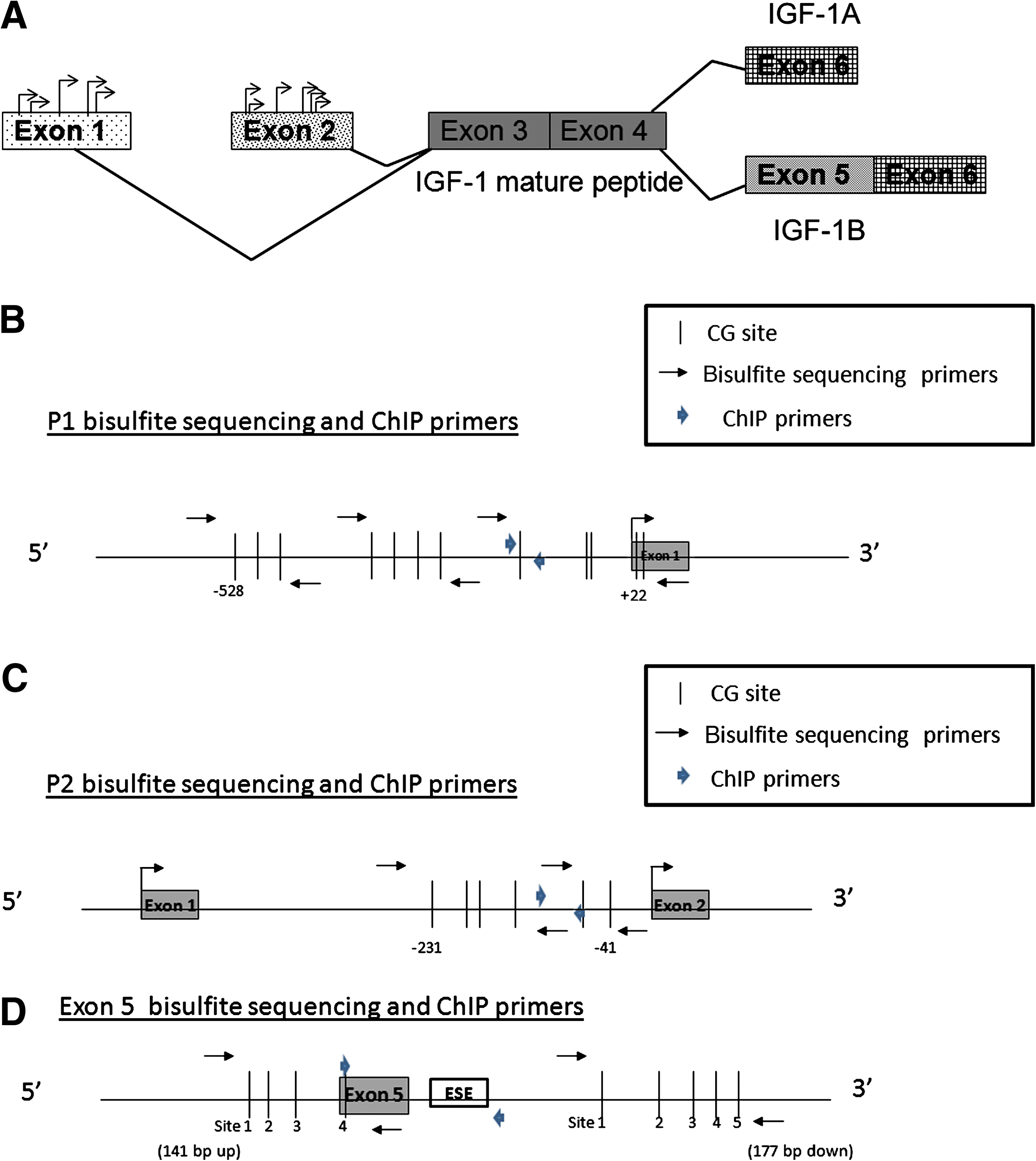
The IGF-1 gene, a highly conserved gene between rats, mice, and humans, is subject to alternative splicing and differential promoter usage (Rotwein et al., 1993), which generate multiple IGF-1 mRNA variants. All the known IGF-1 mRNA variants encode the mature IGF-1 peptide (Fig. 1A). IGF-1 promoter regions are located upstream of exon 1 and upstream of exon 2. IGF-1 mRNA translation generates a pre-pro-protein that is subsequently cleaved to release mature IGF-1 and a carboxy-terminal (E) peptide. IGF-1A and IGF-1B mRNA encode the Ea and Eb peptides, respectively. These E peptides facilitate entry of the mature IGF-1 peptide into neighboring cells (Pfeffer et al., 2009). The Eb peptide itself increases cell proliferation in various tissues (Siegfried et al., 1992; Yang and Goldspink, 2002). Unlike the Eb peptide, the Ea peptide has no known biological activity independent of its role in facilitating IGF-1 action (Pfeffer et al., 2009). As shown in Figure 1A, the different E peptides result from differential splicing of exons 5 and 6. The exon 5-containing variant (named IGF-1B in the rat) accounts for 1–10% of IGF-1 transcripts (Rotwein, 1986), while variants lacking exon 5 (IGF-1A in the rat) account for the majority of transcripts. IGF-1B corresponds to IGF-1Ec in the human (Dai et al., 2010). Recently, a purine-rich region downstream of exon 5 was found to act as an exon-splicing enhancer (ESE) for human IGF-1Ec (Smith et al., 2002). The homologous region in the rat IGF-1 gene, shown schematically in Figure 1D as the putative ESE, is therefore likely to a play a role in alternative splicing leading to IGF-1B mRNA expression in the rat.
(
IGF-1B alternative splicing may involve epigenetic regulation. Epigenetic regulation is the modification of gene expression via covalent modification of DNA and/or histone proteins, without altering the DNA sequence. DNA methylation and histone modifications have both been associated with alternative splicing (Anastasiadou et al., 2011; Lyko et al., 2010; Schwartz and Ast, 2010). In addition, the IGF-1 gene is heavily epigenetically regulated (Fu et al., 2009). Epigenetic modifications may play a role in IGF-1B mRNA regulation by making a particular promoter site more accessible to the transcription machinery, and/or making the ESE more accessible to the spliceosome complex.
An epigenetic approach towards understanding gene expression after experimental TBI is likely to provide useful insight into TBI physiology and potential therapeutics. Indeed, pathophysiologic insights and therapeutic approaches have been suggested by epigenetic studies of psychiatric diseases, cognitive disorders, stroke, and other brain pathologies (Molfese, 2011; Peter and Akbarian, 2011; Qureshi and Mehler, 2010). TBI studies thus far, while largely limited to global (not gene-specific) epigenetic modifications, have also suggested some therapeutic approaches. For example, TBI decreased global histone H3 acetylation in immature (Gao et al., 2008) and mature rat hippocampi (Zhang et al., 2008). Pharmacologic inhibition of histone deacetylation improved histologic (Zhang et al., 2008) and functional outcome after TBI (Shein et al., 2009). Finally, brain epigenetics can be modified by various medications (Dash et al., 2009,2010), and dietary/hormonal interventions (McCarthy et al., 2009; Tremolizzo et al., 2002) that could be implemented in clinical TBI.
IGF-1 mRNA levels increased in the mature and immature brain after TBI (Li et al., 1998; Madathil et al., 2010; Schober et al., 2010). In rats, IGF-1B expression relative to IGF-1A is highest early in development (Beresewicz et al., 2010). Thus, the immature rat brain may have a greater potential to increase IGF-1B levels in response to injury. It is not known if TBI affects levels of IGF-1B mRNA in the brain, nor if IGF-1B mRNA expression is associated with gene-specific epigenetic modifications.
We hypothesized that in the immature hippocampus, TBI would be associated with increased IGF-1B mRNA levels. We also hypothesized that increased IGF-1B mRNA levels would be associated with a pattern of histone modifications and/or DNA methylation of the promoter or the exon 5/ESE region, suggestive of an active chromatin state. Such histone modifications include increased acetylation of histone H3 at lysines 9 and 14 (H3K9ac and H3K14ac), and increased trimethylation of histone H3 at lysines 36 and 4 (H3K36me3 and H3K4me3), at the exon 5/ESE region. While promoter usage has not been linked to any specific 5′ or 3′ mRNA variants, usage of promoter 1 should be associated with exon-1-containing mRNA transcripts. Given that exon-1 mRNA transcripts are considered more likely to have autocrine functions (Musaro et al., 2007), we also hypothesized that increased IGF-1B mRNA levels would be associated with decreased DNA methylation at the P1 promoter region.
To test our hypotheses, we performed controlled cortical impact (CCI) using 17-day-old male rats, an established model of developmental TBI (Schober et al., 2010). The rat CCI model, as first described by Dixon (Dixon et al., 1991), and later modified for use in 17-day-old rats by Adelson (Adelson et al., 1998), produces hippocampal neuronal death and impaired learning and memory (Robertson et al., 2007; Schober et al., 2010). All molecular studies were done using dissected hippocampi. Primers were designed for IGF-1A and IGF-1B mRNA level measurement at post-injury days (PID) 1, 2, 3, 7, and 14. These results guided the selection of PID 3 and 14 time points for epigenetic studies focused at the P1, P2, and exon 5 sites, as further detailed in the methods section.
Methods
Animals
All experimental protocols were approved by the Animal Care and Use Committee at the University of Utah, in accordance with U.S. National Institutes of Health (NIH) guidelines, and carried out at the University of Utah. All surgical procedures were performed using aseptic technique.
Briefly, male Sprague-Dawley rats were obtained from Charles River Laboratories (Raleigh, NC) on post-natal days (PND) 7–10. We chose to study only males to eliminate potential confounding effects of gender. Rats were housed in litters of 10 with the lactating dam until weaning on PND 21–23. After weaning, rats were housed 3–5 per cage and allowed free access to food and water. All cages were kept in a temperature- and light-controlled (12-h/12-h day/night) environment. The two experimental groups were designated CCI and sham groups. The rats were randomized to experimental group on the day of surgery, PND 17. In order to control for maternal rearing characteristics, randomization was distributed evenly within litters.
The rats underwent CCI or sham craniotomy on PND 17. Hippocampal tissue was collected at PID 1, 2, 3, 7, and 14 for molecular studies.
CCI procedure
On PND 17, rats undergoing CCI (n=3–5/litter) were anesthetized with 3% isoflurane for induction, followed by 2–2.5% isoflurane for the duration of surgical preparation, using a VetEquip Bench Top Isoflurane Anesthesia System (Pleasanton, CA). Core temperature was monitored via a rectal probe and maintained at 37±0.5°C using a servo-controlled heating pad. Oxygenation, heart rate, and respiratory rate were monitored via femoral probe pulse oximetry (Mousox®; Starr Life Sciences, Oakmont, PA).
The rat was placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). After shaving, prepping with povidone-iodine, and incising the scalp, a craniotomy (6×6 mm) was performed over the left parietal cortex (centered at a point 4 mm anterior and 4 mm lateral to the bregma) using a high-speed dental drill. Care was taken not to perforate the dura. A temperature probe (2.28 mm outside diameter; Physiotemp Corp., Clifton, NJ) was placed through a burr hole into the left frontal lobe, and brain temperature was maintained at 36±0.5°C. Once the craniotomy was complete, anesthesia was reduced to 1% isoflurane for a 5-min equilibration period. CCI was then delivered (Pittsburgh Precision Instruments, Pittsburgh, PA) to the left parietal cortex (5-mm rounded tip, 4-m/sec velocity, 2-mm deformation, and 100-msec duration; Dixon et al., 1991). Immediately after CCI, isoflurane was increased to 2–2.5%, and the bone flap was replaced and secured with dental cement (Patterson Dental, Salt Lake City, UT). The scalp incision was sutured closed, and triple antibiotic ointment and bupivicaine 0.25% were applied topically to provide local antibiotic and analgesic therapy. Isoflurane was stopped, and the rats were allowed to recover in a temperature-controlled chamber. Once fully awake, the rats were returned to their dams and littermates. Sham rats (n=3–5/litter) underwent identical surgical craniotomy, equilibration, and closure procedures, without CCI.
Tissues
The rats were anesthetized with IP xylazine (8 mg/kg) and ketamine (40 mg/kg), and killed by swift decapitation (n=7–8 per group). After brain removal, the hippocampus was quickly dissected on ice. The left-sided (on the same side as the injury) and right-sided hippocampi from CCI and sham animals were studied separately, as their responses to trauma could be different. Tissue was snap-frozen in liquid nitrogen and stored at −80°C.
RNA isolation and real-time RT-PCR
Hippocampal levels of IGF-1A and IGF-1B mRNA were measured by real-time reverse-transcriptase polymerase chain reaction (RT-PCR). In brief, total RNA was extracted from ground frozen rat hippocampi using the RNeasy Mini Kit (Qiagen, Valencia, CA), treated with DNase I (Ambion, Austin, TX), and quantified by spectrophotometry (Nano-Drop ND-1000; NanoDrop Technologies, Wilmington, DE). Sample integrity was confirmed by gel electrophoresis. cDNA was synthesized from 2 μg of DNase-treated total RNA.
Primer and probe sets for IGF-1A and IGF-1B mRNA variants (Table 1) were designed using Primer Express (Applied Biosystems, Foster City, CA), with the reporter dye FAM and the quencher dye TAMRA. All primers designed were confirmed by PCR and sequencing. Probe and primers were added to Taqman Universal PCR master mix (Applied Biosystems). For each set of reactions, samples were run in quadruplicate. Cycle parameters were 50°C for 2 min, 95°C for 10 min, and then 40 cycles at 95°C for 15 sec and 60°C for 60 sec. Relative quantification of PCR products was based on value differences between the target and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control by the comparative threshold cycle method (Taqman Gold RT-PCR manual; Applied Biosystems).
The listed primers (forward and reverse) were used for real-time RT-PCR, ChIP, and bisulfite sequencing. The listed probes were used for real-time RT-PCR and ChIP for IGF-1A, IGF-1B, GAPDH, P1 (promoter 1), P2 (promoter 2), and exon 5 regions.
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; RT-PCR, reverse-transcriptase polymerase chain reaction; IGF-1A, insulin-like growth factor-1A; IGF-1B, insulin-like growth factor-1B; ChIP, chromatin immunoprecipitation.
DNA isolation and sodium bisulfite sequencing
Genomic DNA was extracted from ground frozen rat hippocampi by overnight proteinase K digestion at 56°C (10 mM Tris [pH 7.6], 25 mM EDTA, 75 mM NaCl, 1% SDS, and 180 μg/mL proteinase K), and centrifugation at 13,000 rpm. Supernatant was vortexed briefly with a saturated NaCl solution, and an equal volume of chloroform was added to each tube. Cold absolute ethanol was added in a 2:1 ratio to the supernatant and re-spun. The pellet was washed with graded series of ethanol dilutions, and the resultant DNA was dried and resuspended. One microliter of RNase (20 mg/mL) was added to each tube and incubated at 37°C for 30 min. DNA was quantified by a NanoDrop spectrometer (ND-1000; NanoDrop Technologies), and subjected to sodium bisulfite modification according to the manufacturer's protocol (CpGenome DNA modification kit; Chemicon International, Temecula, CA) to determine site-specific CG (segments in the DNA sequence where cytosine is next to guanine) methylation. We elected to analyze CG sites that were upstream of the longest known 5′ UTR of transcripts from each promoter. Relative to the cDNA clone accession M15647, the 12 CG sites from −528 to +2 on genomic DNA in promoter site 1 (P1) were determined with 3 primer sets (Table 1 and Fig. 1B). Relative to the cDNA clone accession no. NM_178866, the 6 CG sites from −231 to −41 on genomic DNA in P2 were determined with 2 primer sets (Table 1 and Fig. 1C). Relative to the cDNA clone accession no. NM_001082477, the 4 CG sites on genomic DNA 141 bp upstream of exon 5 initiation, and the CG site within exon 5, were determined with the first primer set (Table 1 and Fig. 1D), while the 5 CG sites on genomic DNA 167 bp downstream of exon 5 initiation were determined with the second primer set (Table 1 and Fig. 1D). The PCR condition for each primer set was determined as described previously (Fu et al., 2009). Briefly, PCR conditions for the primers were 95°C for 10 min, followed by 94°C for 30 sec, annealing at 53°C (sets 1, 4, and 5) or 54°C (sets 2 and 3) for 30 sec, and 72°C for 30 sec for 35 cycles. For each group, 6 animals were analyzed by bisulfite sequencing. The PCR products from bisulfite-treated genomic DNA were cloned into the vector pSC-A (Stratagene, Cedar Creek, TX). Six to eight colonies from each PCR cloning were screened and sequenced according to the manufacturer's instructions for double-stranded plasmid DNA with the BigDye Teminator v3.1 Cycle Sequencing kit (Applied Biosystems).
Chromatin immunoprecipitation assay and real-time PCR
Chromatin immunoprecipitation (ChIP) with anti-acetyl H3/K9, anti-acetyl H3/K14, anti-trimethyl H3/K4 (Millipore, Billerica, MA), anti-dimethyl H3/K9, anti-trimethyl H3/K9, anti-trimethyl H3/K27, and anti-trimethyl H3/K36 (Abcam, Cambridge, MA), was performed as described previously (Fu et al., 2006). Real-time PCR was used to quantitate the amount of DNA from the IGF-1 P1, P2, and exon 5 regions. A nontranscribed, intergenic sequence 250 kb upstream of the IGF-1 gene was used as an internal control, as previously described (Fu et al., 2009), and is shown in Table 1. Relative to the cDNA clone accession numbers M15647, NM 178866, and NM 001082477, three sets of primers and probes for ChIP (Table 1) were designed to detect the proximal promoter regions of exon 1 and exon 2, and the region encompassing exon 5 and the ESE sequence (ACCCAGGAGGGGAACAGG, the region homologous to the human IGF-1Ec ESE), as shown schematically in Figure 1.
Statistical analysis
All data were expressed as mean±standard error of the mean (SEM). To simplify graphical presentation, histone modification data were presented as the percentage of sham animals. For real-time RT-PCR, statistical significance was calculated using four groups: sham (left- and right-sided hippocampi), and CCI (left- and right-sided hippocampi). No differences in mRNA levels were found between the two sham hippocampi and CCI right-sided hippocampi. Therefore subsequent DNA methylation and histone modification studies were analyzed using only two groups: sham (left-sided, or ipsilateral to injury), and CCI (left-sided, or ipsilateral to injury) hippocampi. Real-time RT-PCR mRNA and ChIP data were analyzed using analysis of variance (ANOVA), while DNA methylation data were analyzed using Fisher's exact test (for small sample size) or chi-square test (for larger sample size), using Statview® software (SAS Institute, Cary, NC). A value of p<0.05 was considered statistically significant.
Results
IGF-1A and IGF-1B mRNA
Real-time RT-PCR was used to compare mRNA levels of IGF-1A and IGF-1B between CCI and sham hippocampi after injury. CCI data are shown as percentages of the sham group. GAPDH mRNA expression did not vary between groups at any time point.
IGF-1A and IGF1-B mRNA increased in CCI relative to sham animals as early as PID 1, as shown in Figure 2. IGF-1B peaked at PID 2 (1700±320% of sham), while IGF-1A peaked at PID 3 (280±52% of sham). Hippocampal expression of IGF-1A mRNA remained increased in CCI at least up to PID 14 (165±20% of sham), while the increase in IGF-1B mRNA ceased and became statistically non-significant at PID 14 (241±48% of sham; p=0.06).

Insulin-like growth factor-1A (IGF-1A) and IGF-1B mRNA results. IGF-1A and IGF-1B mRNA in the hippocampus ipsilateral to controlled cortical impact (CCI) was obtained at post-injury days (PID) 1, 2, 3, 7, and 14, and are presented as a percentage of sham mRNA levels. The dashed line represents 100% of sham mRNA. CCI increased mRNA levels of IGF-1B during the first 7 days after injury, while IGF-1A remained high at 14 days after injury. Results are presented as percentages of sham mRNA±standard error of the mean (n=7–8 per group; *p<0.05 relative to sham levels).
Of note, IGF-1B mRNA levels in the injured cortex of CCI rats increased relative to sham levels, to 450±100% of shams (p<0.05, n=5–7/group, unpublished data), suggesting that the increase in brain IGF-1B mRNA after TBI is not limited to the hippocampus.
DNA methylation
Bisulfite sequencing was used to compare CG methylation status between CCI and sham hippocampi at PID 3 and PID 14, using primer sets to target the P1, P2, and exon 5 regions, as shown in Figure 1B, C, and D.
Twelve CG sites were analyzed for methylation status within the P1 promoter region (upstream of exon 1) of the IGF-1 gene. Methylation at the P1 promoter region was increased in CCI rat hippocampi relative to shams at PID 3 (Fig. 3). At PID 14, total P1 methylation in CCI hippocampi was no different from shams (45.6% versus 50.7%, respectively).

Bisulfite sequencing results at the promoter site 1 (P1) region. Bisulfite sequencing, as defined by the bisulfite primer sets shown in Figure 1B, was performed on controlled cortical impact (CCI) and sham hippocampi at post-injury day (PID) 3. CCI results are shown as black bars, and sham results as gray bars. At PID 3, CCI increased total DNA methylation in the P1 region relative to shams. CCI did not affect DNA methylation at the P1 region relative to shams at PID 14. Results are presented as percentage methylation±standard error of the mean (n=7–8/group;
Six CG sites were analyzed for methylation status within the P2 promoter region (upstream of exon 2) of the IGF-1 gene. CG methylation at the P2 region was very low in both groups. Total P2 methylation in CCI hippocampi was no different from sham animals at PID 3 (1.3% versus 0.6%, respectively), or at PID 14 (0.5% versus 0.4%, respectively).
Nine CG sites were analyzed for methylation status in the exon 5 region: four sites upstream and within exon 5, and five sites downstream of exon 5. At PID 3, when IGF-1B mRNA levels were very high, DNA methylation increased in CCI animals relative to sham animals in the regions within and upstream of exon 5. At the same time point, DNA methylation decreased in CCI relative to sham animals downstream of exon 5 (Fig. 4). At PID 14, when IGF-1B mRNA levels had normalized, DNA methylation increased both upstream/within and downstream of exon 5 relative to shams in CCI hippocampi (Fig. 5).

Bisulfite sequencing at the exon 5 region at post-injury day (PID) 3. Bisulfite sequencing, as defined by the bisulfite primer sets shown in Figure 1D, was performed on controlled cortical impact (CCI) and sham hippocampi at PID 3. CCI results are shown as black bars, and sham results as gray bars. CCI decreased total DNA methylation in the region downstream of exon 5 at PID 3. In contrast, CCI increased total DNA methylation in the exon 5/exon 5 upstream region at PID 3. Results presented as percentage methylation±standard error of the mean (n=7–8/group;

Bisulfite sequencing at the exon 5 region at post-injury day (PID) 14. Bisulfite sequencing, as defined by the bisulfite primer sets shown in Figure 1D, was performed on controlled cortical impact (CCI) and sham hippocampi at PID 14. CCI results are shown as black bars, and sham results as gray bars. CCI increased total DNA methylation in the exon 5/exon 5 upstream, as well as in the exon 5 downstream region at PID 14. Results presented as percentage methylation±standard error of the mean (n=7–8/group;
Histone modifications
ChIP was used to compare the chromatin state, in terms of a specified set of histone modifications, associated with the DNA sequences of interest (the P1, P2, and exon 5 regions) between CCI and sham hippocampi. The first set of histone modifications chosen were histone H3 modified by acetylation at lysine 9 (H3K9ac), histone H3 modified by acetylation at lysine 14 (H3K14ac), histone H3 modified by trimethylation at lysine 6 (H3K36me3), and histone H3 modified by trimethylation at lysine 4 (H3K4me3), markers that are classically associated with gene activation. These “activating marks” are represented by lighter bars in Figure 6. The second set of histone modifications were markers classically associated with gene repression (histone H3 modified by trimethylation at lysine 9 [H3K9me3], histone H3 modified by dimethylation at lysine 9 [H3K9me2], and histone H3 modified by trimethylation at lysine 27 [H3K27me3]), represented by darker bars in Figure 6. All results are shown as percentages of sham values.
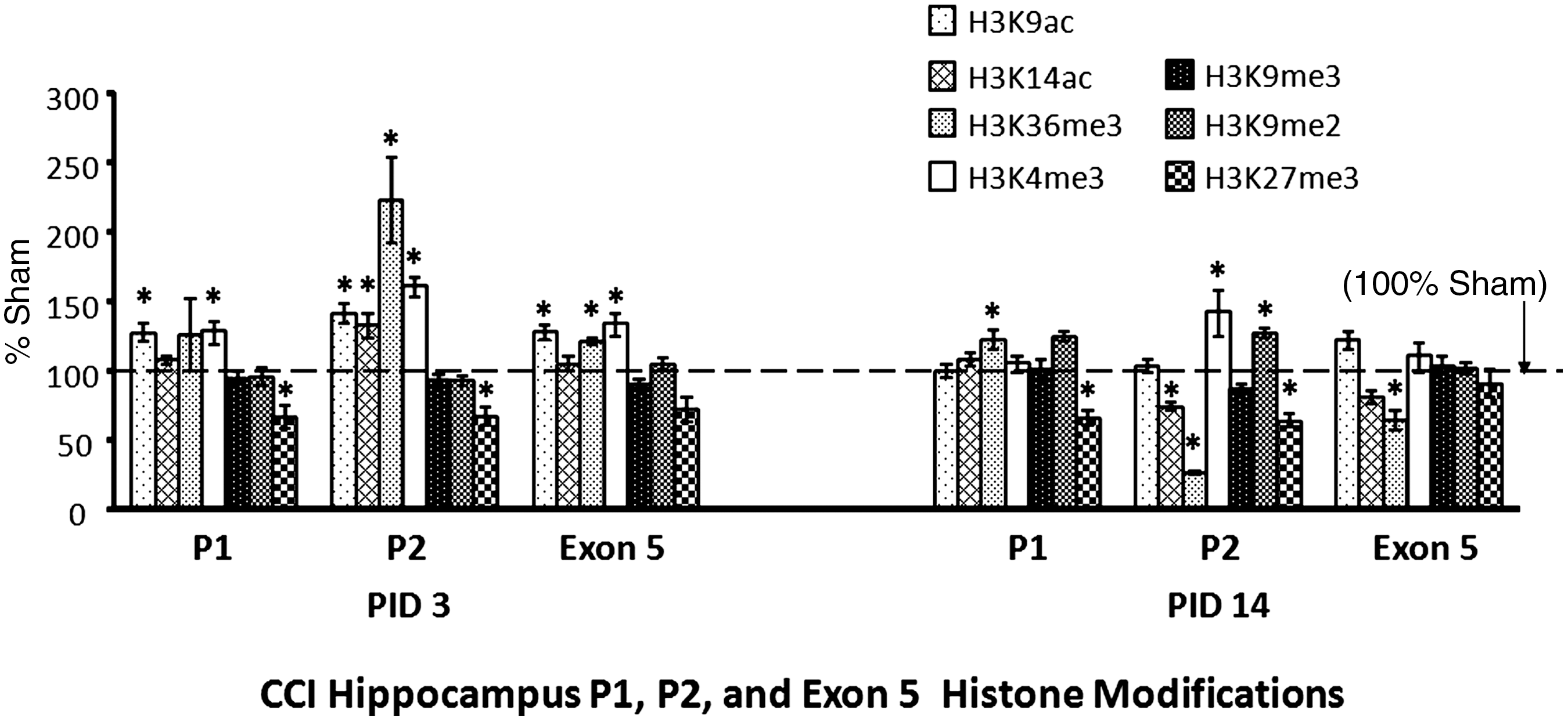
Histone modifications at the promoter sites 1 (P1) and P2 and exon 5 regions. Histone modifications associated with the P1, P2, and exon 5 regions were determined using chromatin immunoprecipitation (ChIP; as defined by the ChIP primer sets shown in Fig. 1B, C, and D) for controlled cortical impact (CCI) hippocampi, as percentages of sham levels, at post-injury days (PID) 3 and 14. The dashed line represents 100% of sham mRNA levels. The four histone modifications frequently associated with gene activation (histone H3 modified by acetylation at lysine 9 [H3K9ac], histone H3 modified by acetylation at lysine 14 [H3K14ac], histone H3 modified by trimethylation at lysine 6 [H3K36me3], and histone H3 modified by trimethylation at lysine 4 [H3K4me3]) are shown by light bars, while those commonly associated with gene repression (histone H3 modified by trimethylation at lysine 9 [H3K9me3], histone H3 modified by dimethylation at lysine 9 [H3K9me2], and histone H3 modified by trimethylation at lysine 27 [H3K27me3]) are shown by dark bars. In the P2 region, CCI increased H3K9Ac, H3K14Ac, H3K36me3, and H3K4me3 at PID 3. At PID 14, however, CCI decreased H3K14Ac and H3K36me3, and did not affect H3K9Ac. CCI continued to increase H3K4me3 at PID 14. Similarly, CCI increased H3K9Ac, H3K36me3, and H3K4me3 in the exon 5/exon-splicing enhancer region at PID 3. At PID 14, however, CCI decreased H3K36me3. Results presented as percentage of sham levels±standard error of the mean (n=7–8 per group; *p<0.05).
CCI increased hippocampal “activating marks” in the P1 region at both time points. Specifically, CCI increased H3K9ac and H3K4me3 occupancy relative to sham animals at PID 3, and increased H3K36me3 at PID 14.
CCI increased four “activating marks” in the P2 region (H3K9ac, H3K14ac, H3K36me3, and H3K4me3) at PID 3, but only one remained increased (H3K4me3) at PID 14. In contrast, at PID 14, two of these marks decreased (H3K14ac and H3K36me3), and another normalized (H3K9ac). In addition, H3K9me2 (a “repressing mark”) was increased in CCI rats at PID 14.
CCI increased three “activating marks” in the exon 5 region at PID 3 (H3K9ac, H3K36me3, and H3K4me3), while none of these remained increased at PID 14. In addition, CCI decreased an “activating mark” (H3K36me3) at PID 14. H3K27me3 (a mark associated with gene repression) did not change direction at any region over time: in the P1 and P2 regions, H3K27me3 was decreased at both time points; in the exon 5 region, H3K27me3 did not differ from shams at PID 3 or PID 14.
Discussion
Progressive cortical and hippocampal tissue loss beyond the first 7 days after TBI is well documented in the immature (Huh and Raghupathi, 2007) and the mature (Smith et al., 1997) brain, continuing even up to 1 year after injury. Such a broad window suggests that prolonging the duration of increased IGF-1B mRNA expression, and thereby the duration of increased Eb peptide production, may blunt neuronal loss after TBI. We speculate that the Eb peptide could not only promote neuronal survival after injury, but also neurogenesis via a mitogenic effect on neural stem cells, similarly to that described in other cell populations (Siegfried et al., 1992; Yang and Goldspink, 2002). It is of course possible, however, that Eb peptide activity late after injury could impede differentiation of neural stem cells. Thus further studies are needed to determine if the presence of the Eb peptide would be beneficial late after TBI.
When IGF-1B mRNA levels were very high, CCI differentially altered DNA methylation in the exon 5/upstream region relative to the downstream region. In contrast, CCI increased DNA methylation in both regions when IGF-1B mRNA levels had normalized. These data are in accord with studies that support a role for DNA methylation in alternative splicing (Lyko et al., 2010), and show that DNA methylation plays a role in marking a genomic sequence as an exon or intron (Chodavarapu et al., 2010). In agreement with a predicted ESE existing in the rat DNA region flanking exon 5 of the IGF-1 gene, Anastasiadou and colleagues reported that CG hypermethylation is frequent in alternatively-spliced sites, and that the methylation frequency was increased in sequences containing multiple putative ESEs (Anastasiadou et al., 2011). Another potential contribution of DNA methylation to IGF-1B mRNA alternative splicing may lie in the association between DNA methylation and the specific histone modifications outlined below, as supported by evidence that a dynamic interaction exists between DNA methylation and the histone code (Jin et al., 2011).
Specific histone modifications at the exon 5 region were associated with increased IGF-1B mRNA levels in CCI hippocampi. When IGF-1B mRNA levels were very high, CCI increased occupancy of the activating histone marks (H3K9ac, H3K36me3, and H3K4me3) in the exon 5 region. These findings suggest increased accessibility of the putative ESE sequence to the spliceosome. When IGF-1B mRNA levels had normalized, none of these activating marks remained increased, and one (H3K36me3) had instead decreased in CCI hippocampi. At the exon 5 site, CCI did not alter any of the histone marks that are associated with repression. In a recent review, Schwartz and associates state that splicing occurs co-transcriptionally and is regulated by transcription-related factors, such as specific histone modifications (Schwartz and Ast, 2010). Further studies are needed to determine if these histone modifications (in particular, those located at the ESE) are necessary for IGF-1B mRNA alternative splicing.
Results from our study suggest that the region homologous to the human ESE may play a role in IGF-1B mRNA upregulation in the rat brain after TBI. Smith and colleagues showed that this 18-nucleotide sequence significantly enhanced in vitro splicing to exon 5 in the presence of phosphorylated (active) alternative splicing factor/splicing factor 2 (SF2/ASF; Smith et al., 2002). The findings of Smith's group suggest that manipulation of IGF-1B mRNA expression may be possible in the future. In human muscle cells, growth hormone administration increased exon 5-containing IGF-1 mRNA 10-fold, suggesting that growth hormone may increase IGF-1B mRNA expression (Smith et al., 2002). In addition, pharmacologic control of SF2/ASF phosphorylation is currently possible (Pilch et al., 2001). While medications that alter epigenetic characteristics are still in their infancy, future drugs that can specifically alter some histone marks or DNA methylation patterns may hold promise for gene regulation.
Epigenetic modifications were not limited to the exon 5 region. When IGF-1B mRNA levels were very high, CCI increased DNA methylation at the P1 site. DNA methylation within a promoter and 5′ region usually represses gene expression (Pokholok et al., 2005), suggesting that transcription of exon 1-containing mRNA was inhibited. When IGF-1B mRNA levels were very high, CCI increased occupancy of histone marks typically associated with gene expression at the P2 site, suggesting that transcription of exon 2-containing mRNA was increased. No further speculation can be made, given the extensive number of transcription start sites in the exon 1 and 2 regions. Further, there is no known association between transcription start site and exon 5 usage. Finally, only one activating mark (H3K4me3) remained elevated at the P2 site when IGF-1B mRNA levels had normalized, and the others (H3K14Ac and H3K36me3) both decreased, and H3K9me2 (a mark associated with gene repression) increased. While no causality can be inferred, these results point to a possible role for the intronic region upstream of exon 2 (intron 1) in IGF-1B mRNA upregulation after injury.
The results of this study are novel and important. To our knowledge, this is the first report of IGF-1B mRNA expression in the brain after TBI. Our results are similar to the limited data available on brain IGF-1B mRNA expression after ischemia. Hippocampal IGF-1B mRNA increased significantly in the adult brain after hypoxic ischemic injury (Dluzniewska et al., 2005). Similarly, hypoxia-ischemia increased IGF-1B mRNA levels in the immature rat brain for the first 7 days after injury (Beresewicz et al., 2010). Interestingly, experimental evidence supports the contention that IGF-1B is more neuroprotective than the IGF-1A isoform (Aperghis et al., 2004), and that unlike IGF-1, its product acts independently of the IGF-1 receptor (Armakolas et al., 2010; Dluzniewska et al., 2005; Gorecki et al., 2007; Siegfried et al., 1992). This independence from the IGF-1 receptor is particularly interesting in light of the fact that we had previously found that IGF-1 receptor mRNA levels decreased early after CCI (Schober et al., 2010) in the 17-day-old male rat hippocampus. In addition, studies in muscle devoid of IGF-1 receptor showed that overexpression of IGF-1B mRNA resulted in activation of pathways both in common and different from those resulting from administration of the IGF-1 mature peptide (Barton et al., 2010). IGF-1B mRNA upregulation could be superior to direct administration of the Eb peptide or IGF-1 peptide alone. However, future studies are needed to determine if increased IGF-1B mRNA upregulation confers neuroprotection after TBI.
As previously reported, TBI is associated with global H3 hypoacetylation in the adult (Zhang et al., 2008) and immature rat brains (Gao et al., 2006), and with global DNA hypomethylation in the brain (Zhang et al., 2007). In contrast, our results are specific to a single gene. For example, we found that CCI was associated with hypermethylation at one region within the IGF-1 gene (exon 5 and upstream), and hypomethylation at the other (downstream of exon 5), in the rat pup hippocampus. Similarly, we found that CCI increased occupancy of H3K36me3 at the P1 region of the IGF-1 gene, but decreased occupancy of H3K36me3 at the P2 region. Finally, CCI did not decrease H3 acetylation throughout the IGF-1 gene. Instead, CCI increased H3 acetylation (i.e., H3K9ac and H3K14ac) in the P2 region at PID 3. Epigenetic changes in the IGF-1 gene are thus unlikely to represent non-specific effects of experimental TBI. Interestingly, epigenetic changes were not limited to the promoter region, emphasizing the importance of taking various gene regions into account.
While our results are limited to male rat pups, we speculate that increased IGF-1B mRNA expression in the brain is a generalizable response to injury throughout development. Indeed, brain IGF-1B mRNA increased both after TBI in the immature brain, and after hypoxic ischemic injury in the mature and immature brain (Beresewicz et al., 2010; Dluzniewska et al., 2005). Data in this study suggest that the 18-nucleotide region shown to be an ESE in the human also acts as an ESE for IGF-1B mRNA in the rat brain after TBI, but mechanistic studies are needed to demonstrate this conclusively. Similarly, the epigenetic associations are intriguing; further research is needed to determine if epigenetic regulation does indeed contribute to IGF-1B mRNA upregulation after TBI.
We report for the first time that hippocampal IGF-1B mRNA levels increase after developmental TBI in the rat, and speculate that it may play an important neuroprotective role in the developing brain after trauma. Increased IGF-1B mRNA levels after CCI were associated with epigenetic modifications that were not limited to the promoter region, but involved the exon 5 region and a putative exon-splicing enhancer as well. Increased use of promoter 2 may be important for IGF-1B upregulation, as may be chromatin modifications and differential DNA methylation in the exon 5 and downstream putative ESE region. While it is not known if IGF-1B mRNA or the Eb peptide is neuroprotective after TBI, our findings suggest that future research in this area is needed.
Footnotes
Author Disclosure Statement
No competing financial interests exist.