
Abstract
Cerebral ischemia is a well-recognized contributor to high morbidity and mortality after traumatic brain injury (TBI). Standard of care treatment aims to maintain a sufficient oxygen supply to the brain by avoiding increased intracranial pressure (ICP) and ensuring a sufficient cerebral perfusion pressure (CPP). Devices allowing direct assessment of brain tissue oxygenation have showed promising results in clinical studies, and their use was implemented in the Brain Trauma Foundation Guidelines for the treatment of TBI patients in 2007. Results of several studies suggest that a brain tissue oxygen–directed therapy guided by these monitors may contribute to reduced mortality and improved outcome of TBI patients. Whether increasing the oxygen supply to supraphysiological levels has beneficial or detrimental effects on TBI patients has been a matter of debate for decades. The results of trials of hyperbaric oxygenation (HBO) have failed to show a benefit, but renewed interest in normobaric hyperoxia (NBO) in the treatment of TBI patients has emerged in recent years. With the increased availability of advanced neuromonitoring devices such as brain tissue oxygen monitors, it was shown that some patients might benefit from this therapeutic approach. In this article, we review the pathophysiological rationale and technical modalities of brain tissue oxygen monitors, as well as its use in studies of brain tissue oxygen–directed therapy. Furthermore, we analyze hyperoxia as a treatment option in TBI patients, summarize the results of clinical trials, and give insights into the recent findings of hyperoxic effects on cerebral metabolism after TBI.
Introduction
T
Increased interest in therapeutic hyperoxia—the elevation of blood oxygen to supraphysiological levels under normobaric (NBO) or hyperbaric (HBO) conditions—has grown in recent years (Alves et al., 2004; Stover, 2008; Vespa, 2008). In vivo imaging and other techniques such as cerebral microdialysis have revealed new insights into the effects of hyperoxia on brain metabolism, and experimental results of this therapeutic approach have been promising (Nortje et al., 2008; Reinert et al., 2004, Rockswold et al., 2010; Rogatsky et al., 2005; Tolias et al., 2004; Vlodavsky et al., 2006; Wang et al., 2010). Nevertheless, a clear benefit for patients must be proven before this therapeutic strategy can be used in clinical practice. As oxygen itself has harmful properties (e.g., vasoconstriction and the formation of reactive oxygen species and radicals), an excessive supply could theoretically aggravate cerebral injuries.
It has been hypothesized that devices for monitoring brain tissue oxygenation are suitable tools to develop a brain tissue oxygen–directed approach to therapeutic hyperoxia. In this review we give an overview of the rationale of monitoring brain tissue oxygenation, the techniques involved, and give insights into the results of studies of brain tissue oxygen–directed therapy. Furthermore, we analyze the strategy of therapeutic hyperoxia, and summarize studies which have examined the impact of HBO and NBO on TBI patients.
Cerebral Oxygen Supply and Metabolism
The brain receives 20% of the cardiac output of blood, although its weight is on average only about 2% of the body's weight. This outstanding demand makes it highly susceptible to a lack of sufficient blood and oxygen supply with subsequent development of cellular hypoxia. Oxygen is inhaled and then transported from the lungs to the brain tissue within the bloodstream. Under normal atmospheric pressure most of the oxygen is bound to hemoglobin and the fraction of dissolved oxygen is very small. At the macroscopic level, the cardiovascular and respiratory systems as well as the amount of hemoglobin play major roles in maintaining the brain's oxygen supply. The oxygen is unloaded from red blood cells into the periphery according to the status of the hemoglobin dissociation curve. The form of this curve is unique and allows easy binding of oxygen to hemoglobin in the lungs, and easy dissolving in the peripheral tissues. The value of pH as well as temperature influence the status of the curve, so a shift of the curve to the right due to increasing acidity in the blood after passage through the tissues results in higher oxygen emission by red blood cells. Oxygen diffusion from capillaries to tissue occurs radially, and was described by Krogh (1919) in a simplified model in which capillaries are arranged in a parallel manner with each capillary supplying a cylindrical tissue volume (Fig. 1). Oxygen content is high at the proximal capillaries and decreases linearly. Fick's laws of diffusion state that the rate of oxygen diffusion is directly proportional to the tension gradient (in the brain this means partial arterial oxygen pressure [Pao2] minus partial oxygen pressure in the brain [P
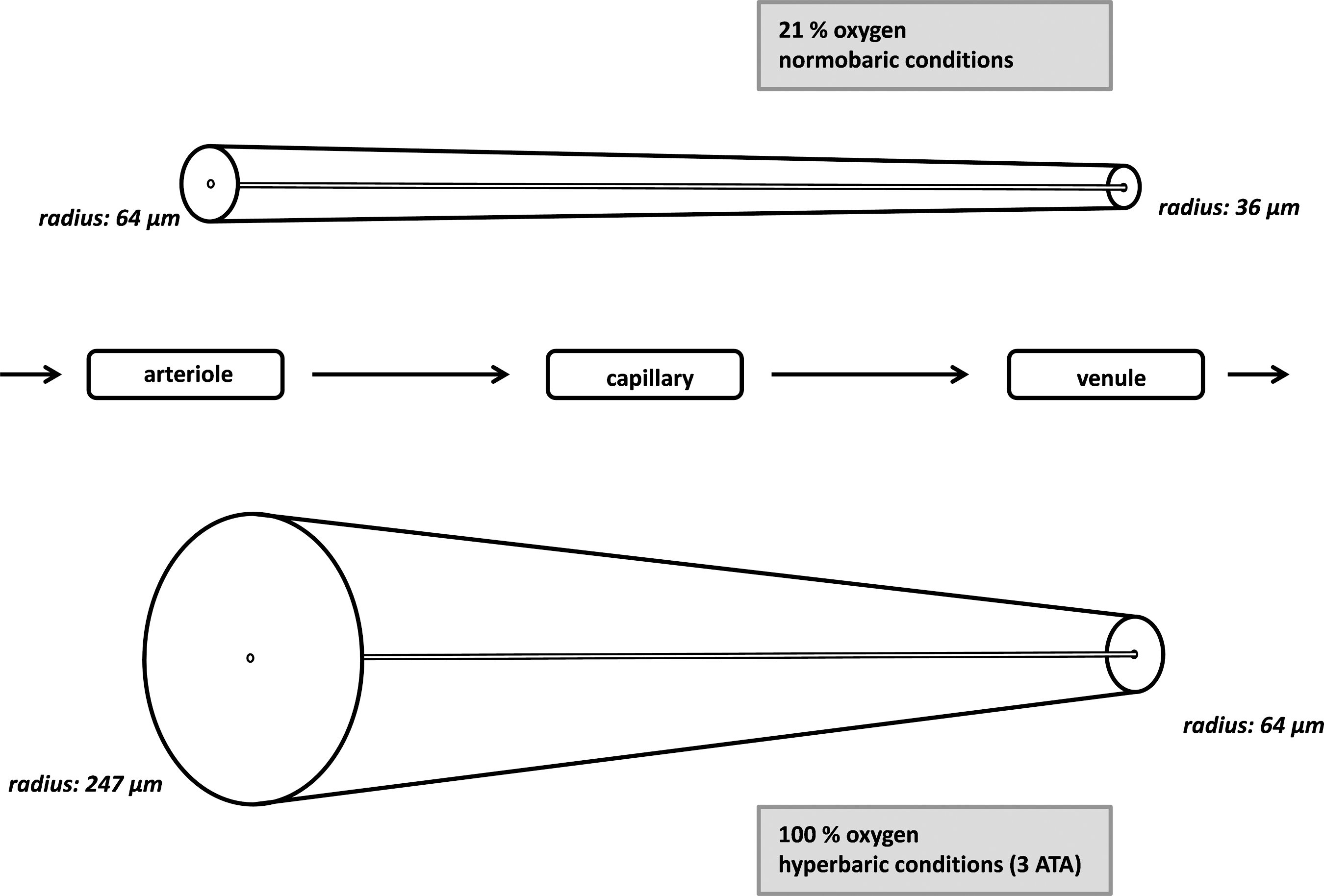
The cylindrical model described by Krogh (1919) demonstrates the association of oxygen tension and diffusion area in capillaries.
The oxygen delivered to cells is used in the aerobic metabolism of glucose through oxidative phosphorylation utilizing energy in form of adenosine triphosphate (ATP). The enzymatic reactions involved are localized in the mitochondria of brain cells. The cerebral metabolic rate of oxygen (CMRO2) reflects this function of mitochondrial activity. Under normal conditions, the normal value in humans is approximately 3.3 mL/100 mL/min (Ito et al., 2005). If there is a significant reduction of CBF or Pa
Pathophysiological changes in cerebral oxygen metabolism after traumatic brain injury
The damage to brain parenchyma caused by TBI is divided into two categories: Focal lesions (e.g., contusions and hemorrhage), caused by direct contact, and global (diffuse) lesions caused by acceleration/deceleration resulting in brain edema and diffuse axonal injury (Werner and Engelhard, 2007). Brain tissue damaged directly at initial trauma is classified as primary brain damage. It is considered irreversible damage to the brain, and therefore is not amenable to neuroprotective therapies. In contrast, secondary brain damage develops through pathological processes in the brain during the time period after the initial trauma. Although these pathological processes have not yet been fully elucidated, secondary brain damage can be influenced by therapeutic interventions, and is the primary objective of current research activities in the field of TBI.
Available data suggest that cerebral ischemia plays a key role in the development of secondary brain damage (Coles, 2004; Werner and Engelhard, 2007). In fact, post-mortem analysis of brain trauma victims showed signs of cerebral tissue infarction in 90% of cases (Graham et al., 1989). The critical threshold for CBF in TBI is considered to be 15 mL/100 mL/min (Cunningham et al., 2005), and alterations in CBF occur in the majority of patients with TBI. Cerebral ischemia after TBI leads to several pathophysiological alterations of cerebral metabolic pathways. The first, acute phase is mainly affected by impaired regulation of CBF, while later stages of the pathophysiological ischemic cascade are characterized by inflammatory processes, finally leading to necrotic or apoptotic cell death.
If ischemia occurs in brain tissue, anaerobic metabolism leads to an accumulation of pyruvate, which is used to regenerate cytoplasmic NADH from NAD+ via lactate dehydrogenase (anaerobic glycolysis). Increased production of lactate results in local acidosis. Ion hemostasis is disturbed, since active transporters, such as the Na+-K+-ATPase transporter, have a high demand for ATP. It is well recognized that cerebral hyperglycolysis commonly occurs after TBI (Bergsneider et al., 1997), and the results of several studies suggest that activation of glucose transporter systems influences cerebral oxygen consumption (Holbein et al., 2009). This may represent a protective mechanism of the brain, as a PET study of TBI patients revealed that brain regions with low OEF were associated with reductions in cerebral glucose metabolism (Abate et al., 2008). Importantly, cerebral hyperglycolysis also occurs in the absence of hypoxia. Cesarini and associates (2002) showed that in patients with aneurysmal subarachnoid hemorrhage and microdialysis monitoring, decreased extracellular concentrations of glucose, and balanced increases in lactate and pyruvate concentrations, were associated with a favorable outcome. Oddo and colleagues (2012) demonstrated that elevated concentrations of cerebral lactate in patients with subarachnoid hemorrhage were more often attributable to aerobic hyperglycolysis than to brain hypoxia (median: 78% versus 11%). Cerebral hyperglycolytic lactate predicted good recovery in these patients, supporting the hypothesis that lactate may also have neuroprotective properties in brain ischemia (Berthet et al., 2009). Cerebral hyperglycolysis in the recovery phase after TBI has been discussed as a mechanism of the brain to encounter the extreme metabolic demand and restore cerebral hemostasis (Cesarini et al., 2002). Therefore, increased production of cerebral lactate in TBI may indicate anaerobic metabolism, as well as cerebral hyperglycolysis augmenting the brain cells' energy supply. Persistent anaerobic metabolism leads to accumulation of sodium and chloride ions in the cell, leading to development of edema through osmotic water influx. In the subsequent phases of the ischemic cascade, cellular mediators including proinflammatory cytokines, prostaglandins, and free radicals are released, which induce chemokines and adhesion molecules. These inflammatory processes lead to infiltration of the injured tissue by cells of the immune system such as macrophages and T lymphocytes. Within hours to weeks, the injured tissue is replaced by scar tissue through production of microfilaments and neutropins by astrocytes (Werner and Engelhard, 2007).
Importantly, neural cells and mitochondria have effective mechanisms to cope with reduced oxygen supply. Experimental studies have shown that values of intracellular oxygen tension as low as 0.2 mm Hg enable mitochondria to sustain cellular respiration (Scheufler et al., 2002). In thromboembolic stroke, the widely accepted concept of the penumbra describes brain tissue which is impaired in functional activity due to an insufficient oxygen supply, but not irreversibly damaged (Fisher, 2004). Although there are substantial differences in the pathophysiology of ischemic stroke and TBI, there is increasing evidence for the existence of a “traumatic penumbra” (Abate et al., 2008; Coles et al., 2004). This brain tissue, also called “tissue at risk,” is most likely to suffer irreversible damage. Monitoring brain tissue oxygenation helps clinicians initiate adequate action when episodes of cerebral hypoxia in this tissue are identified.
Monitoring brain tissue oxygenation
There are several methods described to measure brain tissue oxygenation. Measurement of SjvO2 is a bedside technique allowing measurement of global brain oxygen saturation. An association between Sjv
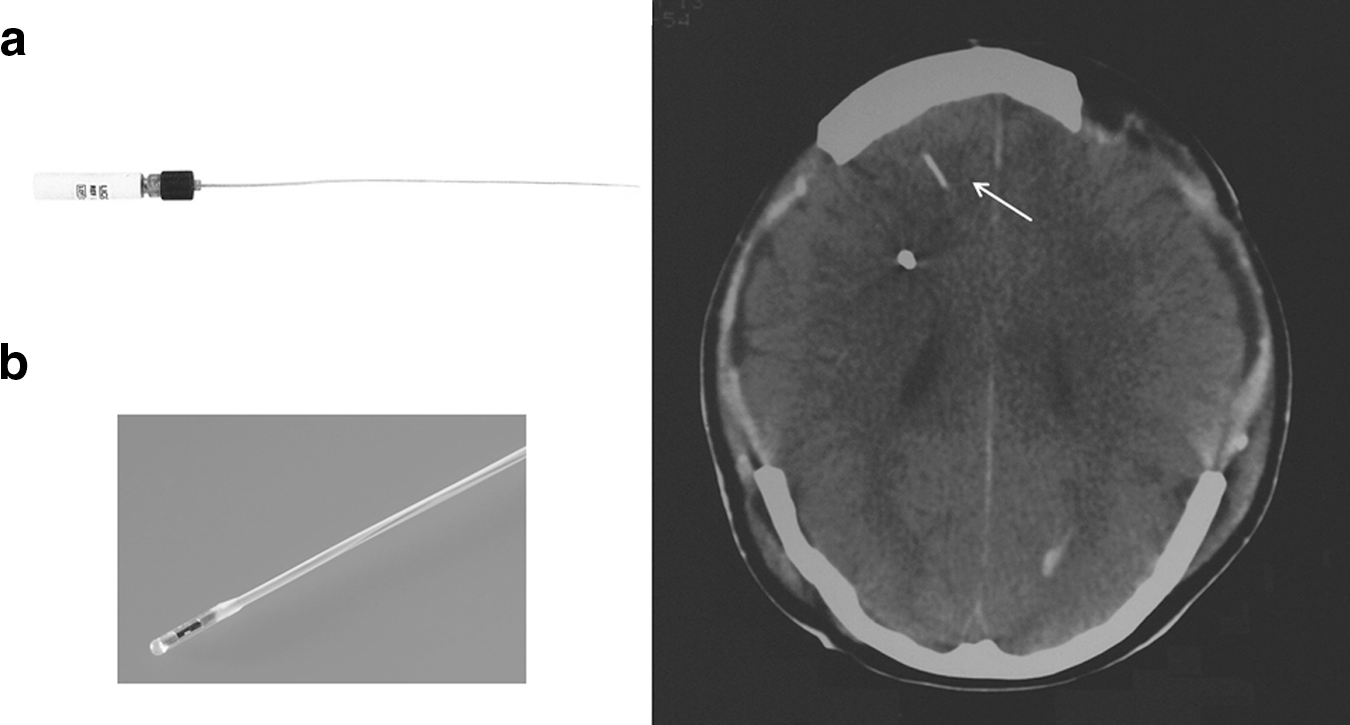
Left: Catheters for monitoring brain tissue oxygenation (
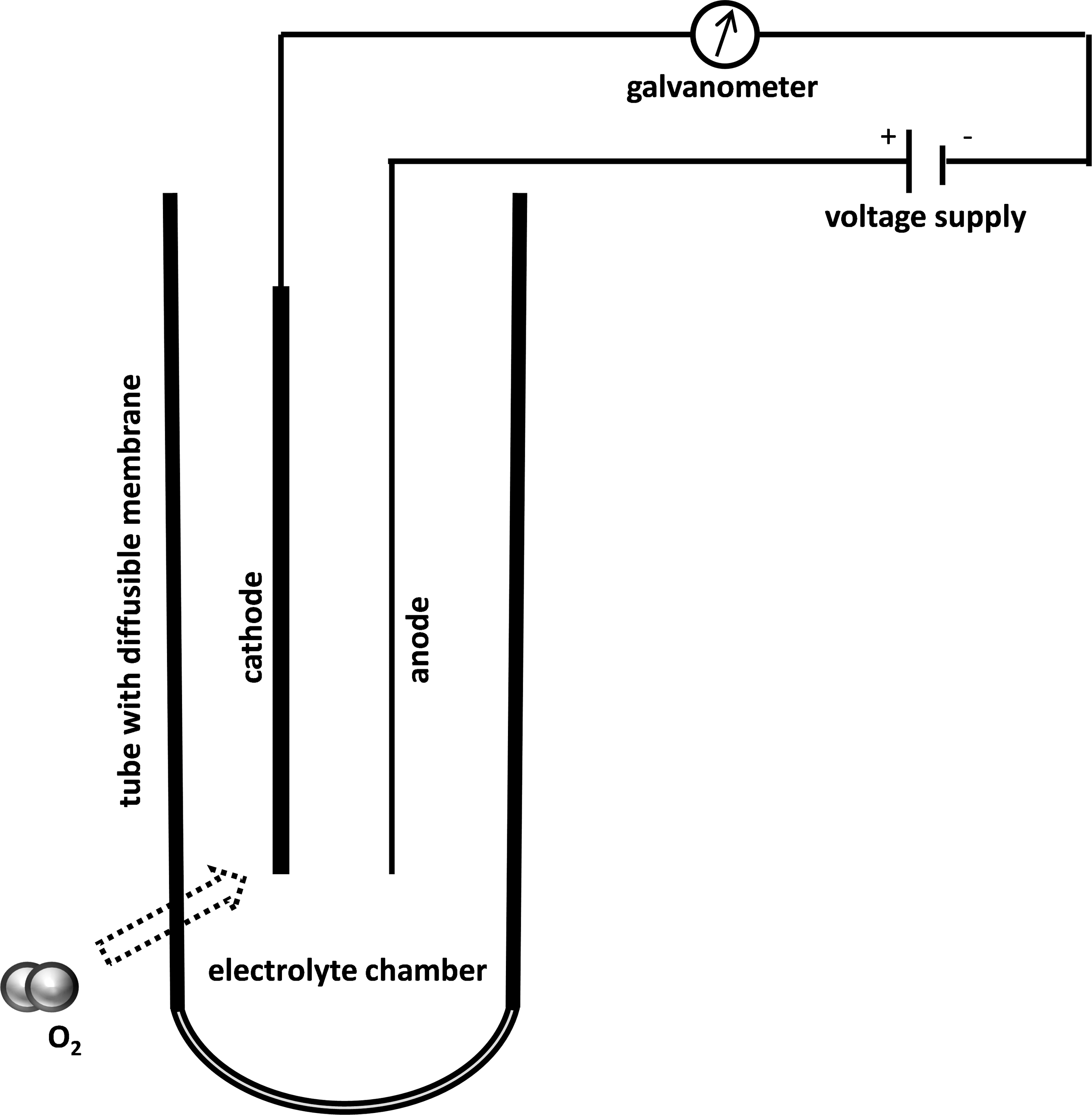
The Clark electrode measures oxygen on a catalytic surface. This technique is oxygen-consuming, but this has no impact in clinical practice.
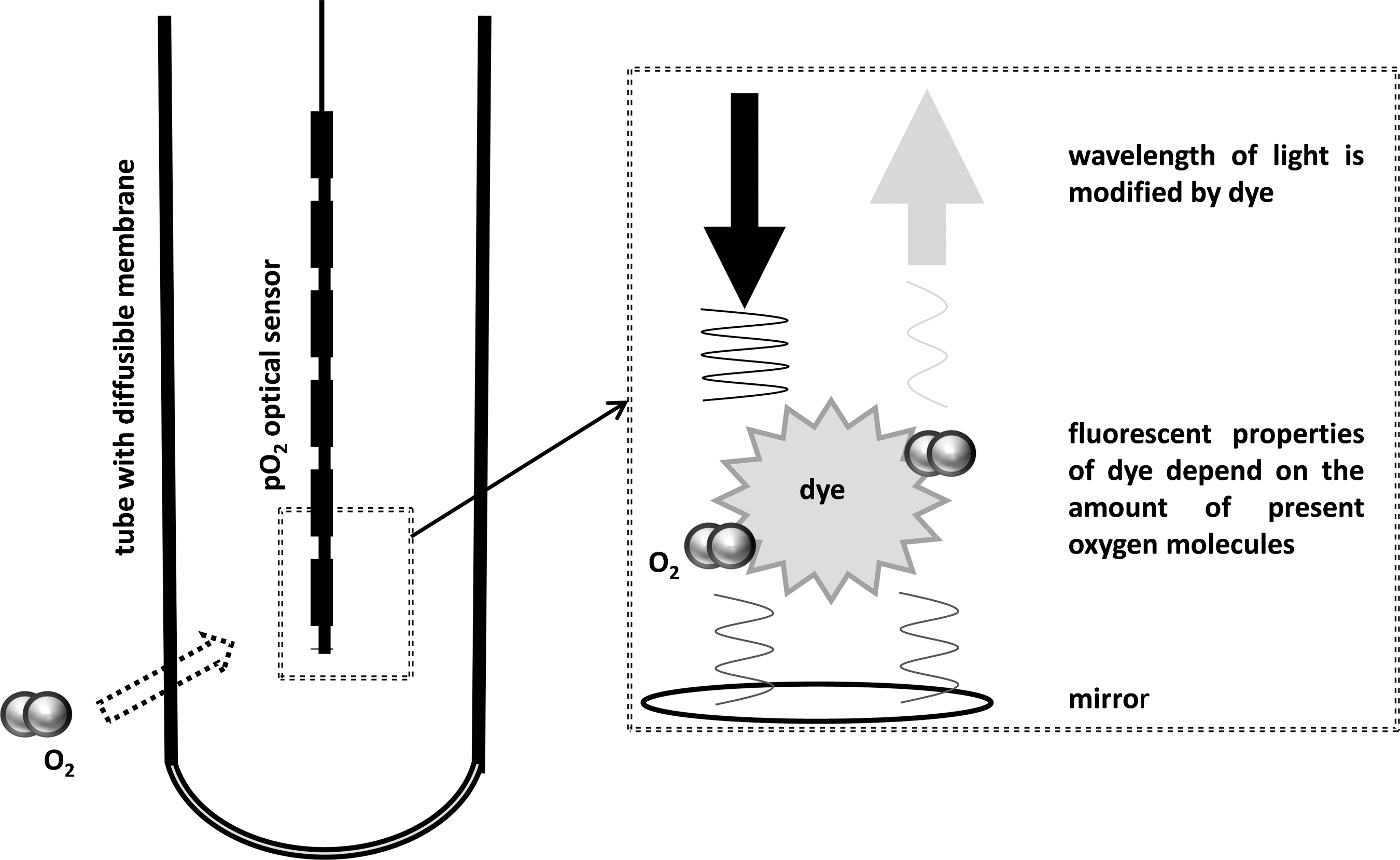
With optical techniques, wavelength alterations by indicator compounds are registered by optical sensors. The presence of oxygen molecules changes optical properties of these compounds. In contrast to the Clark electrode, this technique does not consume oxygen.
The device most widely used is the Licox® probe, distributed by Integra Neurosciences (Plainsboro, NJ). It is a Clark-based device and several studies have confirmed its safety and reliability in clinical studies (Dings et al., 1998; Maloney-Wilensky et al., 2009). In 292 patients monitored with this device, only two adverse events (two iatrogenic hematomas not requiring surgical evacuation) were reported. Originally developed for continuous blood gas monitoring, the Paratrend 7 (Diametrics Medical Inc., St. Paul, MN) was also used to measure brain tissue oxygenation (Hutchinson et al., 2000). Based on the Clark principle, P
The P
Shortly after the introduction of new brain tissue oxygen monitoring devices, several reports clearly demonstrated their impact on the treatment of head-injured patients. Stiefel and colleagues (2005) implanted brain tissue oxygen monitors in 25 patients with severe TBI, and demonstrated that cerebral hypoxia occurred in patients, even though standard guidelines were followed. Brain tissue oxygen was as low as <10 mm Hg in 29% of patients with an ICP<25 mm Hg, and in 27% of patients with a CPP>60 mm Hg. The mortality rate was higher in patients with reduced brain tissue oxygenation. Consistent with these findings, Chang and coworkers (2009) demonstrated in their study of 27 patients with severe TBI that episodes of low brain oxygen content below 20 mm Hg occur frequently, and are independent of ICP. Several studies have demonstrated that brain hypoxia below 10 mm Hg is associated with worse outcomes after TBI (Bardt et al., 1998; Kiening et al., 1997). Van den Brink and colleagues (2000) demonstrated that the mortality rate was over 50% for patients with brain tissue oxygenation below 10 mm Hg for more than 30 min. A meta-analysis by Maloney-Wilensky and colleagues (2009) showed that in TBI, patients without brain hypoxia (defined as P
Management of reduced brain tissue oxygenation
Monitors of P
Further studies are needed to identify the most suitable interventions for restoring P
Studies of brain tissue oxygen-directed therapy
The impact of monitoring brain tissue oxygenation in TBI victims was recognized early, and it was hypothesized that modifying therapy to keep P
Nangunoori and colleagues (2011) recently reviewed the literature regarding studies comparing the impact of brain tissue oxygen–directed therapy to standard ICP/CPP therapy on long-term outcome in TBI patients. Four studies with adequate outcome data to define odds ratios and confidence intervals were identified. Meixensberger and colleagues (2003) included 93 TBI patients undergoing monitoring of P
The results of these studies are summarized in Table 1. The common odds ratio is 2.1 (95% CI 1.3,3.1), suggesting a beneficial effect of brain tissue oxygen–directed therapy on TBI patients. The use of devices for monitoring brain tissue oxygenation was included in the Brain Trauma Foundation's guidelines for the management of severe traumatic brain injury in 2007 (Bratton et al., 2007). Nevertheless, further investigation of the impact of brain tissue oxygen–directed therapy on the treatment of TBI patients is needed, and currently a prospective multicenter randomized controlled trial has started recruiting patients (
GOS, Glasgow Outcome Scale; ICP, intracranial pressure; CPP, cerebral perfusion pressure; P
Effects of hyperoxia on brain metabolism after traumatic brain injury
Several studies have addressed the issue of brain metabolism parameters altered by normobaric hyperoxia treatment. Although modern techniques such as PET scanning have revealed new insights about brain metabolism, this field has to be further elucidated. In a study of patients with severe TBI undergoing multimodal monitoring (Menzel et al., 1999), including cerebral microdialysis, dialysis lactate levels decreased by 40% in patients treated with 100% oxygen for a period of 6 h. Mean P
Baseline lactate level ≤3 mmol/L prior to hyperoxic treatment.
Baseline lactate level >3 mmol/L prior to hyperoxic treatment.
In contrast, the impact of hyperoxia on parameters of cerebral metabolism, as measured by microdialysis parameters, is not clear and displays a heterogenous picture. Different positions of implanted probes (vital brain versus at-risk tissue versus injured brain) in the different patients of various studies may offer one explanation for this discrepancy. Hyperoxia consistently increases measured brain tissue oxygenetion.
L:P, lactate:pyruvate ratio; n/a, not available.
All studies have demonstrated a robust effect of hyperoxia on brain tissue oxygenation, significantly elevating levels from baseline values. The effect of this dramatic increase on brain tissue is unclear, and recent studies have focused on this topic using sophisticated imaging techniques. Diringer and colleagues (2007) have examined the effects of 100% oxygen for a period of 1 h on cerebral metabolism and CBF using PET scanning techniques. Five patients with TBI were included in this study, and no differences were seen in CBF, OEF, and global CMRO2 during the application of 100% oxygen. These results suggest a lack of metabolic benefit from hyperoxia for the global brain tissue. Nevertheless, Nortje and colleagues (2008) used 15O-PET scanning and found results indicating that some areas of brain may benefit from hyperoxic treatment. Eleven patients with severe TBI were included in their study. After completing baseline PET scanning, 60–80% oxygen was administered, and following a stabilization period of about 60 min, PET scanning was repeated. Consistent with the findings of Diringer and colleagues (2007), no significant differences were observed in whole brain measurements for CBF, OEF, and CMRO2. Interestingly, in areas in which baseline scans revealed a reduced CMRO2 of less than 37 μmol×100 mL−1×min−1, hyperoxia increased the CMRO2 from 23 to 30 μmol×100 mL−1×min−1. No significant changes were observed for CBF and OEF. These results suggest a preferential metabolic benefit with hyperoxia for at-risk tissue.
Figaji and colleagues (2010) studied the effects of hyperoxic challenges of 15 min on 28 children (age<15 years) with severe TBI. Hyperoxia increased Pa
Hyperbaric Oxygen Treatment
If oxygen is inhaled under normal atmospheric pressure, the amount of dissolved oxygen molecules in plasma is low and most of the blood oxygen is bound to hemoglobin. Increasing the amount of inhaled oxygen increases the amount of dissolved oxygen in plasma, but this increase is limited to approximately 10% of the total amount of oxygen in blood when 100% oxygen is inhaled. Inhaling oxygen under increased pressure enables further solution of oxygen in plasma unbound to hemoglobin, therefore the total amount of oxygen in the blood is elevated by approximately 30% when HBO treatment is carried out at a pressure altitude of 3 absolute atmospheres (ATA; Table 3).
Under normal atmospheric conditions oxygen is mainly bound to hemoglobin. An increase in inspired oxygen as with normobaric oxygen treatment results in only small changes of oxygen dissolved in blood plasma. In contrast, hyperbaric oxygen treatment can elevate the amount of total oxygen in blood substantially. All values are for an environment with a temperature of 37°C and hemoglobin levels of ∼15 g/dL.
ATA, absolute atmosphere; Ptotal total pressure; P
Studies of HBO in experimental models of TBI have shown several neuroprotective effects (Daugherty et al., 2004; Palzur et al., 2008; Rogatsky et al., 2005; Vlodavsky et al., 2006; Voigt et al., 2008; Wang et al., 2010; Zhou et al., 2007), resulting in reduced mortality and improved neurological outcomes. Nevertheless, HBO is not yet ready to be implemented in clinical practice, as a clear benefit for patients with TBI remains to be proven. Three clinical trials were carried out to examine the effects of HBO treatment on outcomes of patients with TBI (Artru et al., 1976; Ren et al., 2001; Rockswold et al., 2001; Table 4). Of these, only the study by Rockswold and colleagues meets today's standards of a controlled randomized prospective trial. A total of 168 patients with TBI and a GCS score<9 were included and were randomized to HBO or standard treatment within 6–24 h after admission. HBO treatment for 1 h was carried out every 8 h for a period of 2 weeks or until the patient followed commands or was pronounced brain dead. Outcome analysis showed a reduction of mortality from 32% to 17% in patients treated with HBO. Subgroup analysis demonstrated that patients with an initial ICP of>20 mm Hg and with a GCS score of 4–6 had the greatest benefit (mortality reduction from 42% to 17% and 48% to 21%, respectively). Nevertheless, outcome analysis 18 months after trauma did not show any significant differences in GOS scores between groups. Differences between guideline therapies at the time the study was carried out and the guidelines accepted today do not allow a complete comparison to today's situation. Nevertheless, the results of this study do suggest that HBO therapy might be beneficial in the treatment of TBI victims. HBO treatment modalities have to be further investigated since they seem to have a significant impact on the efficacy of this treatment. Results of a study in a rat model of focal cerebral ischemia suggest that application of HBO beyond a time window of 6 h after stroke onset may aggravate histological and clinical ischemic injury due to possible detrimental effects of hyperoxic treatment (Lou et al., 2004).
Three clinical trials of hyperbaric oxygen therapy (HBO) in patients with TBI have been carried out. The only study meeting today's standards for controlled prospective randomized trials was carried out by Rockswold and colleagues (1992). Patients had been enrolled from 1983–1989 in this study, so guidelines for the treatment of TBI patients have changed substantially since then.
GOS, Glasgow Outcome Scale.
Rockswold and colleagues (2010) recently published results of a study comparing the effects of HBO and NBO on cerebral metabolism, ICP, and oxygen toxicity in patients with severe TBI. Sixty-nine patients were enrolled and received either standard care treatment, 3 h of NBO, or HBO for 1 h. Lactate:pyruvate ratios were reduced in patients treated with HBO and NBO (10% and 3% compared to control group, respectively), but CMRO2 was significantly increased, by 37% at 6 h after HBO treatment, whereas NBO therapy had no effect on CMRO2 compared to controls. Additionally, HBO therapy reduced elevated ICP compared to controls, while NBO had no impact on ICP. The authors showed that the greatest benefit for cerebral metabolism, especially CMRO2, was achieved when measured brain tissue oxygen exceeded a level of 200 mm Hg. This level was reached in 51% of patients treated with HBO, and in 5% of patients treated with NBO, indicating a more robust effect of HBO treatment than NBO treatment. These results are consistent with findings from experimental studies on ischemic stroke. In a model of transient focal cerebral ischemia, HBO was more effective than prolonged NBO in reducing infarct size and improving functional outcome, although treatment onset of HBO was delayed (Beynon et al., 2007).
Nevertheless, HBO has several disadvantages compared to NBO. Logistical challenges with increased transfer time of patients and limited availability of chambers are major restrictions. In contrast, NBO therapy could be initiated earlier after trauma by emergency medical personnel with only minimal risk. Further studies on both treatment modalities are needed, and perhaps the sequential administration of both therapies will prove to have a positive impact on the clinical course of patients with TBI.
Side effects of hyperoxic treatment
Several side effects of hyperoxia are known and this has to be considered when discussing hyperoxic treatment with NBO or HBO as an option in patients with TBI. Hyperoxia is associated with systemic side effects on the lungs, heart, and gastrointestinal tract (Bostek, 1989). It is assumed that lung damage is caused by a high production of reactive oxygen species (ROS; Pagano and Barazzone-Argiroffo, 2003). In healthy individuals, inhalation of 95% oxygen leads to tracheobronchitis within 4–22 h, but terminating inhalation results in complete resolution of symptoms within a few days (Clark and Lambertsen, 1971). In animals exposed to prolonged hyperoxia, histopathological analysis of lung tissue showed similar changes to those seen in acute respiratory distress syndrome (ARDS), such as high-permeability edema, pulmonary vascular lesions, and eventual pulmonary fibrosis (Jones et al., 1984; Nash et al., 1967). When using HBO, oxygen is applied under increased pressure, therefore the systemic hyperoxic side effects are combined with side effects induced by the elevated pressure, such as barotrauma of the lungs and ears. Seizures with HBO have been reported with an incidence of 0.03% (Hampson and Atik, 2003), but these are self-limiting and cause no permanent damage (Clark and Lambertsen, 1971).
In addition to systemic side effects, hyperoxia is also associated with negative effects on brain tissue itself (Brucken et al., 2010; Oter et al., 2005). It is known to induce cerebral vasoconstriction in healthy individuals, leading to a decrease of CBF (Bulte et al., 2007). Exactly how NBO-induced vasoconstriction affects ischemic brain tissue is unclear, with results of several studies suggesting a possible beneficial effect. Shin and associates (2007) demonstrated an augmentation of CBF through NBO in ischemic brain tissue of rats subjected to a model of focal cerebral ischemia. In patients with severe TBI, Rangel-Castilla and colleagues (2010) found that hyperoxia-induced vasoconstriction led to an improved autoregulatory index, suggesting that this mechanism might allow cerebral vessels a better response to transient hypotension.
A further concern is the development of free oxygen radicals through hyperoxic treatment. Elevated levels of free oxygen radicals can cause loss of neurons and induce neurological deficits (Balentine, 1966; Mickel et al., 1990; Nakashima et al., 1999; Oh and Betz, 1991). In an experimental model of global cerebral ischemia in gerbils, NBO of lasting 3 and 6 h resulted in high lipid peroxidation of brain tissue, and in a mortality rate three times higher than in non-treated animals (Mickel et al., 1987). In the hippocampi of rats subjected to experimental TBI (Ahn et al., 2008), 3-nitrotyrosine staining was elevated after a 1-h period of hyperoxic ventilation, demonstrating an oxidative damage to proteins. In contrast, other groups did not find any evidence of increased oxidative damage in experimental models of brain damage when hyperoxia was applied (Mink and Dutka, 1995; Schabitz et al., 2004; Singhal et al., 2002; Sunami et al., 2000).
In patients with TBI, Puccio and colleagues (2009) examined the effects of NBO for a period of 2 h on markers of oxidative stress, including lipid and protein peroxidation, antioxidant defenses, and glutathione. Analysis of these parameters in samples of patient cerebrospinal fluid (CSF) revealed no differences compared to samples from normoxic-treated patients. Rockswold and colleagues (2010) found no differences in levels of F2-isoprostane (a marker of oxidative damage) in CSF samples of TBI patients treated with NBO or HBO compared to controls.
As hyperoxic treatment seems to have beneficial as well as detrimental effects on brain tissue, positive effects may be diminished by negative effects. Finding the right balance is probably not based on an “all or nothing” principle; it is likely dependent on many factors, including therapeutic time window and dosage regimen. Further investigation is needed to identify pathophysiological mechanisms and optimum treatment modalities of therapeutic hyperoxia in order to evaluate the potential of this therapy and reduce its side effects. Threshold values have to be defined for hyperoxic treatment, since application of more oxygen than needed to have a neuroprotective effect would involve needless oxygen toxicity. Monitoring brain tissue oxygenation guides the clinician in avoiding hypoxia in cerebral tissue. It might also be a tool to identify excessive amounts of oxygen in brain tissue, so the term “brain oxygen tissue-directed therapy” would mean avoiding both hypoxia and detrimental hyperoxia.
Conclusion
The use of devices for monitoring brain tissue oxygenation allows identification of hypoxic episodes in brain tissue of TBI patients. Although controlled clinical trials have not yet been carried out, the available study results suggest that a brain tissue oxygen–directed therapy involving adjustments in therapeutic modalities to keep brain tissue oxygen levels above certain thresholds may improve mortality and functional outcomes in these patients. Additionally, the response of P
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.