
Abstract
Vascular endothelial growth factor (VEGF) plays a role in angiogenesis and has been shown to be neuroprotective following central nervous system trauma. In the present study we evaluated the pro-angiogenic and neuroprotective effects of an engineered zinc-finger protein transcription factor transactivator targeting the vascular endothelial growth factor A (VEGF-ZFP). We used two virus delivery systems, adeno-virus and adeno-associated virus, to examine the effects of early and delayed VEGF-A upregulation after brain trauma, respectively. Male Sprague-Dawley rats were subject to a unilateral fluid percussion injury (FPI) of moderate severity (2.2–2.5 atm) followed by intracerebral microinjection of either adenovirus vector (Adv) or an adeno-associated vector (AAV) carrying the VEGF-ZFP construct. Adv-VEGF-ZFP–treated animals had significantly fewer TUNEL positive cells in the injured penumbra of the cortex (p<0.001) and hippocampus (p=0.001) relative to untreated rats at 72 h post-injury. Adv-VEGF-ZFP treatment significantly improved fEPSP values (p=0.007) in the CA1 region relative to injury alone. Treatment with AAV2-VEGF-ZFP resulted in improved post-injury microvascular diameter and improved functional recovery on the balance beam and rotarod task at 30 days post-injury. Collectively, the results provide supportive evidence for the concept of acute and delayed treatment following TBI using VEGF-ZFP to induce angiogenesis, reduce cell death, and enhance functional recovery.
Introduction
T
The VEGF-ZFP transcription factor has been previously shown to accelerate cutaneous wound healing in mice, promote angiogenesis in preclinical models, 9 restore VEGF-A deficits in sensory neurons, improve motor and sensory nerve conduction velocities in models of diabetic neuropathy, promote axon growth in primary neurons, 13 –16 enhance recovery after spinal cord compression injury, 17 and improve recovery after modeled ischemia. 18 Here we sought to evaluate two treatment paradigms after TBI in rats. The first approach was to target the acute stages of secondary injury through use of recombinant adenoviral (Adv) vectors. Adv vectors have high transcription yield but short duration of transgene expression. 19 The second approach was to evaluate outcome following prolonged and delayed VEGF-A expression using recombinant adeno-associated viral (AAV) vectors, which exhibit slower but sustained transgene expression. 20 We report that Adv-VEGF-ZFP and AAV2-VEGF-ZFP treatment reduces the presence of TUNEL positive cells, improves microvascular recovery, and contributes to improved functional outcome measures after injury.
Methods
All surgical procedures were performed in accordance with guidelines established by the Animal Care Committee at St. Michael's Hospital in accordance with the standards set by the Canadian Council on Animal Care.
Viral vector constructs
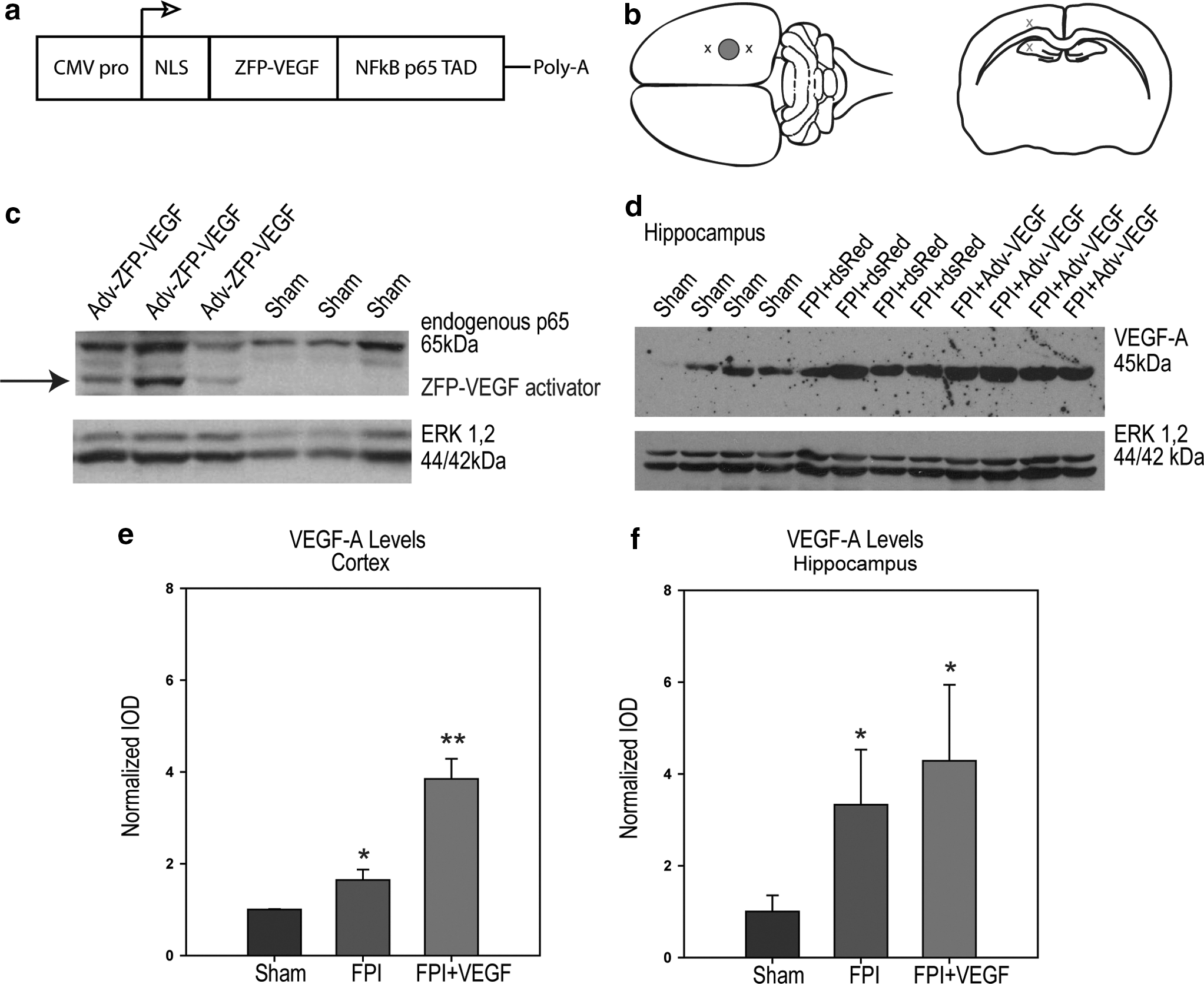
We used two viral constructs, adenovirus and adeno-associated virus, to examine the effects of early and delayed VEGF-A upregulation on outcome after FPI, respectively. The VEGF-A- ZFP activator and controls were provided as viral vectors by Sangamo BioSciences (Richmond, CA) and have been previously described. 8,14 A diagram of the VEGF-ZFP expression cassette is illustrated in Figure 1a. 32E-p65 is a 378 amino acid multidomain protein that is composed of three functional regions: (a) the nuclear localization signal (NLS) of the large T-antigen of SV40; (b) a designed three-finger zinc-fingered protein (32E) that binds to a 9 base-pair target DNA sequence (GGGGGTGAC) present in the human VEGF-A promoter region; and (c) the transactivation domain from the p65 subunit of human NF-κB, which is identical to VZ+434, subcloned into pVAX1 (Invitrogen, San Diego, CA) with expression driven by the human cytomegalovirus (CMV) promoter. 14
(
Adenoviral (Ad5-32Ep65 or Ad5-DsRed) and AAV (AAV2-32Ep65 or AAV2-GFP) vectors were packaged by transfecting T-REx-293 cells (Invitrogen, San Diego, CA). T-REx-293 cells in ten-stack cell factories were inoculated with Adv vectors at a multiplicity of infection (MOI) of 50 to 100 particles per cell. When adenoviral mediated cytopathy effect (CPE) was observed, cells were harvested and lysed by three cycles of freezing and thawing. Crude lysates were clarified by centrifugation.
AAV2 vectors were produced in five-stack cell factories. T-Rex 293 cells were seeded and grown 3 days prior to transfection. The calcium phosphate method was used for transfection. At day 3 post-transfection, the cells were harvested and AAV2 was purified by two rounds of cesium chloride density gradient centrifugation. The cesium chloride was removed by dialysis against phosphate buffer saline (PBS) with additional sodium chloride to 200 mM and Pluronic F 68 (Sigma Aldrich, St. Louis, MO) to 0.01%. Infectious titers of the Ad vectors were quantified using the Adeno-X Rapid Titer kit (Clontech, Mountain View, CA). The genome copy number of AAV2 was determined using Taqman polymerase chain reaction (PCR; Applied Biosystems, Foster City, CA). 21
Fluid percussion injury and vector delivery
In the current study, we used the unilateral rat fluid percussion injury (FPI) model. 22 –24 In brief, male Sprague-Dawley rats (350–400 g) were anesthetized with 2.0–2.5% isoflurane. Temperature was maintained at 37°C by a thermal heating blanket. A craniotomy (∼2 mm diameter) was performed in the right lateral hemisphere such that the medial edge of the craniotomy was ∼2 mm from the midline suture, midway between bregma and lambda. A polyethylene luer fitting with inner diameter of ∼1.5 mm was fixed to the opening with cyanoacrylate adhesive and dental acrylic and cured for 20 min. Prior to FPI, the luer fitting was filled with 0.9% isotonic saline and attached to the FPI device. Rats were subject to an extradural fluid percussion injury with the weighted pendulum set to an angle of 27°, resulting in a single peak pressure wave of 2.2–2.5 atm. The duration of the waveform response due to fluid percussion was recorded as 12–15 msec. Bone wax was used to seal the hole in the skull and scalps were sutured prior to recovery in a temperature-controlled chamber.
FPI rats were randomized to receive Adv-VEGF-ZFP, AAV2-VEGF-ZFP, Adv-DsRed, or AAV2-GFP at 20 min following fluid percussion injury. Intracerebral microinjections were made by stereotaxic measurements at a depth of 1.5 mm into the cortex, rostral and caudal to the location of fluid percussion injury application. Hippocampus injections were performed at a depth of 3.75 mm rostral and caudal to the injury epicenter (Fig. 1b). Coordinates for rostral and caudal virus delivery were −2 mm and −6 mm from the bregma, respectively, based on coordinates by Paxinos and Watson. 25 The focal injury area spanned approximately the distance from −3 mm to −5 mm from the bregma. Two μL of either Adv or AAV2 containing the VEGF-ZFP (5×1010 pfu/mL) was administered per injection site for a total of 8 μL per treated rat at a rate of 0.5 μL/min similar to the treatment paradigm used by Liu and colleagues 8 in a model of spinal cord compression injury.
Western blot analysis
Rats were sacrificed at 72 h after sham surgery or fluid percussion injury for Western blot analysis of VEGF-A and VEGF-A activator expression (n=6 per treatment group). Brain tissue samples were collected from the injured cerebral cortex and hippocampus in iced artificial cerebral spine fluid (ACSF; 126 mM NaCl, 3 mM KCl, 1.4 mM KH2PO4, 2.4 mM CaCl2, 1.3 mM MgSO4, 26 mM NaHCO3, 20 mM glucose) bubbled with carbogen (95% O2, 5% CO2). The extracted tissue blocks included the injury epicenter as well as the rostral and caudal injection sites. Samples were homogenized in lysis buffer containing protease inhibitors (50 mM Tris-HCl, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM NaF, 1 μg/mL aprotinin, 1 μg/ mL leupeptin, 1μg/mL pepstatin). Protein quantification was determined by the modified Lowry method. 26 Samples were normalized for equal loading (40 μg/lane) and were electrophoresed on 12% SDS-PAGE gels and transferred overnight to nitrocellulose membranes (Bio-Rad, Mississauga, Canada). Blocking of membranes was performed in 5% non-fat milk blocking solution for 1 h at room temperature (RT). Immunoblots were probed for VEGF-A using rabbit polyclonal anti-VEGFA antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Primary incubations were performed overnight at 4°C at a dilution of 1:200 in blocking solution. Secondary antibody incubation was performed for 45 min with horse-radish peroxidase (HRP) conjugated goat anti-rabbit (1:5000 dilution) secondary antibodies. Expression of the VEGF-A activator was assessed using a rabbit polyclonal anti-NFκBp65 antibody (Santa Cruz Biotechnology) at a dilution of 1:200 in blocking solution. Secondary antibody incubation was performed for 45 min with HRP-conjugated goat anti-rabbit (1:5000 dilution) secondary antibodies. We chose total ERK1,2 expression as a loading control as others have demonstrated that total ERK expression does not change after brain trauma while phosphorylated activation of ERK (p-ERK1,2) expression is altered. 27 –29 Rabbit polyclonal anti-ERK1,2 antibody (Sigma Aldrich, Oakville, Canada) was used as a loading control (1:15,000 dilution) and visualized with HRP-conjugated goat anti-rabbit (1:3000 dilution) secondary antibodies. Washes in three changes of Tris-buffered saline Tween-20 (TBST) were performed between incubations. Bands were visualized using a chemiluminescence kit (Perkin Elmer, Waltham, MA) and exposure to X-ray film. Blots were scanned at 300 dpi and densitometry analysis performed with ImageJ software (rsbweb.nih.gov/ij/). Protein levels were normalized to levels of ERK1,2 expression. Post-injury normalized integrated optical density (IOD) values were expressed as changes relative to naïve levels of expression.
TUNEL quantification and immunohistochemistry
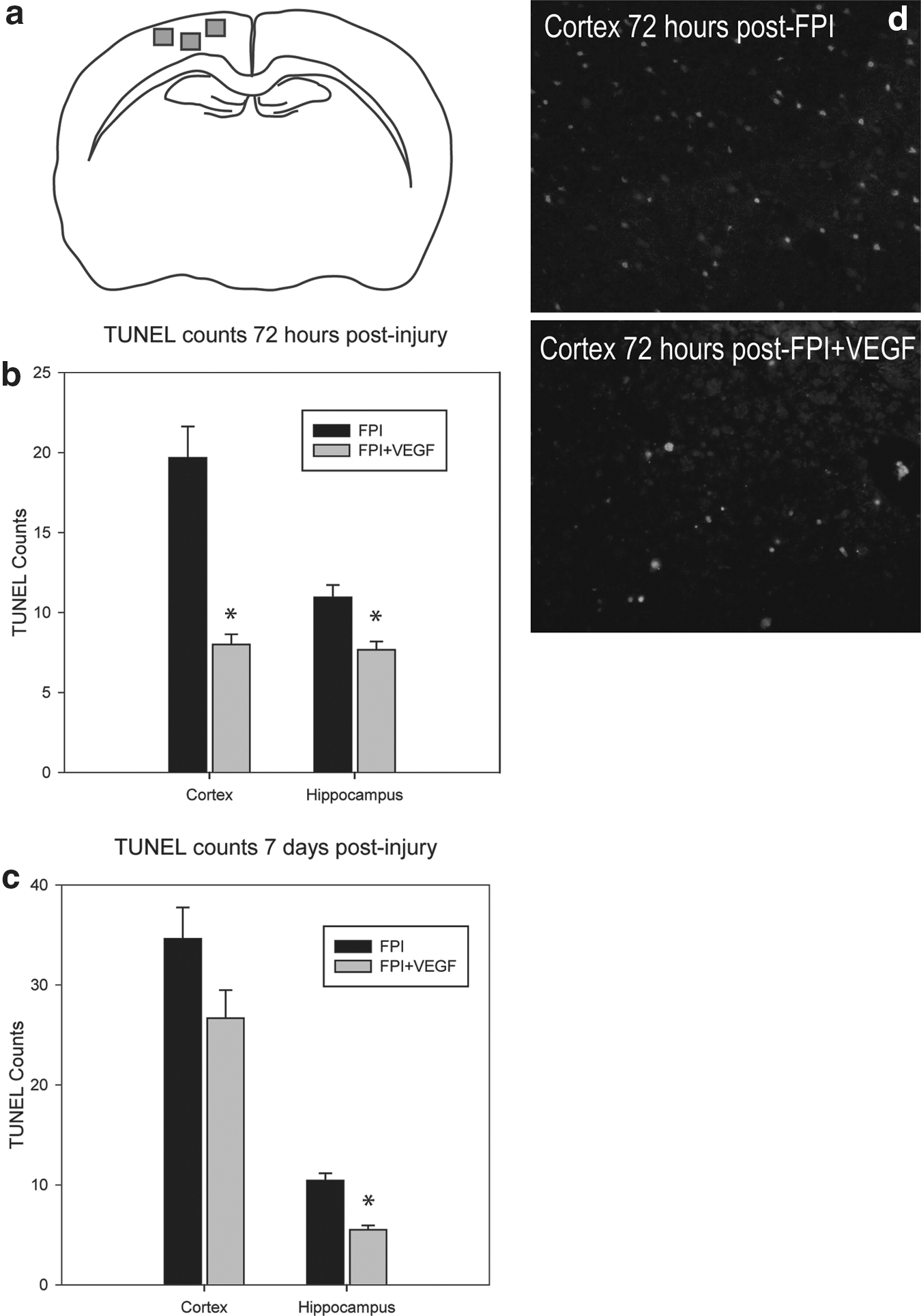
Adv-VEGF-ZFP–treated animals and Adv-DsRed controls were transcardially perfused with 0.9% saline followed by 4% paraformaldehyde at 72 h and 7 days post-injury (n=6 per time point/treatment group). Brains were post-fixed overnight in 4% paraformaldehyde with 0.5 M acetate solution prior to paraffin embedding. Paraffinized coronal brain sections of 10 μm thickness were used for in situ fluorescein TUNEL (Roche Applied Science, Laval, Canada) according to the manufacturer's instructions. TUNEL positive cells were quantified in the cortex from three 20x objective fields with dimensions, 408× 326 μm, immediately below and adjacent to the epicenter of fluid percussion application (Fig. 3a). Four non-adjacent sections per rat were sampled from −2.5 mm and −3.8 mm from bregma. Ipsilateral hippocampus TUNEL counts were made from the CA1, CA3, and dentate gyrus. All TUNEL quantification was performed in a blinded manner.
Double-labeled immunohistochemistry of DsRed or GFP expression in conjunction with cell-specific markers was assessed in fresh frozen tissues at 3 days or 30 days post-injury, respectively. Following 4% paraformaldehyde transcardial perfusion and post-fixation, brains were cryoprotected in 30% sucrose in PBS for 48 h at 4°C. Brains were snap frozen in 2-methylbutane and embedded in Cryomatrix (Fisher Scientific, Ottawa, Canada). Coronal brain sections were obtained on a cryostat at a thickness of 16 μm. Tissue sections were pretreated with 10% normal goat serum (NGS) and 0.01% Triton-X blocking solution for 1 h at RT in PBS prior to primary antibody incubation. Microwave antigen retrieval in 0.01 M sodium citrate buffer was used for anti-CNPase labeling. Primary antibody incubations were performed with monoclonal anti-NeuN (Millipore, Billerica, MA), GFAP (Sigma Aldrich), CNPase (Millipore), OX-42 (Sigma Aldrich), RECA-1 (Abcam, Cambridge, MA), or alpha smooth muscle actin (Cedarlane, Burlington, Canada) for identification of neurons, astrocytes, oligodendrocytes, microglia/macrophages, endothelial cells, and arterioles, respectively. All primary antibodies were used at a dilution of 1:400 overnight at 4°C in blocking solution. Alexa 488 or Alexa 555 goat anti-mouse secondary antibodies (Invitrogen, Burlington, Canada) were used for visualization. Secondary antibodies were incubated at a dilution of 1:1000 in blocking solution for 1 h at RT. Three washes were made in 0.1 M PBS between each step. Hoechst (Invitrogen) was used as a nuclear counterstain. Negative controls were run as described, with the omission of primary antisera.
Microangiography
Assessment of cerebral microvascular density in sham-, FPI+Adv-VEGF-ZFP–, and FPI+DsRed–treated animals was performed as previously described using MICROFIL® (Flow Tech, Carver, MA). 1 Briefly, rats (n=6 per treatment group) were anesthetized with ketamine at 2 weeks post-injury and transcardially perfused with blue silicone compound MV-120, diluent, and curing agent at an initial rate of 180 mL/h for 5 mL then at 55 mL/h for a volume of 10 mL with a KDS120 syringe pump (KD Scientific, Holliston, MA). The silicone compound was cured for a minimum of 90 min prior to brain extraction and storage in 70% ethanol. Following graded alcohol dehydration, brains were transferred to methyl salicylate for tissue clearing. A brain slicer (Zivic Instruments, Pittsburg, PA) producing 2 mm coronal slices was used to section the cerebral cortex. Slices were mounted on slides, cover-slipped, and imaged with a Nikon Eclipse 90i microscope using the extended depth of focus (EDF) module to create an in-focus image from a series of Z-plane stacks.
Electrophysiology
Hippocampus CA1 field excitatory post-synaptic potentials (fEPSPs) were recorded from sham-, FPI+Adv-VEGF-ZFP–, and FPI+Adv-DsRed–treated animals at 3 days (n=7 per treatment group). Brains were dissected in iced ACSF bubbled with 5% CO2:95% O2. A vibratome was used to section brains transversely at a thickness of 400 μm with each brain yielding approximately three to five recordable slices. A concentric bipolar microelectrode was placed along the Schaffer collateral pathway, and fEPSP recordings were taken from the somatic CA1 layer using microelectrodes (2–3 MΩ) backfilled with 150 mM NaCl. For in/out ramp recordings, stimulation pulses of constant current (0.1–1.5 μA, 0.1 msec) were generated by a Grass S88 stimulator (Grass Instrument, West Warwick, RI) and delivered through an isolation unit every 30 sec. The evoked field potentials were recorded using an Axopatch 200B amplifier and data stored and analyzed with the pCLAMP 10 software (Molecular Devices, Sunnyvale, CA). Long-term potentiation (LTP) was induced by theta-burst stimulation (TBS). Baseline recordings at 40% maximum intensity values derived from in/out curves were used for LTP studies. The TBS protocol consisted of five bursts at 200 msec intervals (5 Hz). Each burst consisted of five pulses at 100 Hz. TBS was applied three to four times with 45 sec intervals to induce LTP. Amplitude responses were recorded every 30 sec up to 660 sec following TBS. LTP was calculated as the mean value of the LTP amplitude from t=210 to t=660 sec following TBS divided by the initial baseline amplitude. The mean value derived from brain slices of a single rat was used as the representative unit of measurement for the animal.
Behavioral testing
Sham, FPI+AAV2-GFP, and FPI+AAV2-VEGF-ZFP rats were tested on a balance beam and rotarod task at 7, 10, 14, 17, 21, and 30 days after injury (n=9 per treatment group). The Morris Water Maze (MWM) test was used to examine spatial memory at 10, 14, 17, 21, and 30 days after injury (n=9 per treatment group). The balance beam consisted of a wooden dowel elevated 60 cm above a water bath. The diameter of the beam was 1 cm. The time spent on the beam (latency) was recorded over a 2 min period. Each rat was given three trials per testing day. For the rotarod task, the initial starting speed was 1 revolution per minute (rpm). At 45 sec the rotating drum reached maximum speed of 15 rpm. The total length of each rotarod trial was 2 min. The distance, time on the rod, and final rpm speed were recorded. Water maze testing consisted of rats being placed in one of four randomized quadrants in a 178 cm diameter pool. Rats were given 90 sec to locate a submerged and fixed location platform (NW quadrant) with a diameter of 10 cm. The time to platform was recorded. Rats that did not find the platform in the allotted time period were given a default time of 90 sec and placed on the platform for 10 sec. Three MWM trials were given per testing session.
Statistical analysis
Densitometry analysis of VEGF-A expression in Western blot analysis was performed using a one-way ANOVA and post-hoc SNK test. TUNEL counts were analyzed using a t test for comparisons between FPI and FPI+VEGF-ZFP treatment. Microvascular density analysis of the cortex and hippocampus was analyzed using a one-way ANOVA and post-hoc Tukey test. Field excitatory postsynaptic potentials (fEPSP) were analyzed using a one-way ANOVA and SNK-post hoc. The maximum evoked population spike amplitude obtained at 150 μA stimulation was used for analysis. LTP analysis was performed by comparing the post-tetanic stimulation amplitudes immediately following TBS by one-way ANOVA and post-hoc SNK test. The mean LTP values (mean PSA/baseline amplitude from 210–660 sec post-TBS) were compared between treatment groups by one-way ANOVA and post-hoc SNK test. The diameter of Reca-1 positive capillaries in the AAV2 study was analyzed by one-way ANOVA and Dunn's post-hoc test. Behavioral tests were analyzed using a two-way repeated measures ANOVA. The dependent variable for the MWM was the latency to platform finding. For the rotarod task, the dependent variable was the distance traveled. On the balance beam we evaluated two dependent parameters, the time spent on the beam and the total number of hindlimb foot faults. A SNK test was used for post-hoc analysis when values from the two-way repeated measures ANOVA were significant. All error bars are expressed as standard error of mean (SEM). Statistical significance was assumed at a value of p≤0.05.
Results
Mortality rates following FPI and treatment
A total of four rats were excluded from analysis in this study. Two rats died following prolonged apnea with application of the FPI procedure. Two rats injected with Adv-VEGF-ZFP were euthanized 2 days post-injection due to excessive weight loss and lethargy. A post-mortem analysis was not performed on these rats.
VEGF-A protein expression is increased following intracerebral Adv-VEGF-ZFP injection
Rats injected with Adv-VEGF-ZFP expressed the VEGF-ZFP activator at 3 days following virus administration (Fig. 1c). The antibody also detected the presence of endogenous p65 in the protein homogenate. Upregulation of VEGF-A expression by the VEGF-ZFP activator was observed in the cortex and hippocampus when compared to the levels seen following FPI and treatment with the vector control (Fig. 1d,e). The increase in VEGF-A expression was significantly different in the cortex following injury and VEGF-ZFP (p<0.001; Fig. 1e). Post-hoc analysis indicated that there was a significant increase in VEGF-A expression following FPI alone (p<0.001), suggesting an endogenous VEGF-A mediated response to trauma. However, the expression of VEGF-A between FPI+DsRed and FPI+VEGF-ZFP was also significantly different (p<0.001), indicating that Adv-VEGF-ZFP delivery resulted in increased expression of VEGF-A beyond the endogenous levels of post-traumatic expression. Expression levels of VEGF-A were also significantly different in the hippocampus at 72 hours post-injury (p<0.001; (Fig. 1f). However, post-hoc analysis indicated that the increase in VEGF-A expression between FPI+DsRed– and FPI+VEGF-ZFP–treated animals was not statistically significant (p=0.19).
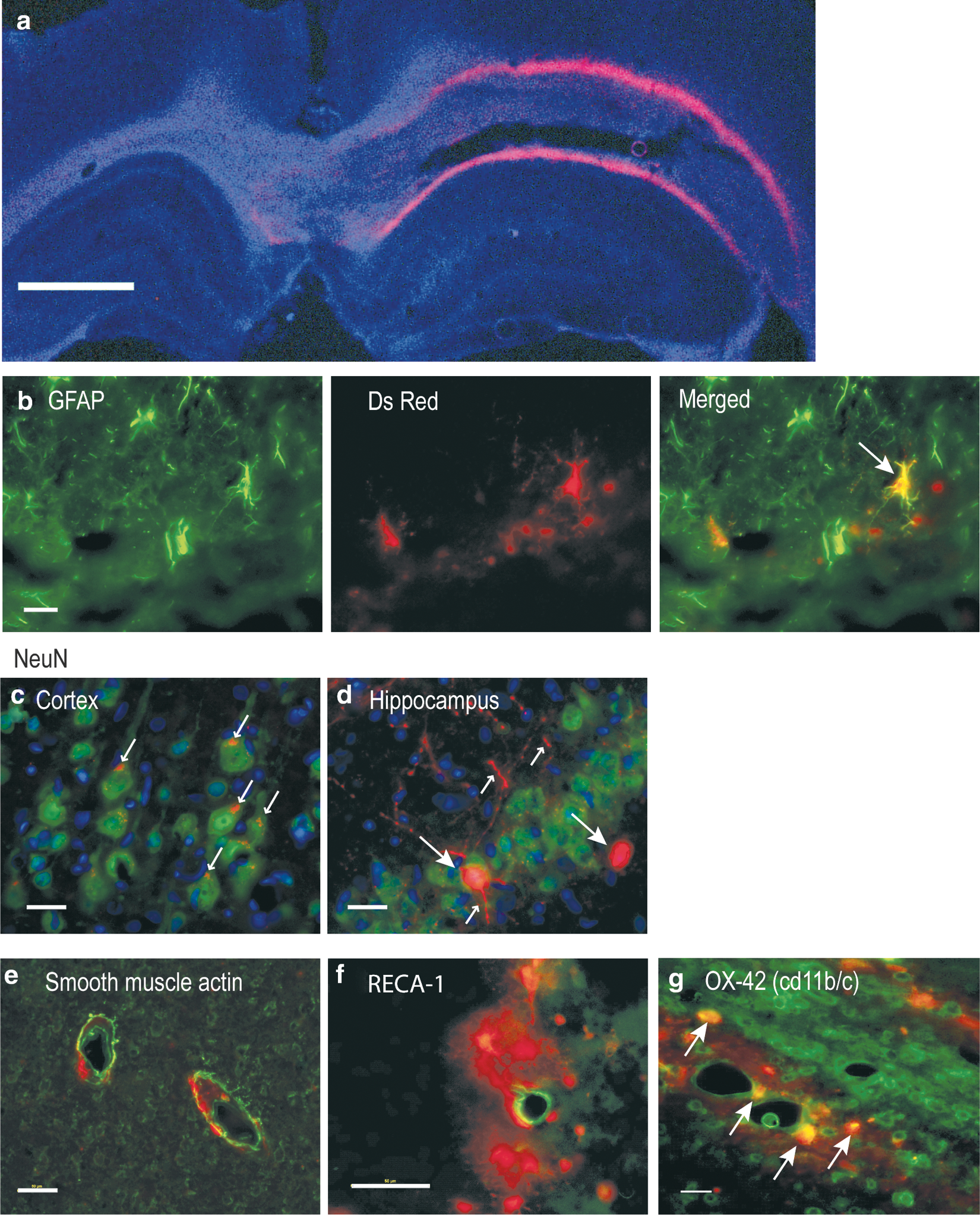
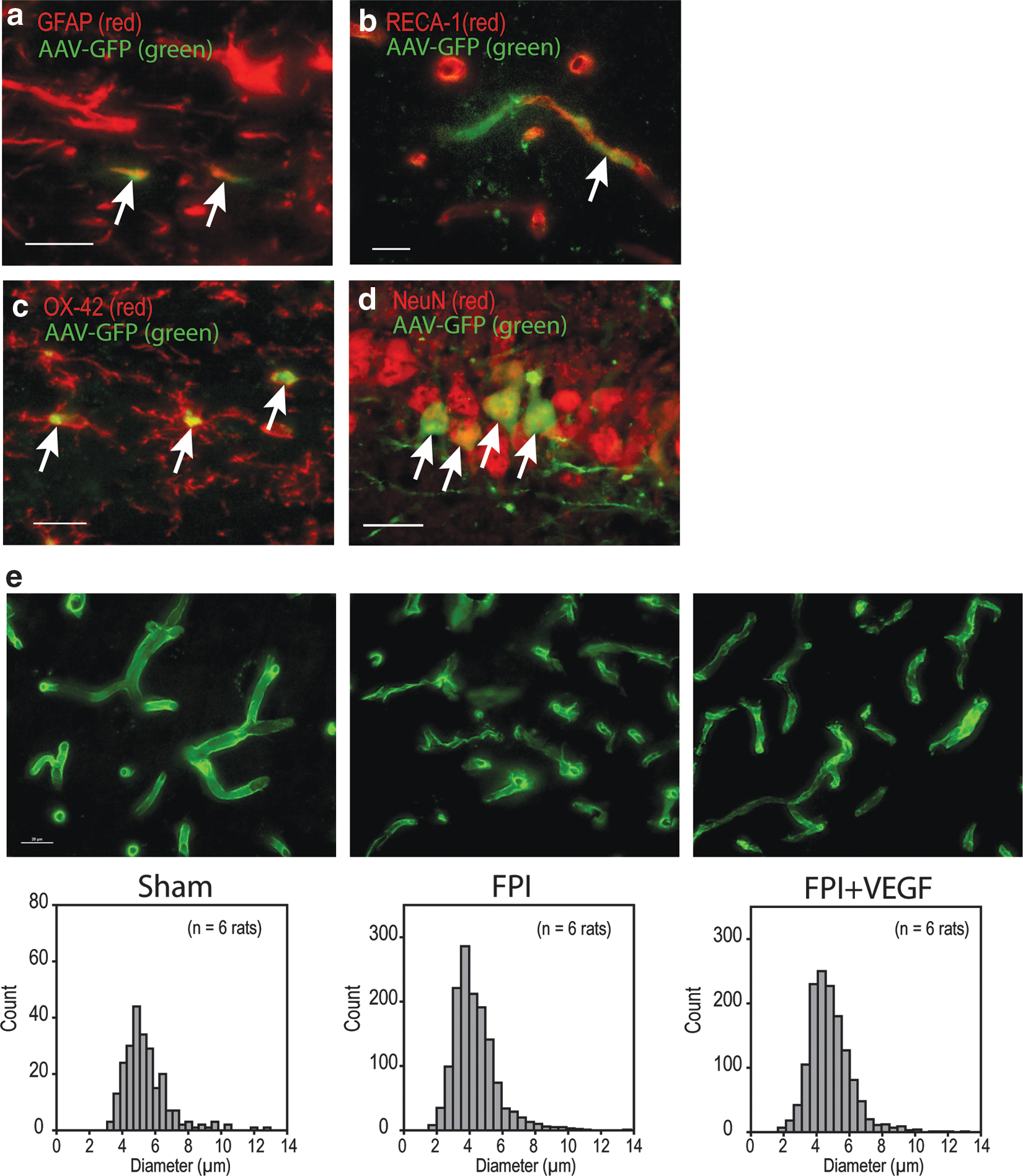
Qualitative localization of Adv-VEGF-ZFP transfection was determined by double-labeled immunohistochemistry of DsRed with cell specific markers at 72 h post-injection. Expression was localized to the hemisphere of intracerebral injection. Interestingly, despite injections into the cortex and hippocampus, DsRed expression was most pronounced around the periphery of the corpus callosum (Fig. 2a). Double-labeling with NeuN, GFAP, CNPase, OX-42, RECA-1, and smooth muscle actin indicated a heterogeneous population of infected cells. GFAP-expressing cells co-localized with DsRed (Fig. 2b). In the cortex NeuN co-localization with DsRed was punctate within cell bodies while expression in CA1 hippocampus neurons was more pronounced and also present in neuronal processes (Fig. 2c,d, respectively). We were unable to perform double-labeled immunohistochemistry with oligodendrocyte marker CNPase in frozen tissues since microwave antigen retrieval resulted in the loss of DsRed expression. For similar reasons we were unable to perform double-labeled immunohistochemistry in GFP expressing tissues. Despite close proximity of expression among endothelial and arterial markers, RECA-1, and smooth muscle actin, there was no observable co-localization with DsRed (Fig. 2e,f). Finally, a subset of OX-42 positive cells was found to co-localize with DsRed (Fig. 2g).
(
Adv-VEGF-ZFP treatment reduces presence of TUNEL at 3 and 7 days post-injury
The number of TUNEL positive cells at 3 days post-injury was significantly different between FPI+DsRed and FPI+Adv-VEGF-ZFP (p<0.001). The mean number of TUNEL positive cells in the injured cortex was 19.7±2.0 in FPI+DsRed rats and 8.0±0.6 in FPI+Adv-VEGF-ZFP treated rats (Fig. 3b). Representative images are shown in Figure 3d. In the hippocampus a significant reduction in TUNEL positive cells was observed at 3 days post-injury (p<0.001). The mean number of TUNEL positive cells was 10.9±0.8 in FPI+DsRed rats and 7.7±0.5 in FPI+Adv-VEGF-ZFP–treated rats.
(
At 7 days post-injury, a similar trend in neuroprotective effects was observed. There was a reduction in the number of TUNEL positive cells in the cortex that approached statistical significance (p=0.07). In the hippocampus, mean TUNEL counts were 10.4±0.7 cells in the FPI+DsRed group versus 5.5±0.5 cells in FPI+Adv-VEGF-ZFP rats (p<0.001; Fig. 3c).
Adv-VEGF-ZFP treatment improves microvascular density in the cortex at 2 weeks post-injury
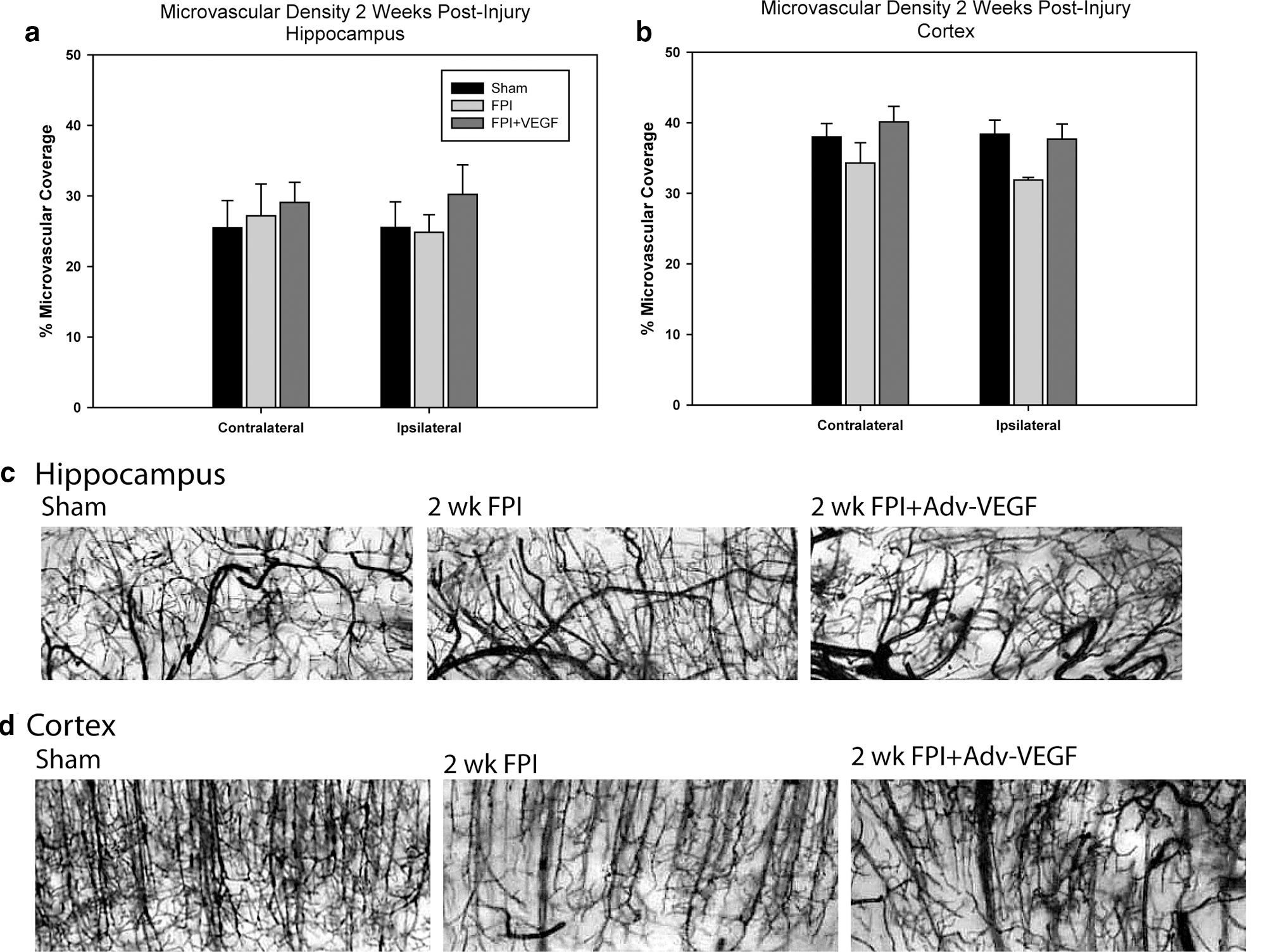
In the hippocampus we observed no statistically significant difference in microvascular density in either the contralateral or ipsilateral hemisphere (Fig. 4a). Representative images are shown in Figure 4c. In the cortex the mean density values were not significantly different in the contralateral cortex with 38.0±1.9%, 34.3±2.9%, and 40.1±2.2% area coverage in sham-, FPI+DsRed–, and FPI+VEGF-ZFP–treated animals, respectively. The injured ipsilateral cortex density values approached significance by one-way ANOVA analysis (p=0.052), with mean density values of 38.4±2.0%, 31.9±0.4%, and 37.7±2.1% area coverage in sham,- FPI+DsRed–, and FPI+VEGF-ZFP–treated animals, respectively (Fig. 4b). Although there was a numerical trend toward increased microvascular density, there were morphological differences between sham- and VEGF-ZFP–treated animals. There were noticeably larger caliber vessels, which appeared in the VEGF-ZFP–treated animals relative to sham- and FPI+DsRed–treated rats. Representative images are shown in Figure 4d.
(
Adv-VEGF-ZFP treatment improves CA1 fEPSP but not long-term potentiation 72 h after injury
Schaffer collateral stimulation and field EPSP from the CA1 pyramidal neurons were recorded from the ipsilateral cortex at 3 days post-injury and treatment (Fig. 5a). There was a notable decline in the fEPSP amplitude following FPI (representative traces shown in Fig. 5b). Mean fEPSP values at maximum stimulation of 150 μA were 3.7±0.2 mV, 2.3±0.3 mV, and 3.0±0.3 mV in sham-, FPI+DsRed–, and FPI+VEGF-ZFP–treated animals, respectively (p=0.007). Post-hoc analysis indicated a significant decline in fEPSP amplitude in FPI rats relative to sham (p=0.006; Fig. 5c). There was also a significant improvement in fEPSP amplitude in VEGF-ZFP–treated rats relative to FPI+DsRed (p=0.050).
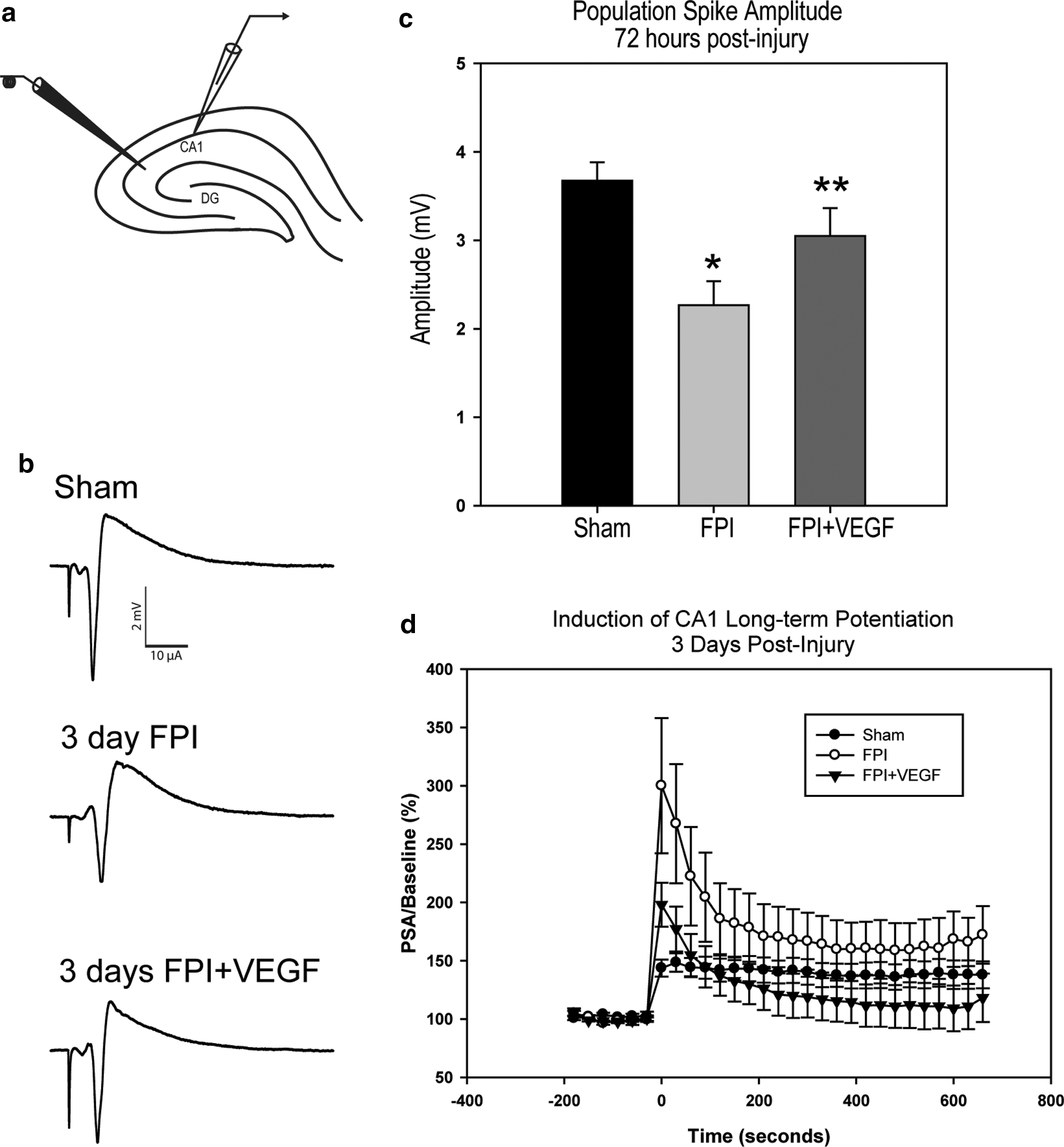
(
We examined the effect of VEGF-ZFP treatment on LTP at 72 h post-injury. Interestingly, we observed a large increase in the immediate post-tetanic potentiated (PTP) response following theta-burst stimulation. The mean amplitude values at time=0 sec immediately following TBS were 143.6±7.3%, 333.8±58.0%, and 198.0±11.2% in sham, FPI+DsRed, and FPI+VEGF-ZFP, respectively (p=0.007; Fig. 5d). Post-hoc analysis indicated a significant increase in the PTP response in the FPI+DsRed treatment group relative to sham animals (p=0.006) and FPI+VEGF-ZFP animals (p=0.026). There was no significant difference between sham and FPI+VEGF-ZFP treatment groups (p=0.36). The mean LTP response at t=210 sec to t=660 sec following TBS was also significantly different between treatment groups (p<0.001). Post-hoc analysis indicated a significant increase in the mean LTP response in FPI+DsRed animals relative to sham animals (164.2±1.2% vs. 138.6±0.4% for FPI+DsRed and sham, respectively; p<0.05). The mean LTP values in the FPI+VEGF-ZFP treatment group was 115.0±1.2% and was significantly lower than the sham group (p<0.05).
Cellular localization of AAV2-GFP in the brain following intracerebral microinjection
Following our initial results using the Adv-VEGF-ZFP, we sought to evaluate the effects of VEGF-ZFP using a vector with prolonged expression and lower level of pathogenicity relative to Adv vectors. AAV delivered GFP was only detected by 14 days post-injury (data not shown), indicating that the time to infection, transcription, and translation of the VEGF-ZFP construct occurs at a later period following initial injury. GFP expression localized to the hemisphere of the site of injection. GFP co-localized minimally in cells expressing GFAP (Fig. 6a) and RECA-1 (Fig. 6b). GFP did not co-localize with OX-42. However, expression localized within OX-42 expressing cell bodies, suggesting possible phagocytosis (Fig. 6c). Morphology of OX-42 cells was consistent with a reactive ramified phenotype unlike the activated ameboid macrophage phenotype observed following Adv administration. 30 GFP was detected in numerous NeuN positive cells in the CA1 pyramidal layer (Fig. 6d). GFP expression was evident in axonal and dendritic processes as well. Despite co-localization with cellular markers for astrocytes, endothelial cells, microglia, and neurons, GFP-expressing cells generally had very weak expression of cell-specific markers.
(
AAV2-VEGF-ZFP increases post-traumatic capillary diameter and reduces endothelial fragmentation
We evaluated endothelial density by examining expression of RECA-1 in capillaries of the cerebral cortex at 30 days post-injury and treatment. In sham animals, the morphology of RECA-1 positive capillaries was tubular and continuous (Fig. 6e). The mean diameter of sham capillaries was 5.44±0.09 μm and a total number of 242 RECA-1 positive objects were counted. In FPI+GFP animals, there was clear fragmentation of capillaries as evidenced by the higher total number of RECA-1 positive objects counted within a sampling frame (total of 1397 objects). Further, the mean diameter of capillaries in FPI+GFP animals was 4.33±0.04 μm. One-way ANOVA analysis indicated a significant difference in the capillary diameter between treatment groups (p<0.001). Post-hoc analysis indicated that the mean diameter of capillaries in FPI+GFP rats was significantly smaller than that of sham rats (p<0.05). In the FPI+VEGF-ZFP treatment group, the total object count was 1386. The mean diameter of capillaries was significantly larger (4.66±0.04 μm) relative to FPI+GFP animals (p<0.05).
AAV2-VEGF-ZFP improves select behavioral outcome measures following FPI
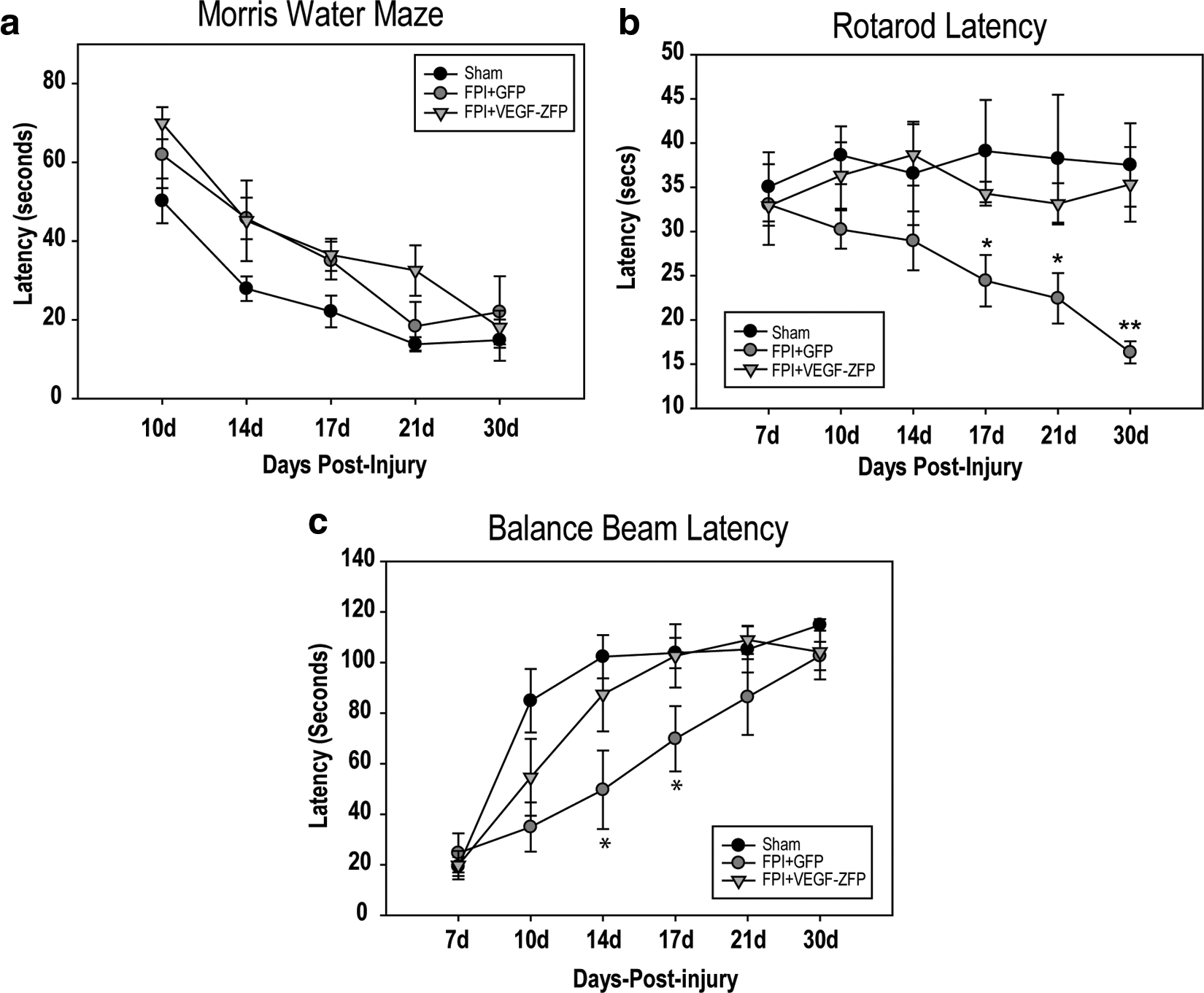
We evaluated the effects of AAV2-VEGF-ZFP treatment on behavioral activity using the rotarod, balance beam, and Morris Water Maze tasks. Results for the MWM indicated a significant difference among treatment groups (p=0.001; Fig. 7a). Post-hoc pair-wise comparisons indicated a significant difference between sham and FPI+GFP (p=0.002) as well as between sham and FPI+VEGF-ZFP (p=0.008), but not between injury and treatment comparisons.
(
On the rotarod task, a significant difference was found between treatment groups (p=0.047) as well as a significant interaction between treatment and testing day (p=0.037). Sham and FPI+VEGF-ZFP latencies did not vary significantly over the testing period, with a least square means latencies of 37.5±3.0 sec and 34.3±3.2 sec, respectively, and were not significantly different. We observed a significant decrease in the time spent on the beam in the FPI+GFP group relative to sham rats (p=0.040). On days 17, 21, and 30, there was a significant difference between FPI+GFP and sham animals (p=0.035, 0.021, and 0.002, respectively). The difference in latency at 17 days post-injury indicated a trend toward improved function in VEGF-ZFP–treated rats and was significantly different at 30 days post-injury (p=0.008).
There was a significant difference in latencies on the balance beam due to treatment (p=0.026; Fig. 7c). There was a significant difference in mean latencies between FPI+GFP and FPI+VEGF-ZFP at 14 (p=0.010) and 17 days (p=0.045) post-injury.
Discussion
In the present study we evaluated the neuroprotective and angiogenic effects of the VEGF-ZFP activator following modeled TBI in rats. We examined the effects of high yield, short duration adenoviral vectors on cell survival, and microvascular recovery. Using an AAV2 vector, we examined the effects of a sustained and delayed treatment and evaluated the effects of AAV2-VEGF-ZFP on capillary integrity and behavioral outcome up to 30 days post-injury.
Following FPI we observed an endogenous increase in VEGF-A expression in the cortex and hippocampus, suggesting an innate reparative or neuroprotective response to trauma. Introduction of the Adv-VEGF-ZFP significantly increased the endogenous levels within the cortex but did not significantly increase VEGF-A levels within the hippocampus. The quantification of TUNEL demonstrated a proportionally greater reduction of TUNEL in the cortex relative to the modest reduction in TUNEL in the hippocampus following Adv-VEGF-ZFP treatment. The limited effect of VEGF-ZFP on reducing TUNEL positivity in the hippocampus is in agreement with the nonstatistically significant increase in VEGF-A expression in the hippocampus following ZFP treatment. It is possible that the saturated level of VEGF-A expression with FPI alone may have minimized benefit from the VEGF-ZFP treatment. Another possibility may be due to low viral transduction efficiency in hippocampus neurons relative to transduction.
While VEGF-ZFGP treatment improved fEPSP amplitude, consistent with TUNEL quantification in the hippocampus, it was interesting to note that VEGF-ZFP treatment reduced the post-tetanic potentiated response but did not affect LTP. Our group and others 31 –33 have previously shown both in vitro and in vivo that brain injury results in an NMDA-dependant stoichiometric shift in the AMPA receptor populations from calcium impermeable, to those which are GluR2 lacking, or calcium permeable. The implications of this shift are such that surviving neurons are more sensitive to secondary insult. The electrophysiological data indicating an exaggerated PTP response following FPI supports the notion that the CA1 neurons were in a vulnerable state and prone to excitotoxic insult. The blunting of this heightened PTP response through VEGF-ZFP treatment suggests a possible interaction between VEGF signaling and post-synaptic receptor makeup as an underlying mechanism that governs the neuroprotective effects of VEGF. Schumann and colleagues 33 reported differences between the hippocampus and cortex in the expression pattern of NMDA receptor subunits, NR1, NR2A, and NR2B, and the GluR1 AMPA subunit following trauma, which may further explain our observed differences in response to VEGF-ZFP with respect to TUNEL counts. 33 VEGF administration has been shown to reduce neuronal excitation in motor neurons 34 and has recently been shown to act through modulation of NMDA receptor activity in CA1 neurons. 35
While the timing of treatment differed with the use of short duration Adv compared to the delayed but sustained expression by the AAV2 vectors, there were modest improvements in microvascular recovery observed in both treatment models. There were also associated improvements in behavioral outcome. However, the effects of VEGF-ZFP did not translate to uniform improvement in all behavioral parameters. The lack of significant effect on Morris Water Maze results with AAV2 treatment is in line with the modest effects on neuroprotection observed in the hippocampus of Adv-VEGF-ZFP–treated rats. There was no significant difference between GFP- and VEGF-ZFP–treated animals, suggesting that the VEGF treatment did not adversely affect nor improve spatial memory in this treatment paradigm. Given the cellular and region-specific tropism of the Adv and AAV vectors, the behavioral outcome assays that we evaluated may not have fully elucidated the behavioral consequences of VEGF-ZFP treatment. Further study is warranted in this regard.
Despite some improvements in outcome, it is also important to note the heightened inflammatory response following use of Adv and AAV as delivery vehicles for the VEGF-ZFP treatment. The morphology and clustered organization of OX-42 cells with macrophage phenotype suggest the possibility of an immune response toward the adenovirus. In both instances there was increased macrophage and reactive microglial presence in the vicinity of DsRed and GFP expression. While inflammation represents a double-edged sword in TBI, 36,37 it is possible that the influence of pro-inflammatory cytokines negated some of the neuroprotective and reparative effects conferred by the VEGF-ZFP treatment. The apparent internalization of GFP expressing cells by resident microglia and the reduced expression of various cell-specific proteins suggest that AAV-infected cells may undergo some transformation, perhaps accounting for the initiation of a mild immune response. The loss of expression of cell markers such as NeuN and RECA-1 in virus-infected cells is further indication that the titration of the viral load and sequestration of the virus to non-pathogenic levels requires further refinement in order to maximize the benefits of virally delivered gene therapy.
Despite the highest detected immunofluorescence levels of DsRed in the periphery of the corpus callosum delivered by the Adv, we did not evaluate the effects on white matter function or protection after lateral FPI. This remains to be investigated, as the behavioral improvements we observed could also be contributed to by white matter sparing.
Conclusion
Numerous preclinical studies have targeted early molecular cascade events associated with secondary injury progression aimed at promoting preservation of tissue after injury. However, translation of these studies effective to clinical treatment has been less successful. 38 –40 Based on our work, a gene therapy approach using an engineered zinc-finger protein activator shows promise as a potential treatment for traumatic brain injury. Moreover, the treatment paradigm presented in this study is novel from a therapeutic timing perspective since the delayed but sustained expression mediated by AAV vectors targets reparative mechanisms beyond the acute stages of secondary injury where the majority of neuronal injury takes place. The improvement conferred by the AAV2-VEGF-ZFP indicates a larger window of opportunity for intervention. While there are still challenges in gene therapy application, particularly with respect to potential complications with inflammation in TBI, the results are an encouraging proof-of-principle for the potential effectiveness of VEGF-ZFP therapy.
Footnotes
Author Disclosure Statement
S.K. Spratt, R. Surosky, G. Lee, D. Ando, and M. Giedlin are employees of Sangamo BioSciences, Inc., which provided the adenovirus and adeno-associated virus zinc-finger protein constructs used in this study.