
Abstract
Acute membrane damage due to traumatic brain injury (TBI) is a critical precipitating event. However, the subsequent effects of the mechanical trauma, including mitochondrial and lysosomal membrane permeability (MOMP and LMP) remain elusive. The main objective of the current study was to assess the role of a putative membrane-resealing agent poloxamer 188 (P188) in MOMP and LMP in response to a well-defined mechanical insult. Using an in vitro cell shearing device (VCSD), mechanical injury resulted in immediate disruption of membrane integrity in cultured primary neurons, and neurons were treated with P188 or a cathepsin B inhibitor (CBI) after VCSD 10 min. The protective effect of P188 on cultured primary neurons was first detected visually with a light microscope, and measured by MTT assay and LDH assay. The validity of monitoring changes in mitochondrial membrane potential (ΔΨm) was measured by JC-1 staining, and Western blot for cytochrome c and truncated Bid (tBid) in purified mitochondria was also performed. In addition, lysosomal integrity was detected by blotting for cathepsin B and tBid in purified lysosomes. Our results showed post-injury P188 treatment moderated the dissipation of ΔΨm in mitochondria, and inhibited VCSD-induced cytochrome c release from mitochondria as well as cathepsin B from lysosomes. Cathepsin B inhibition (CBI) could also increase cell viability, maintain mitochondrial membrane potential, and repress VCSD-induced release of cytochrome c from mitochondria to cytosol. Both P188 and CBI treatment decreased the cytosolic accumulation of tBid in supernatant of purified lysosomes, and the amount of mitochondrial localized tBid. These data indicate injured neurons have undergone mitochondrial and lysosomal membrane permeability damage, and the mechanism can be exploited with pharmacological interventions. P188's neuroprotection appears to involve a relationship between cathepsin B and tBid-mediated mitochondrial initiation of cell death.
Introduction
T
Poloxamer 188 (P188) is a non-ionic, nontoxic, amphiphilic co-polymer (MW: ∼8400), including a central hydrophobic molecule that is flanked on both sides by two hydrophilic chains of polyoxyethylene. 5 With various clinical applications as a surfactant, P188 is capable of sealing damaged cell membranes. 6 The putative membrane-resealing agent P188 may repair injured (permeable) cells after controlled cortical impact in mice. 7 Using cortical and hippocampal neuron cultures, P188 was shown to protect neurons from excitotoxic or oxidative stress-related necrosis and electroporation by inserting the membrane and inhibiting membrane peroxidation. 5 Recently, in vitro models of mechanical injury have been developed via the utilization of an in vitro cell shearing device (VCSD). Although originally devised to evaluate endothelial cell shearing, its utility for evaluating primary neuronal cell stress and the protection provided by P188 was not evaluated. Not only could this approach allow for an assessment of the role of P188 in cultured primary neurons after VCSD but also it could potentially allow the evaluation of the effects of the shear injury on mitochondrial and lysosomal membrane permeability.
Mitochondria play an important role in the regulation of cell death and survival. The failure in mitochondrial function leads to collapse of membrane potential and disruption of membrane integrity. 9 The fluorescent probe 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl benzimidazolo-carbocyanine iodide (JC-1) was used to investigate mitochondrial membrane permeability (ΔΨm) in primary neurons. 10 JC-1 enters the mitochondria as a monomer, forms aggregates that have a fluorescence emission spectra peak at 590 nm at high ΔΨm (red), and becomes a monomer again with a fluorescence emission peak at 529 nm (green) at low ΔΨm. 11 The role of mitochondria in apoptosis has now been well documented as activation of an executioner caspase, caspase-3 is mediated by mitochondria. 9 Briefly, cytoplasmic cytochrome c, apaf-1, and pro-caspase-9 form the “apoptosome,” a macromolecular assembly, leading to caspase-9 activation. In this turn, caspase-9 activates caspase-3, thereby triggering activation of cell death pathway. 12,13 Moreover, cytosolic p22 Bid is activated by limited proteolysis in the loop region to generate the p15 Bid (t-Bid), which translocates to mitochondria. 14
In addition, lysosomes may play a vital role not only in degrading macromolecules, but also in their transport to the deposition site. 15 Cathepsin B, as one of the major lysosomal cysteine proteases, may be important in the intracellular protein catabolism in neurons, and may play an important role in the apoptosis and necrotic programmed cell death. 16 Cathepsin B has now been reported to be involved in apoptotic regulation via cleaving Bid. 17,18 A lysosomal–mitochondrial axis theory of programmed cell death has also been proposed. 17 Therefore, obtaining purified lysosomes should represent an important step in the study of cathepsin B release from lysosomes, in order to exploit lysosomal membrane permeability (LMP) after VCSD. Meanwhile, isolation of highly purified lysosomes using percoll gradients with a variety of calcium concentrations has been reported. 15
Based on the above-mentioned findings, we postulate that P188 might participate in reducing VCSD-induced immediate disruption of cultured primary neurons, and attenuating VCSD-induced disruption of mitochondria and lysosomal membrane integrity. To confirm this hypothesis, the effect of P188 on cultured neurons was first observed visually with a light microscope, and measured by 3-[4,5-dimethylthiazol-2-yl-]-2,5-diphenyltetrazolium bromide (MTT) assay and lactate dehydrogenase (LDH) assay. The present study was then designed to assess the validity of monitoring changes in mitochondrial membrane potential (ΔΨm) with JC-1 staining, and to perform Western blot analysis for cytochrome c and tBid detection in purified mitochondria. Additionally, lysosomal integrity was assessed by blotting for cathepsin B and tBid in supernatant (cytosolic fraction) and in pellet (lysosomal fraction) of purified lysosomes. Finally, to further investigate the mechanisms of neuroprotection by P188, we used a selective cathepsin B inhibitor (CBI), 18,19 and determined the effects of cathepsin B inhibition on cell viability, membrane potential, and mitochondrial function following VCSD.
Methods
Primary cortical neuron culture
All experimental procedures followed protocols were in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committee at Soochow University and Fudan University. A primary cortical neuron culture method was used as described previously. 9 Cortical tissues were obtained from embryonic day 18 Sprague-Dawley (SD) rats (Soochow University). After incubation with 0.125% trypsin (Invitrogen) for 10 min, cells were then re-suspended in a solution of Neurobasal media (Invitrogen) containing 2% B27 (Invitrogen), 2 mM glutamine (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 25 μM glutamate (Sigma), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). Then cells were seeded in 24-well plates (Falcon) coated with 0.01% (w/v) poly-l-lysine (Sigma) at a density of 1.25–2.0×105 cells/cm2 and put into a standard incubator (Taibai Espec) maintained at 37°C in 95% air, 5% CO2. A half medium was changed every 3 days. The purity of neurons was confirmed by immunostaining for MAP2 (a neuronal-specific marker), and indicated that 90%–95% of cells in cultures were MAP2 positive. The remaining cells exhibiting morphological characteristics of glial cells were verified by GFAP immunostaining.
In vitro cell injury protocol
Cortical neurons cultured on coverslips were subjected to shear stress by using an in vitro cell shearing device (VCSD, Multi Channel Systems, Germany) on the 7th day after plating as previously described. 18,20 Prior to experimentation, the cells were rinsed with 1×Eagle's Balanced Salt Solution (EBSS) (Life Technologies) two times to remove any unattached cells. Based on the previously demonstrated loading-rate dependence of cell injury, we varied the loading rate over a wide range. This was achieved by increasing the applied shear stress rapidly from zero to variable peak value. Based on the results obtained from magnitude and rate dependence studies, the combination of 100 dyn/cm2 peak shear stress and 20 dyn/cm2 of steady shear stress for 200 msec was used. 21 Cells were mechanically loaded, and P188 (50, 100, and 150 μM, pluronic acid F-68, Sigma) or CBI (0.5 μg/μL, Santa Cruz Biotechnology) was added into the cell medium at 10 min following VCSD, gently agitating the medium to distribute the applied surfactant evenly. Sham controls and untreated injured cells were treated identically including the gentle agitation. Microscopic observations were performed with Nikon eclipse TE2000-U microscope.
Assessment of neuronal viability
The viability of the cells in neuronal monocultures was assessed by their ability to take up thiazolyl blue tetrazolium bromide (MTT). 22 Neurons were treated with P188 (50, 100, and 150 μM) or CBI (0.5 μg/μL) dissolved in 0.1% dimethyl sulfoxide (DMSO) at 10 min following VCSD. Sham (uninjured) neurons were also treated with 0.1% DMSO or P188 (100 μM) or CBI (0.5 μg/μL). The MTT solution (5 mg MTT/ml medium) was added to each well at 6 h and 24 h after VCSD at the final concentration of 250 μM and incubated for 3 h. The media was removed, and cells were dissolved in 0.1% DMSO. Optical density was determined using a spectrophotometer (SpectraMax 340PC384, Molecular Devices) at 550 nm test and 690 nm reference wavelengths. Neurons were stained with trypan blue, and viability was determined using the trypan blue exclusion assay. After incubation with the indicated conditions, neurons were incubated with 0.4% trypan blue in HBSS for 2 min at room temperature. Neurons were observed under the microscope and counted as stained and nonstained cells on a hemacytometer separately, then viable cell ratios were calculated according to the following formula: viable cell ratio (%)=(nonstained cell number/total cell number)*100%. 23
Lactate dehydrogease assay
To further examine the protective effect of P188 on the VCSD-induced neuronal cell death, we utilized the lactate dehydrogenase (LDH) assay. Neurons were treated with P188 (100 μM) or CBI (0.5 μg/μL) dissolved in 0.1% DMSO 10 min following VCSD. Sham (uninjured) cells were treated with 0.1% DMSO or P188 (100 μM) or CBI (0.5 μg/μL). The release of LDH into the culture media was measured 6 h after VCSD using a LDH assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, PR China). Briefly, after VCSD treatment, the supernatant of the cell culture was reserved. Neurons were rinsed with PBS and lysed with 1% Triton X-100 at 37°C for 30 min. Then samples of supernatants and cell lysates were prepared following the manufacturer's instructions. The absorbance value (A) at 440 nm was determined with an automatic multiwell spectrophotometer (Bio-Rad Laboratories, Hercules, CA). LDH leakage was calculated as follows: LDH leakage (%)=(A positive/A positive blank)/(A negative/A negative blank)×100%. 24
Mitochondrial membrane potential measurements
Total cells were incubated with 0.1% DMSO or P188 (100 μM) or CBI (0.5 μg/μL) at 10 min after VCSD, and cells were loaded with the dye JC-1 6 h and 24 h later. Briefly, total cells were collected into 1.5 mL tubes and incubated at 37°C for 20 min with 5mg/L JC-1 (Beyotime Biotech, China), then washed twice with PBS and placed in fresh medium without serum. Images were viewed and scanned by fluorescence microscope at 514 excitation and 529 emission for green, and at 585 excitation and 590 emission for red. The ratios of red/green fluorescent densities were calculated. 25,26
Preparation of mitochondrial and cytosolic fractions
Isolation of mitochondria was performed as described previously. 13,19 Briefly, each sample was homogenized in 0.3 mL ice-cold buffer A containing 250 mM sucrose, 1 mM EDTA, 50 mM Tris-HCl, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 0.28 U/mL apotinin, 50 μg/mL leupeptin, and 7 μg/mL pepstatin A (pH adjusted to 7.4 with NaOH) and homogenized with a glass Pyrex microhomogenizer (30 strokes). The homogenate was centrifuged at 1000 g at 4°C for 10 min, and the resultant supernatant was transferred to a new tube to be centrifuged at 12,000 g at 4°C for 20 min to obtain the mitochondrial pellet and supernatant. The supernatant was centrifuged at 100,000 g for 1 h at 4°C to generate the cytosolic fraction. The mitochondrial pellet was washed three times in buffer B containing 250 mM sucrose, 1 mM EGTA, 10 mM Tris-HCl (pH 7.4), spun at 12,000 g at 4°C for 10 min, and then lysed in Western blot lysis buffer.
Measurement of lysosomal permeabilization
Highly purified lysosomes free of mitochondria were isolated as described. 15 Each sample was homogenized in the sucrose solution, and the suspension was centrifuged at 750 g for 10 min. The supernatant was collected and the pellet was resuspended in 10 mL sucrose solution (0.3 M), then rehomogenized and centrifuged. The final pellet was discarded and the two supernatant fractions were pooled. The pooled supernatant was further subfractionated on an isotonic percoll gradient. After percoll centrifugation, the lysosomal fraction was collected into 25 mL tubes from the bottom of the rotor tube using a Pasteur pipette. The fractions were diluted with 3 volumes of sucrose solution (0.3 M). The diluted suspension was centrifuged at 10,000 g for 10 min. The supernatant was collected and the pellet was resuspended in the sucrose solution and recentrifuged at 10,000 g for 10 min. The washed pellet was finally suspended in a small volume of sucrose solution (0.3 M) and used as the lysosomal fraction. A 20-μL aliquot of each supernatant or pellet was subjected to SDS-PAGE and analyzed by immunoblot for cathepsin B.
Electron microscopic examination
The isolated lysosomal fraction was immediately fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer at pH 7.2 for 12 h and postflxed with 1% osmic acid in the cacodylate buffer for 1 h, as described. 15 The fraction was routinely dehydrated in grade ethyl alcohol and propylene oxide and embedded in a mixture of epoxy resin (812), then cured overnight at 60°C. Sections (70 nm thick) were cut with glass knives on an ultracut microtome after staining with uranyl acetate and lead citrate and examined with a Philips CM-120 electron microscope.
Western blot analysis
The procedure used was published previously. 19,27 Samples were homogenized in Western blot analysis buffer containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% (v/v) Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 1 mM PMSF, 0.28 kU/L aprotinin, 50 mg/L leupeptin, 1 mM benzamidine, and 7 mg/L pepstain A (all chemicals from Sigma-Aldrich). The homogenate was then centrifuged at 12,000 g for 15 min at 4°C and the supernatant was retained and preserved at −80°C for later use. Protein concentration was determined using a BCA kit (Pierce). Each sample was subject to electrophoresis on 10% SDS-PAGE gel. Proteins were transferred to nitrocellulose membranes on a semidry electrotransferring unit (Bio-Rad), and incubated with antibodies against cathepsin B (Santa Cruz Biotechnology), cytochrome-c (BD PharMingen), HSP-60 (BD PharMingen), COX (Santa Cruz Biotechnology), anti-lysosomal associated membrane protein 1 (LAMP1) (BD Pharmingen), anti-PDI (Affinity BioReagents), and Bid (Santa Cruz Biotechnology) in Tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% nonfat dry milk overnight at 4°C. After the overnight incubation with the primary antibodies, membranes were washed and incubated with horseradish peroxidase-conjugated second antibody in TBST for 3 h. Immunoreactivity was detected with enhanced chemoluminescent autoradiography (ECL kit, Amersham). The membranes were reprobed with β-actin (Sigma) after striping. The signal intensity of primary antibody binding was quantitatively analyzed with Sigma Scan Pro 5 and was normalized to a loading control, β-actin.
Statistical analysis
The data were expressed as mean±SD, and were analyzed using GraphPad Prism 4 Software (La Jolla, CA). Statistical analysis was performed using single way analysis of variance (ANOVA), followed by Tukey's post hoc test to determine significance values between different experimental groups.
Results
VCSD-induced neuronal injury is protected by P188 and CBI
To determine whether P188 could reduce VCSD-induced immediate disruption of cultured primary neurons, the effect of P188 on cultured neurons was first observed visually with a light microscope. As shown in Figure 1, some cells showed shrinkage, whereas others rounded up. Some injured cells became partially detached from the substratum at 24 h post-injury as detected visually with a light microscope. Also, beading of the plasma membranes in both the cell bodies and axonal processes was apparent. Control cells did not show any of these morphological changes (Fig. 1a), but a subpopulation of mechanically-injured cells without P188 treatment exhibited some structural and morphological changes after 24 h (Fig. 1b). Neuronal cells treated with 100 μM P188 (Fig. 1c) at 24 h showed remarkable recovery of the morphology characteristics.
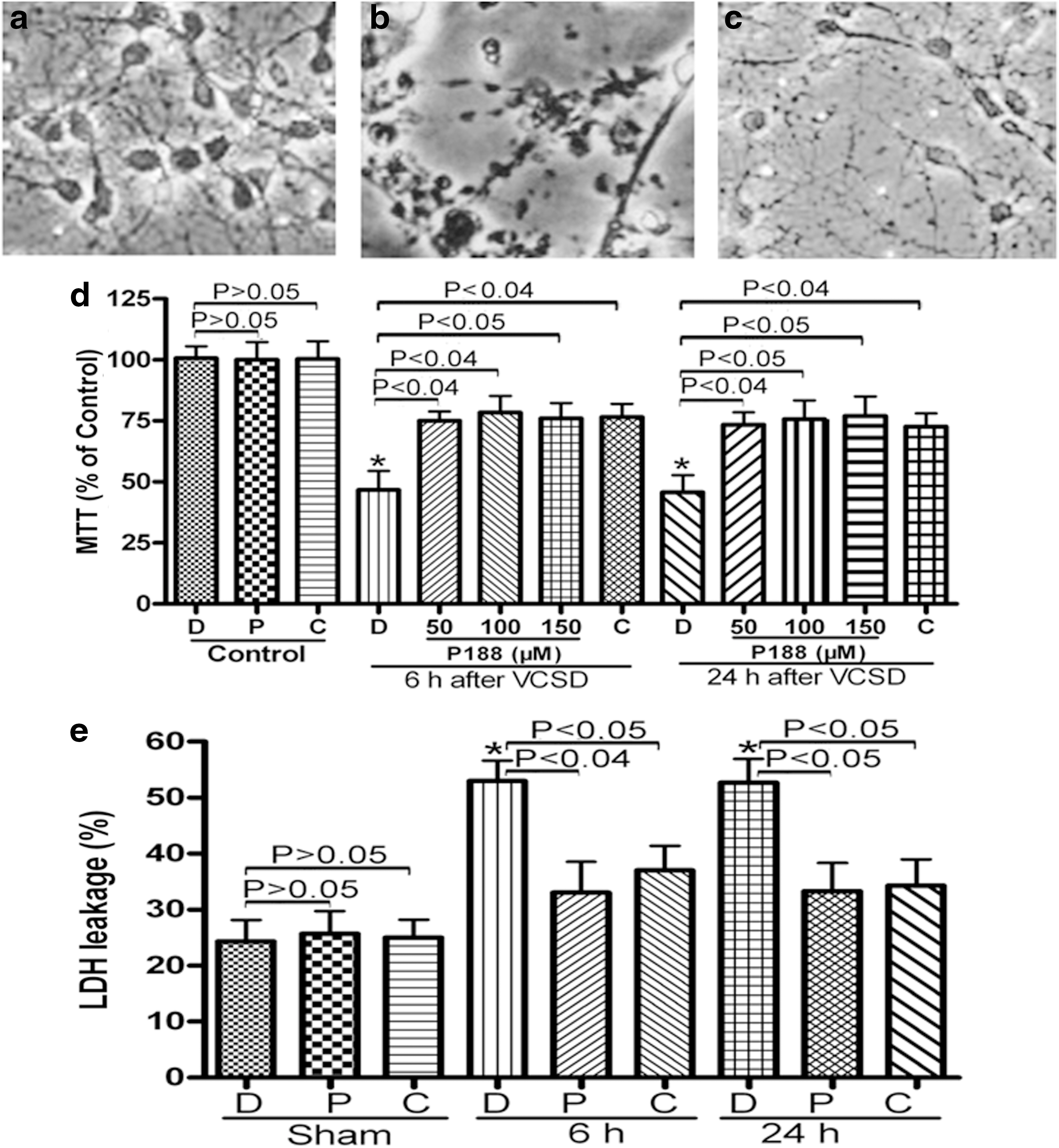
VCSD-induced neuronal injury is protected by P188 and CBI.
To verify that P188 has a protective effect on neuronal injury in vitro, and further investigate the mechanisms of neuroprotection by P188, using a selective cathepsin B inhibitor (CBI), we determined the effects of P188 and CBI on cell viability by MTT assay. Assessments of cell viability demonstrated that there were no significant differences in the effects of both P188 and CBI on sham neurons between DMSO (100.67±8.5%) and (96.67±12.02% P188-treated group, 100.33±12.66% for CBI-treated group) treatments (p>0.05, Fig. 1d). Approximately 50% of neuronal cells were dead at 6 h (46.67±13.43% for DMSO+VCSD group versus 100.67±8.5% for Sham group; p<0.01), and at 24 h (45.67±12.22% for DMSO+VCSD group versus 100.67±8.5% for Sham group; p<0.01) after VCSD (Fig. 1d). P188 (50, 100, and 150 μM) treatment significantly attenuated cell death in neurons at 6 h (75±6.56% for 50 μM P188+VCSD group, 78.33±12.01% for 100 μM P188+VCSD group, 76±11.04% for 150 μM P188+VCSD group; p<0.05), and at 24 h (73.33±9.02% for 50 μM P188+VCSD group, 75.67±13.32% for 100 μM P188+VCSD group, 77±14.01% for 150 μM P188+VCSD group; p<0.05) after VCSD in a dose dependent fashion (Fig. 1d). Treatment with CBI (0.5 μg/μL) could also increase cell viability at 6 h (76.67±9.29% for CBI+VCSD group versus 46.67±13.43% for DMSO+VCSD group; p<0.04) and at 24 h (72.67±9.29% for CBI+VCSD group versus 45.67±12.22% for DMSO+VCSD group; p<0.04) following VCSD (Fig. 1d).
In order to further strengthen our findings, we explored the effect of P188 and CBI on neuronal cell death using an LDH assay. There were no significant differences in the effects of P188 (100 μM) and CBI (0.5 μg/μL) on sham neurons between DMSO and drug (P188 or CBI) groups (p>0.05; Fig. 1e). A significant increase in LDH leakage was observed at 6 h and 24 h after VCSD (p<0.01; Fig. 1e), compared to the sham control group. Injury-induced LDH leakage at 6 h and 24 h was significantly attenuated by P188 and CBI (P<0.05; Fig. 1e), respectively.
Injury-induced the dissipation of ΔΨm in mitochondria is moderated by P188 and CBI
JC-1 easily penetrates cells and healthy mitochondria. A green fluorescent JC-1 probe exists as a monomer at low membrane potentials; however, at higher potentials, JC-1 forms red-fluorescent ‘J-aggregates.’ The ratio of red/green JC-1 fluorescence is dependent only on the mitochondrial membrane potential and not on other factors that may influence single-component fluorescence signals, such as mitochondrial size, shape, and density. 28 In this study, JC-1 could aggregate in normal mitochondria and present red fluorescence (Fig. 2a). Exposure of neuronal cells to VCSD resulted in dissipation of ΔΨm, which was shown as increased green fluorescence by JC-1 staining (Fig. 2b). The ratio of red/green fluorescence was also used to demonstrate dissipation of ΔΨm in mitochondria. Representative photographs showed that the ratio of mitochondrial JC-1 aggregate (red) to cytosolic JC-1 monomer (green) was reduced at 24 h after VCSD treatment (Fig. 2b, e), compared to the healthy control cultures (Fig. 2a, e). This decline at 6 h and 24 h after VCSD was significantly reversed by post-injury P188 (Fig. 2c, e) and CBI (Fig. 2d, e), respectively. As shown in Figure 2e, no significant differences in the effects of P188 and CBI on sham neurons were observed between DMSO and drug (P188 or CBI) groups (p>0.05), suggesting P188 and CBI do not affect JC-1 fluorescence.
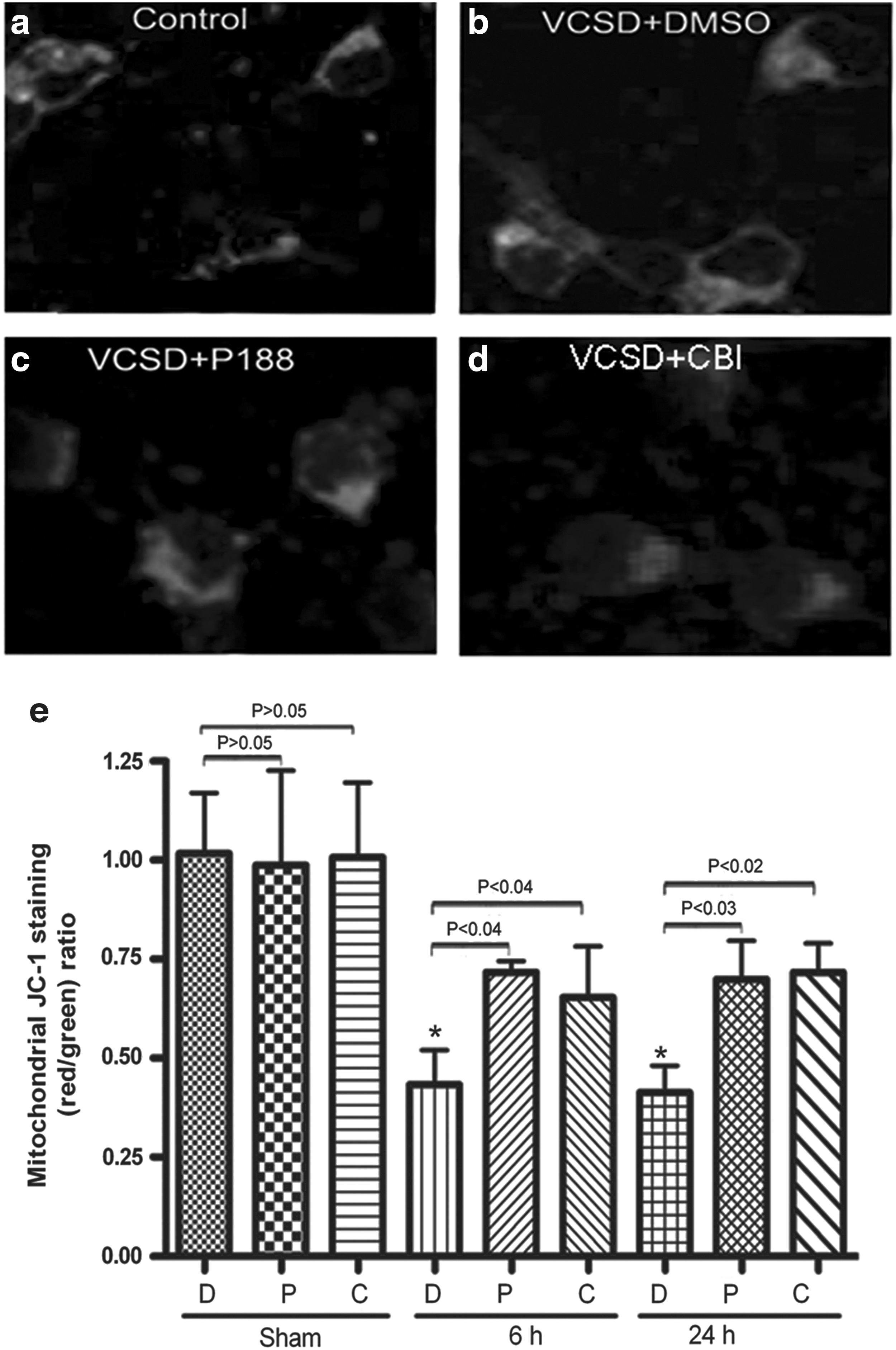
Injury-induced the dissipation of ΔΨm in mitochondria is moderated by P188 and CBI.
VCSD-induced release of cytochrome c from mitochondria to cytosol is inhibited by P188 and CBI
We next assessed blotting for cytochrome c in purified mitochondria. Western blot analysis showed that a significant reduction in protein levels of cytochrome c in mitochondrial fraction (p<0.02; Fig. 3a, b), and a concomitant increase in cytochrome c levels in the cytosolic fraction (p<0.05; Fig. 3a, c) were observed at 6 h and 24 h after VCSD. As shown in Figure 3, no significant differences were observed between DMSO and drug (P188 or CBI) groups (p>0.05), thus confirming the effects of P188 and CBI, and rule out any direct effect on cytochrome c. Treatment with P188 (100 μM) significantly inhibited VCSD-induced release of cytochrome c from mitochondria to cytosol at 6 h and 24 h after injury (P<0.05; Fig. 3a–c). Treatment with CBI also inhibited VCSD-induced release of cytochrome c from mitochondria to cytosol at 24 h (p<0.05; Fig. 3d–f).
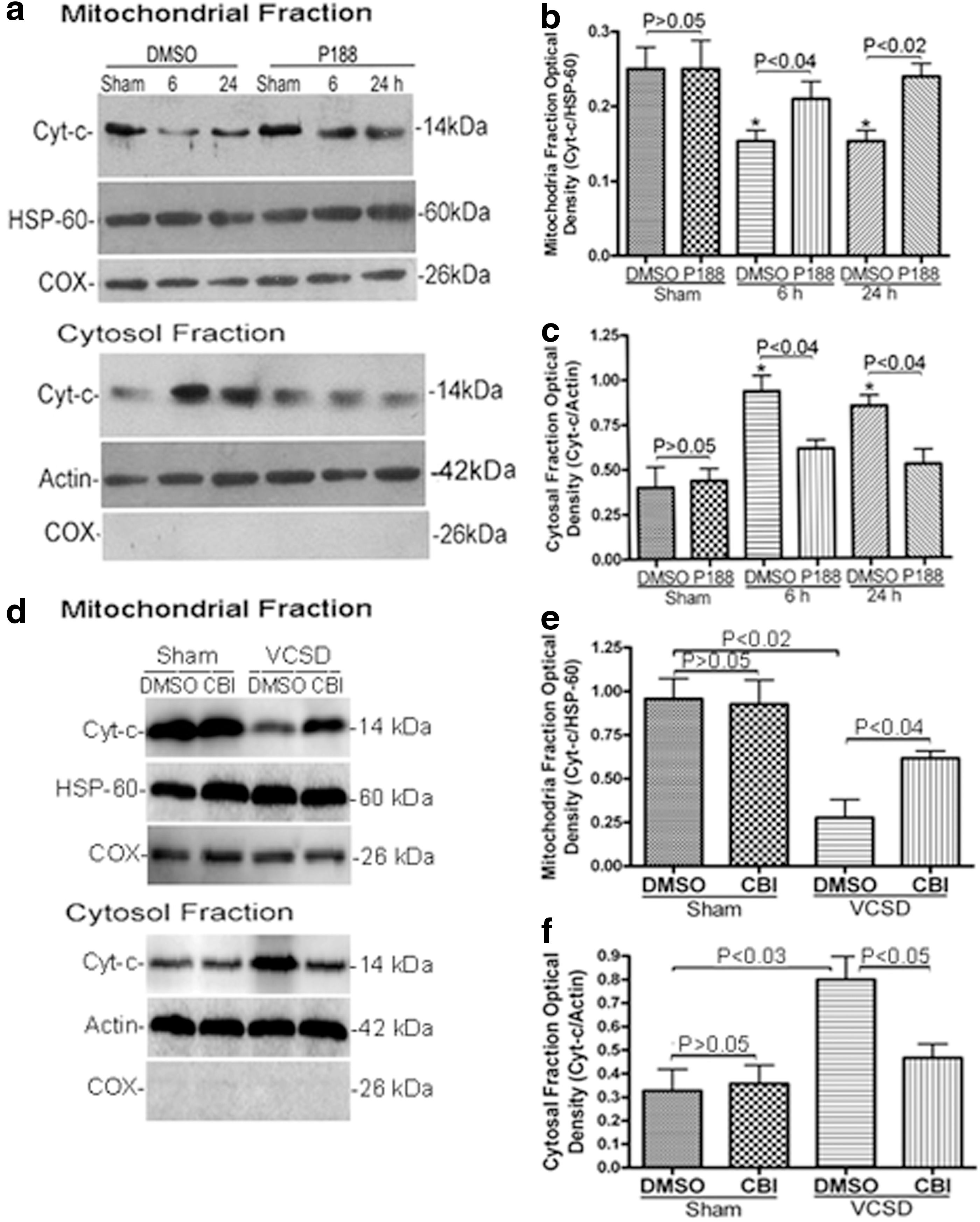
VCSD-induced release of cyt-c from mitochondria to cytosol is inhibited by P188 and CBI.
P188 attenuates VCSD-induced lysosomal membrane permeabilization damage
To verify whether the isolated lysosomes were highly purified, samples of intact lysosomes were first analyzed by electron microscopy. Representative electron micrographs (x 6500) are depicted in Figure 4a. Examination showed largely enriched vesicles with electron dense material along with a few mitochondrial particles. Next, we verified that isolated lysosomes (the marker LAMP1) were free of the endoplasmic reticulum marker PDI and the mitochondria protein cytochrome c oxidase (Fig. 4b). This confirmed that the lysosomal preparation was highly purified with little contamination by mitochondria.

Treatment with P188 attenuates VCSD-induced lysosomal permeabilization damage.
Next, lysosomal permeabilization was assessed by blotting for cathepsin B in supernatant (cytosolic fraction) and pellet (lysosomal fraction). Reduction of cathepsin B in the cytosolic fraction was observed, together with a concomitant increase in cathepsin B levels of lysosomal fraction at 6 h (p<0.01) and 24 h (p<0.02) after VCSD (Fig. 4c, d). Post-injury P188 (100 μM) treatment significantly reversed the effects on cahepsin B release at 6 h (p<0.02) and 24 h (p<0.04) after VCSD (Fig. 4c, d). As shown in Figure 4d, no significant difference in the effects of P188 on sham neurons was observed between DMSO and P188 treatment (p>0.05), confirming the effects of P188 and rule out any direct effect of P188 on cathepsin B.
Both P188 and CBI treatment decrease the cytosolic accumulation of tBid in supernatant of lysosomes, and the amount of mitochondria localized tBid
We then detected the levels of tBid in purified lysosomes and mitochondria by Western blot. After injury, an increase in tBid levels of cytosolic fraction (supernatant of lysosomes) was observed (p<0.02), whereas no tBid was found in the pellet of lysosomes (lysosomal fraction) at 24 h post-VCSD (Fig. 5a, b). In marked contrast, an increase in tBid levels of cytosolic fraction after VCSD was inhibited by 100 μM P188 (p<0.04) and 0.5 μg/μL CBI (p<0.05).
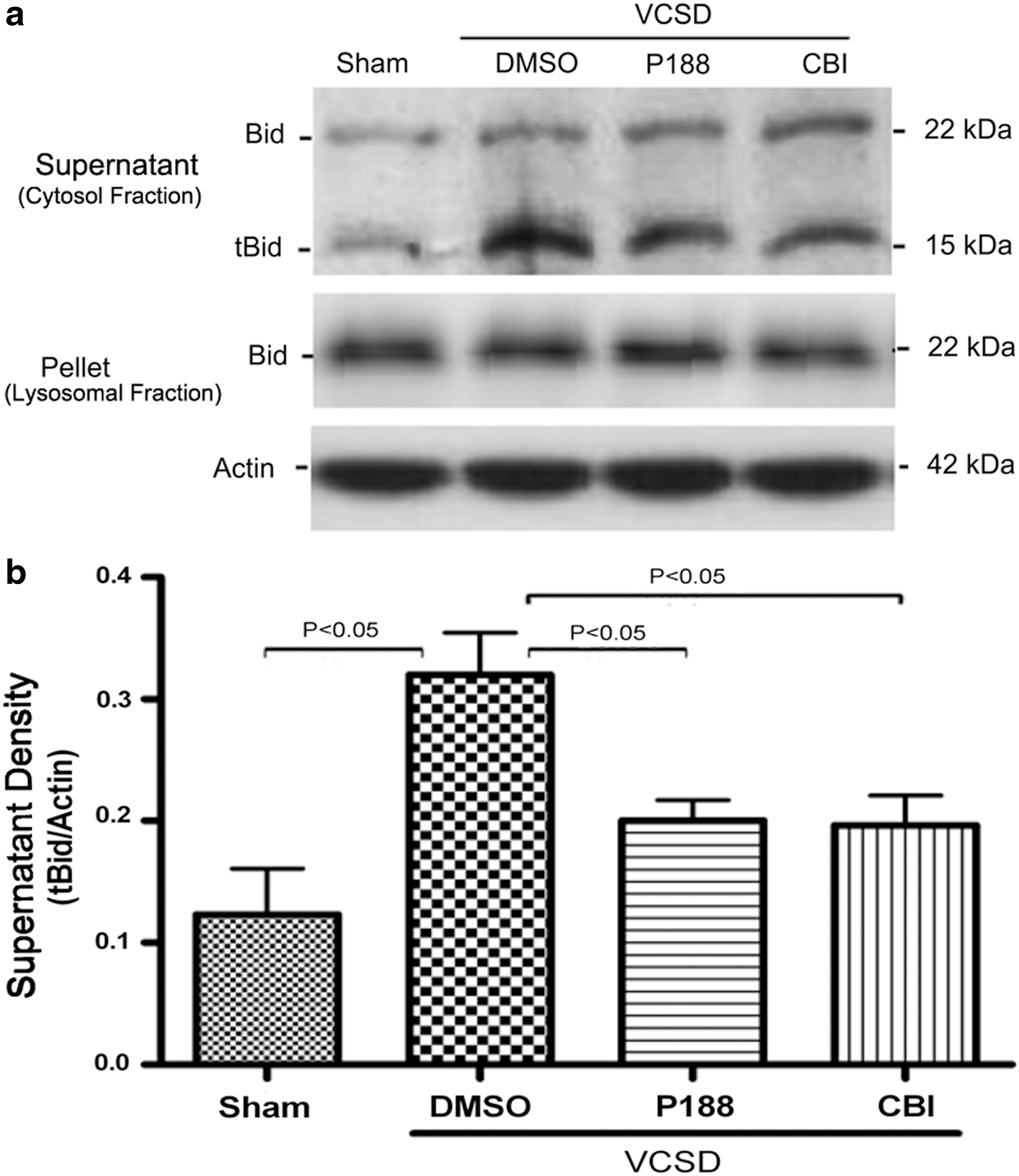
Both P188 and CBI prevent VCSD-induced elevation of tBid in supermatant of lysosomes (cytosolic fraction).
Furthermore, as shown in Figure 6, the levels of tBid in mitochondrial fraction were elevated to sham at 24 h after VCSD (p<0.03), whereas no tBid was found in the cytosolic fraction. Elevation in mitochondrial tBid following mechanical injury was significantly inhibited by 100 μM P188 (p<0.05) and 0.5 μg/μL CBI (p<0.04), respectively.
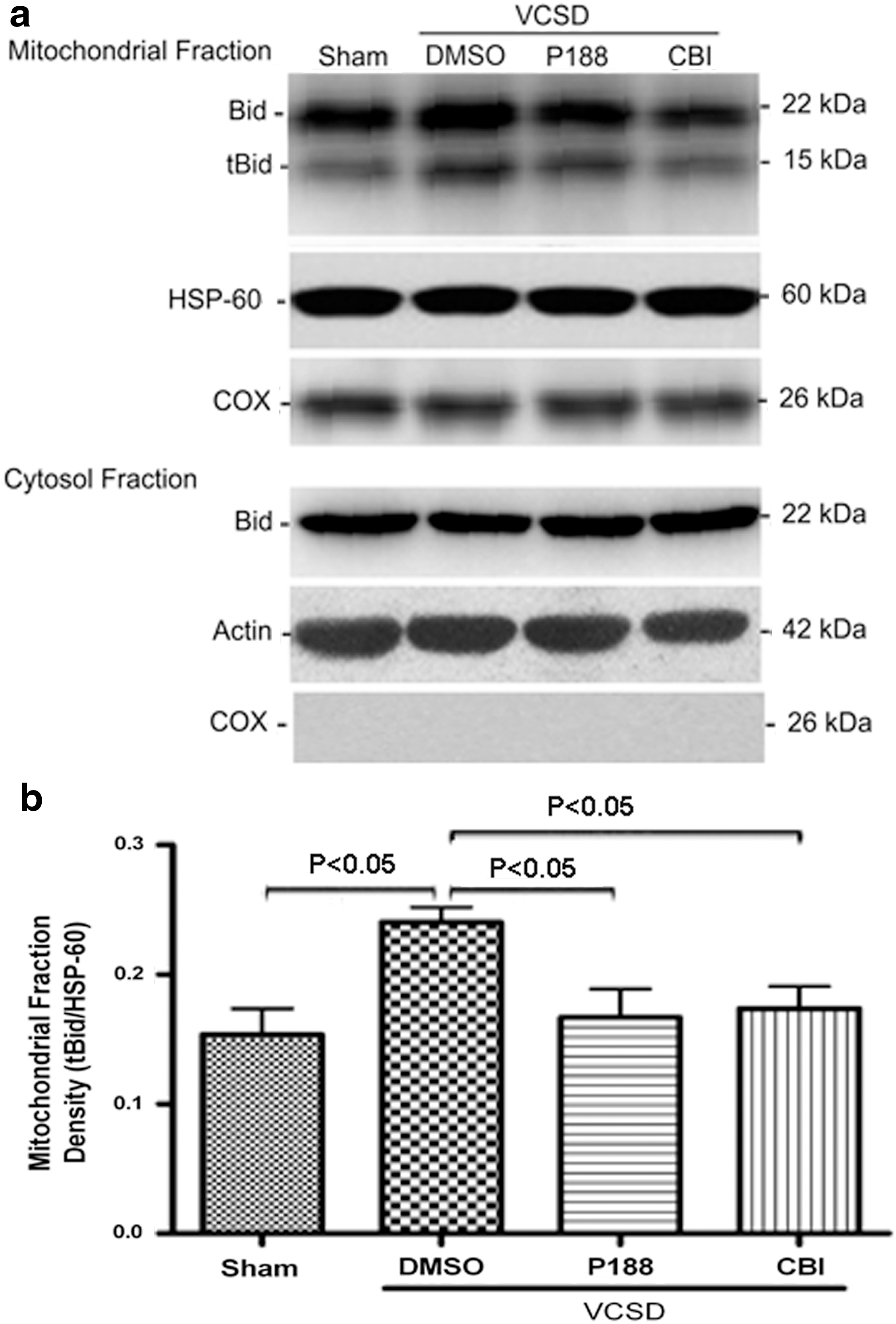
Both P188 and CBI counteract VCSD-induced elevation of tBid in mitochondrial fraction.
Discussion
This study reports that: 1) post-injury P188 attenuate cell death and the morphology characteristic of injured cultured primary neurons, 2) P188 moderates in vitro cell shearing device (VCSD)-induced dissipation of ΔΨm in mitochondria, and inhibit VCSD-induced release of cytochrome c from mitochondria to cytosol, 3) P188 reduces VCSD-induced lysosomal membrane permeabilization damage, 4) cathepsin B inhibition (CBI) could also maintain mitochondrial membrane potential, inhibit VCSD-induced release of cytochrome c from mitochondria to cytosol, and increase cell viability, 5) both P188 and CBI treatment decrease the cytosolic accumulation of tBid in supernatant of lysosomes, and the amount of mitochondrial localized tBid.
Management of traumatic brain injury (TBI) can pose enormous challenges to the health team. 29 One possible consequence of brain injury is the loss in plasma membrane integrity and delayed damage. 1,7 Previous experiments have tested poloxamer 188 following in vivo traumatic brain injury and these studies have indicated a therapeutic effect of the treatment. 7 The VCSD configuration applied to cells cultured on a rigid substrate is not intended to mimic explicitly the in vivo, three-dimensional tissue deformation that occurs during injury. 8,20 Rather, the mechanical loading profile was developed and used to replicate the characteristics of cellular injury observed in vivo. 4 Cellular mechanical injury does not result in immediate irreversible cell lysis and cell death, but membrane damage that, if left untreated, leads to cell death over the ensuing 24 h after injury. 21,30 Poloxamer 188 was used previously to study the effects of shear stress on neuronal membrane integrity; 31,32 however these studies did not investigate the effect of poloxamer 188 on mitochondria or lysosomal membrane integrity that are shown in this study.
Acute membrane damage due to traumatic injury is a critical precipitating event. However, plasmalemma resealing may not be sufficient for survival of injured cells. 7 In the current study, cultured primary neurons treated with post-injury P188 showed a remarkable recovery of the morphology characteristic of sham (uninjured) cells. Moreover, P188 treatment significantly attenuated cell death and LDH leakage in neurons following VCSD. From our data, we conclude that other mechanisms, except cell rescue, mediate the protective effects of P188 in cultured primary neurons. We predict that membrane damage due to the initial traumatic event can be responsible for the initiation of the delayed pathological events, including disruption of mitochondrial and lysosomal membrane integrity after traumatic injury. The main aim of the present study was to assess the role of P188 in mitochondrial and lysosomal membrane permeability (MOMP and LMP) in response to a well-defined mechanical insult-VCSD, and to further investigate the mechanisms of P188's neuroprotection. To our knowledge, this study is the first to document neuroprotection by P188 in response to trauma-induced mitochondrial and lysosomal membrane damage in cultured primary neurons. First, characterizing alterations in ΔΨm is of great diagnostic value in predicting cell fate after VCSD because mitochondria are known to play a major role in the regulation of cell death and survival. 33 JC-1 is the most reliable fluorescent probe compared to Rhodamin123 and DiOC6 (3) for assessing changes of ΔΨm. 34 Using JC-1 staining in vitro, post-injury P188 treatment could moderate the dissipation of ΔΨm in mitochondria, thus indicating the protective effect of P188 on trauma-induced mitochondrial membrane damage. Next, cytochrome c (cyt-c) is a diffusible electron carrier located in the intermembrane space of mitochondria. 35 We recently reported that a significant increase is observed in the percentage of high cytosolic cyt c after TBI. 18 In the current work, the relatively lower cytosolic cyt c levels detected in P188-treated samples demonstrated that a correlation exists between P188 and the preservation of mitochondrial function. Moreover, once cyt c enters the cytosol, it associates with apoptotic peptidase activating factor 1 (Apaf-1) and dATP to form an apoptosome and activate pro-caspase-9, which in turn activates other downstream caspases (e.g., caspase-3) to initiate apoptosis. 35
Several recent studies indicate that lysosomal permeabilization with release of lysosomal proteases into the cytosol participates in cell death. 36,37 Cathepsin B is one of the most abundant and well-studied lysosomal cysteine proteases. 16 However, the molecular mechanisms of lysosomal membrane breakdown and the release of cathepsin B into the cytosol in the cultured primary neurons following VCSD are still poorly understood. In the present study, highly purified lysosomes free of mitochondria were isolated, and verified by electron microscopy and blotting for LAMP1 (the lysosomes marker), PD1 (the endoplasmic reticulum maker), and cytochrome c oxidase (the mitochondria protein), and then lysosomal integrity was assessed by blotting for cathepsin B in supernatant (cytosolic fraction) and pellet (lysosomal fraction). Our findings that an increase of cathepsin B release from lysosomes to cytosol was observed after VCSD, and post-injury P188 treatment reversed the effects on cathepsin B release, indicates P188 could attenuate VCSD-induced lysosomal membrane damage.
The lysosomal apoptotic pathway, either cathepsin-dependent or -independent, is believed to proceed through mitochondria. 38 Lysosomal cathepsins that are released into the cytosol after lysosomal membrane destabilization can cleave various protein substrates, thereby propagating apoptosis. 39 Cathepsin B has been reported to be involved in apoptotic regulation, not by direct proteolytic activation of executioner caspases, but through the cleavage of p22 Bid to generate p15 tBid. 40 Using a selective cathepsin B inhibitor (CBI), we found that both P188 and CBI treatment decreased the cytosolic accumulation of tBid in supernatant of lysosomes (Fig. 5). However, the function of cathepsin B and tBid in apoptotic induction after VCSD is still a matter of controversy.
The current study sought to investigate further the mechanisms of P188's neuroprotection against VCSD-induced LMP and MOMP damage. We found that, besides P188, cathepsin B inhibition (CBI) also inhibited VCSD-induced release of cytochrome c from mitochondria to cytosol, maintained mitochondrial membrane potential, and increased cell viability following VCSD. Both P188 and CBI treatment decreased the cytosolic accumulation of tBid in supernatant of lysosomes, and the amount of mitochondrial localized tBid. These data imply that neuroprotection by P188 appears to be involved in the relationship between cathepsin B and tBid-mediated mitochondrial initiation of cell death. Therefore, it was convincingly shown that VCSD-induced destabilization of lysosomal membrane and release of lysosomal cathepsin B into the cytosol might initiate the lysosomal apoptotic pathway, which is dependent on mitochondria destabilization. This is in agreement with previous literature data. 38 Although increased levels of cytosolic cytochrome c and cathepsin B, along with increased Bid cleavage after VCSD in this study, indicate that injured neurons have undergone mitochondrial and lysosomal membrane permeability damage, and such mechanisms are also exploited with pharmacological interventions, these data do not establish a directly causative link between cathepsin B and tBid-mediated mitochondrial initiation of cell death. Further studies are required to evaluate this in more detail.
In conclusion, we demonstrate that post-injury P188 treatment attenuates the morphology characteristics of injured cells, and cell death in primary cultured neurons. Additionally, treatment with P188 restores mitochondrial and lysosomal membrane integrity after VCSD. However, our data do not conclusively show that: 1) P188's primary protective mechanism is through inhibition of Bid-mediated cell death, 2) mitochondrial and lysosomal membrane resealing is the only, or even the major mechanism explaining the observed neuroprotection by P188. Calcium is known to play a role in endogenous membrane repair mechanisms. 41 Increase in [Ca2+]i following mechanical injury (VCSD) was blocked by calcium chelation EGTA and by repairing the cell membrane with P188. 32 The data suggest that, mimicked by resealing the membrane and stopping calcium influx with EGTA treatment, P188 is preventing the initiation of the secondary cascades. Thus, we conclude that attenuating neuronal outer membrane damage would be expected to reduce the subsequent activation of pathology cascades, which could lead to mitochondrial and lysosomal damage. Further studies are needed to investigate the exact mechanism by which P188 repairs the damaged mitochondrial and lysosomal membrane following VCSD in primary cultured neurons.
Footnotes
Acknowledgments
This work was supported by the National Science Foundation of China (No. 81271379, No. 81172911, No. 81172897), and the Shanghai Forensic Key Laboratory Foundation (No. KF1005).
Author Disclosure Statement
No competing financial interests exist.