
Abstract
Traumatic brain injury (TBI) is the leading cause of death and disability in children and young adults. Neuroprotective agents that may promote repair or counteract damage after injury do not currently exist. We recently reported that stimulation of the purinergic receptor subtype P2Y1R using 2-methylthioladenosine 5′ diphosphate (2MeSADP) significantly reduced cytotoxic edema induced by photothrombosis. Here, we tested whether P2Y1R stimulation was neuroprotective after TBI. A controlled closed head injury model was established for mice using a pneumatic impact device. Brains were harvested at 1, 3, or 7 days post-injury and assayed for morphological changes by immunocytochemistry, Western blot analysis, and wet/dry weight. Cerebral edema and expression of both aquaporin type 4 and glial fibrillary acidic protein were increased at all time points examined. Immunocytochemical measurements in both cortical and hippocampal slices also revealed significant neuronal swelling and reactive gliosis. Treatment of mice with 2MeSADP (100 μM) or MRS2365 (100 μM) 30 min after trauma significantly reduced all post-injury symptoms of TBI including edema, neuronal swelling, reactive gliosis, and AQ4 expression. The neuroprotective effect was lost in IP3R2-/- mice treated with 2MeSADP. Immunocytochemical labeling of brain slices confirmed that P2Y1R expression was defined to cortical and hippocampal astrocytes, but not neurons. Taken together, the data show that stimulation of astrocytic P2Y1Rs significantly reduces brain injury after acute trauma and is mediated by the IP3-signaling pathway. We suggest that enhancing astrocyte mitochondrial metabolism offers a promising neuroprotective strategy for a broad range of brain injuries.
Introduction
It has also been well established that TBI causes breakdown of the BBB, which is followed by vasogenic edema formation, 7,8 increased intracranial pressure, decreased cerebral blood flow, and subsequent ischemia. 9 –11 Cytotoxic edema, because of the loss of adenosine triphosphate (ATP)-maintained ion homeostasis, generally precedes vasogenic edema. 12,13 TBI impairs oxygen delivery to the brain, decreasing mitochondrial function and energy production. 14 –17 Central to the regulation of water homeostasis are aquaporins, a family of membrane water channels. 18 Aquaporin 4 is the primary water channel expressed within astrocytic end feet surrounding the brain vasculature. It is presumed to play a key role in edema formation, especially during a state of inhibited energy production. Inhibition of glial mitochondrial metabolism increases astrocyte swelling and necrotic cell death, which has been implicated as a primary cause of cytotoxic brain edema. 19 –22
Astrocytes, the most abundant cell type in the human brain, 23,24 play a vital role in supporting and protecting neuronal function and in modulating brain energy metabolism. 25,26 P2Y1R activation in astrocytes provides a mechanism whereby local extracellular signals can rapidly elevate release of Ca2+ through increased production of IP3. 27,28 IP3-mediated Ca2+ release increases mitochondrial Ca2+ and, consequently, increases respiration and ATP production. 29 –32
Our laboratory demonstrated that both in vitro and in vivo neuroprotection could be enhanced by increasing astrocyte mitochondrial metabolism via P2Y1Rs. 33,34 The goal of this study was to investigate the therapeutic effects of stimulating astrocyte mitochondrial metabolism using 2MeSADP and MRS2365, two P2Y1R ligands, as a novel therapy to treat patients with TBI.
Methods
Controlled closed skull injury model
A pneumatic impact device was used to generate a moderate TBI leaving the skull and dura matter intact. To achieve this, C57BL/6 mice were anesthetized with isoflurane (3% induction, 1% maintenance) in 100% oxygen. A body temperature of 37°C was maintained using a temperature-controlled heated surgical table. A small midline incision was made on the scalp using aseptic surgical techniques. A 5-mm stainless steel disc was positioned on the skull and fixed using superglue on the right parietal bone between bregma and lamda over the somatosensory cortex. The mouse was then positioned on a stage directly under the pneumatic impact tip. A calibrated impact was delivered at 4.5 m/sec at a depth of 2 mm, which generates a moderate injury in the mouse. Apneic episode after injury and righting reflex after removal of the mouse from anesthesia were timed and recorded (supplementary Fig. 3; see online supplementary material at
Mice were placed in a Thermo-Intensive Care Unit (Braintree Scientific model FV-1; 37°C; 27% O2) and monitored until fully awake and moving freely. Thirty minutes after injury or sham (uninjured), mice were treated with either vehicle (saline) or 100 μM 2-methylthioladenosine 5′ diphosphate (2MeSADP) or MRS2365 [[(1R, 2R, 3S, 4R, 5S)-4-[6-Amino-2-methylthio)-9H-purin-9-yl]-2,3-dihydroxybicycl-[3.1.0]hex-l-yl]methyl] diphosphonic acid mono ester trisodium salt (100 μm) intravenously. Brains were harvested at 1, 3, and 7 days post-TBI. At selected survival times, mice were anesthetized under isoflurane, sacrificed, and prepared accordingly for the assay to be performed. For immunostaining, mice were perfused with 4% paraformaldehyde in 5% sucrose and post-fixed overnight followed by placement in 30% sucrose for 48 h. Brains were then stored at −80°C until sliced.
IP3R2 knockout mice
A breeding pair of IP3R type 2 knockout mice were provided to our laboratory by Dr. Ken McCarthy with Dr. Ju Chen's permission. The mice were made in Dr. Chen's laboratory, the details of which can be found in Li and associates. 35
Edema formation
To quantify cerebral edema after TBI, brain water content was measured. Briefly, mice were anesthetized using 3% isoflurane 1, 3, or 7 days post-TBI or sham procedure. Mice were decapitated and the brain removed. Samples were placed in glass bottles and weighed (wet weight). The brains were dried at 105°C for 72 h and reweighed (dry weight). Brain water content was calculated as the difference between wet and dry weights using the formula ([wet weight-dry weight]/wet weight)×100%.
Nissl staining
Standard procedures were used for detection of Nissl body found in the cytoplasm of neurons to identify the neuronal changes. Briefly, brains were harvested as described above and sectioned at 25 μm and placed on gelatin-coated slides. The slides were dried at 37°C overnight. Slides were hydrated with graded alcohols to distilled water, 0.1% cresyl violet was applied for 7 min, followed by a wash in distilled water. The slides were then dehydrated, cleared in xylene, and a cover-slip applied. Images were acquired on a Zeiss AX10 microscope using a 40×objective. To analyze the soma area, three serial sections from each animal were stained as described.
After staining, brightfield images were collected from the impacted cortex and CA3 region of the impacted side of the brain at 40×. Image J was then used as follows: The find edges function key was used to determine the boundaries of each cell, the images were then despeckled, and the measure function was used to determine the area of each soma. The data were then averaged for each region imaged, and this was done for each of the three sections per animal. The resulting data were then averaged among all the animals in this experimental series.
GFAP staining
Slides were washed in phosphate buffered saline (PBS), permeabilized with 0.2% Triton-X 100 in PBS. Sections were then blocked in 5% goat serum. Glial fibrillary acidic protein (GFAP) antibody was diluted (1:1000) in goat serum and applied to the sections for 1 h at 37°C. The sections were then washed and secondary goat anti-rabbit IgG antibody was applied at 1:200 for 30 min at 37°C. Nuclei were stained with Hoechst 33342 (100 μg/mL). Slides were washed with PBS, and Vectashield was used to mount the cover-slip. Images were acquired on Nikon C1si microscope using a 60×1.1 NA water immersion objective. The 10×images were also collected and were used for the analysis of the number of reactive astrocytes per field (1.26 mm×1.26 mm).
Western blot analysis for GFAP and AQ4
Mice were anesthetized using isoflurane and subsequently decapitated. The brain was removed and placed on ice for dissection into impacted and non-impacted hippocampus and cortex. The isolated tissue was rapidly homogenized in chilled homogenization buffer (0.32M sucrose, 1mM ethylenediaminetetraacetic acid, 1M Tris-HCL pH=7.8) on ice using a Wheaton glass dounce (20 strokes). The homogenate was transferred to a 2 mL tube and centrifuged at 1000 g for 10 min at 4°C; the supernatant was collected and analyzed. Protein concentration was determined by the bicinchoninic acid assay using a 1:50 dilution. For each sample, 100 μg of protein was aliquoted and Laemmli buffer containing β-mercaptoethanol added; the sample was placed in a heat block for 3 min at 95°C. Samples were loaded on a 12% gel and ran at 80 V for 20 min followed by 40 min at 130 V.
Samples were transferred to nitrocellulose membrane at 100V for 1 h. The membrane was blocked with 5% milk in Tris-buffered saline with Tweet (TBS-T) for 30 min. GFAP (1:1000-Imgenex IMG-5083-A) or AQ4 (1:500; Santa Cruz SC-9888) was added and placed at 4°C overnight. The membrane was washed with TBS-T three times for 10 min. Secondary antibody for GFAP (Donkey anti-rabbit horseradish peroxidase [HRP] conjugated (ImmunoJackson Laboratories; 711-035-152; 1:20000) or AQ4 (donkey anti-goat HRP conjugated; Santa Cruz; sc-2020; 1:5000) was applied at room temperature for 1 h. The membranes were washed with TBS-T for 15 min (3 times) and developed using the Western Lightning® Plus-ECL kit (PerkinElmer, Inc) following manufacturer's directions.
Statistical analysis
One-way analysis of variance (ANOVA) was used to compare the differences among three or more groups. The Student t test was used to compare the difference between two groups. The significance level was set at p<0.05. Data are presented as mean±standard error of the mean (SEM). GraphPad Prism software (GraphPad Software Inc.) was used to perform statistical analyses.
Results
P2Y1R stimulation reduces whole brain vasogenic edema formation after TBI
Edema formation is a classic indicator of TBI that is thought to occur in two stages. First, cytotoxic edema forms because of the loss of intracellular ATP and the disruption of ion homeostasis. 36,37 Within minutes to hours, the second stage of edema is initiated when vascular fluid flows into the extracellular space (vasogenic edema) and increases intracranial pressure, generally with neurodestructive consequences. 38 –41 To test the impact of P2Y1R stimulation on both stages of edema formation, we first developed a controlled closed head injury model for mice using a pneumatic impact device. Whole brain vasogenic edema formation was measured as an increase in brain tissue water content using the wet/dry weight method as described previously. 42
One-way ANOVA analysis of time post-injury and treatment group revealed a significant increase in water content after TBI compared with sham-treated mice at all time points measured (Fig. 1). There was an 8.06% increase in water content after TBI at 24 h post-injury compared with sham mice (sham 72.41%±0.75, n=5; 24 h TBI 80.47%±1.56, n=8). Three days post-injury, a 10.35% increase was observed in TBI mice (82.76%±1.8; n=5) compared with sham mice, and the increase was maintained 7 days post-injury (82.62%±1.3; n=5) with a 10.21% increase above sham mice. When test mice were injected in the tail vein with the purinergic agonist 2MeSADP (100 μM, 100 μL) 30 min post-injury, there was a significant reduction in the percent of whole brain water content compared with untreated TBI mice at day 1 (74.69±0.65, n=5), day 3 (78.38±1.18, n=5) and day 7 (79.08±1.01, n=5) (Fig. 1). In addition, measurement of hemisphere size also demonstrated a significant increase following TBI and was reversed with 2Me SADP treatment (Supp. Fig. 1).
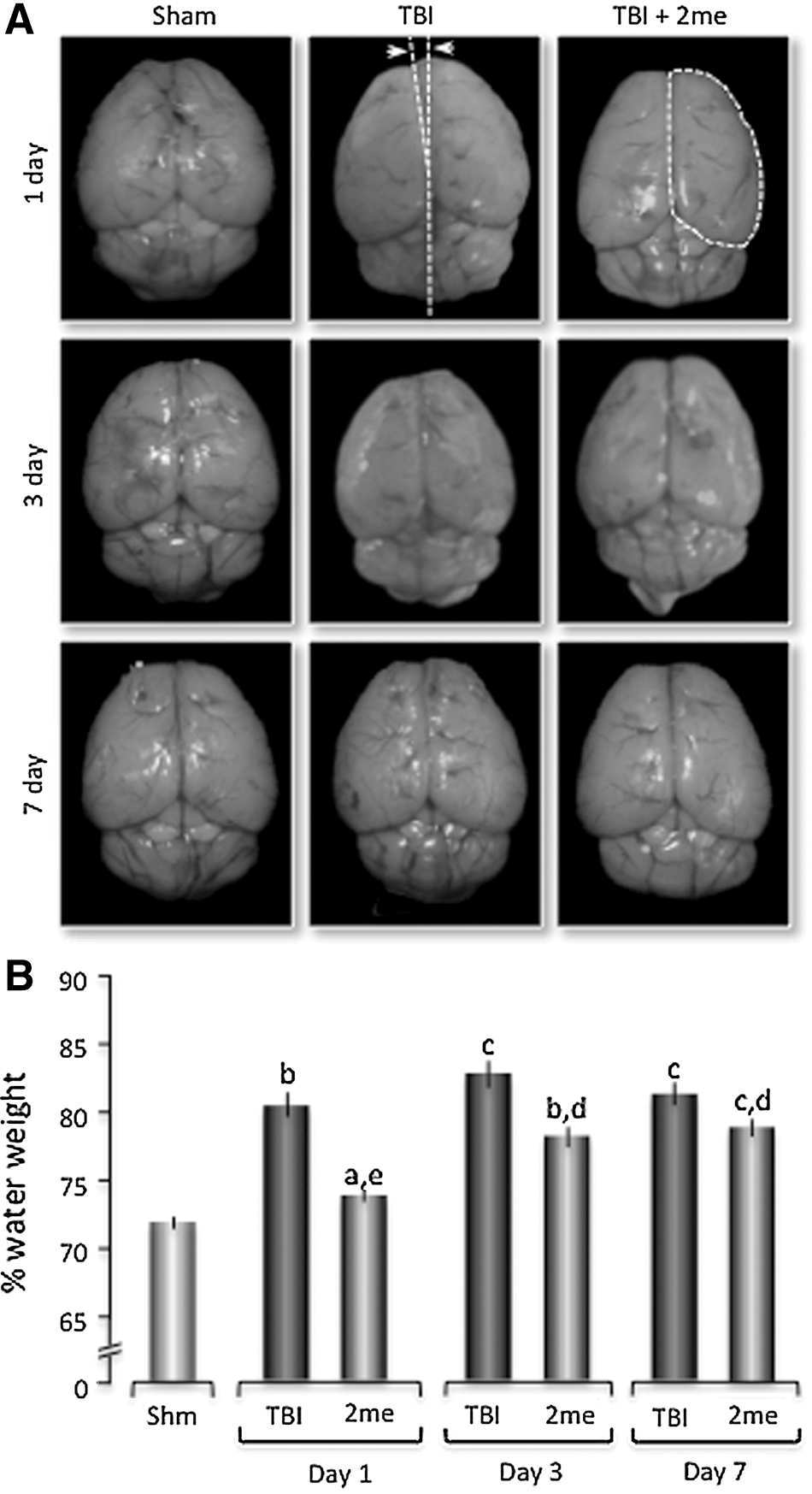
P2Y1R stimulation (2MeSADP) reduces edema formation in a traumatically injured brain.
P2Y1R stimulation reduces neuronal cytotoxic edema formation in the hippocampus and cortex after TBI
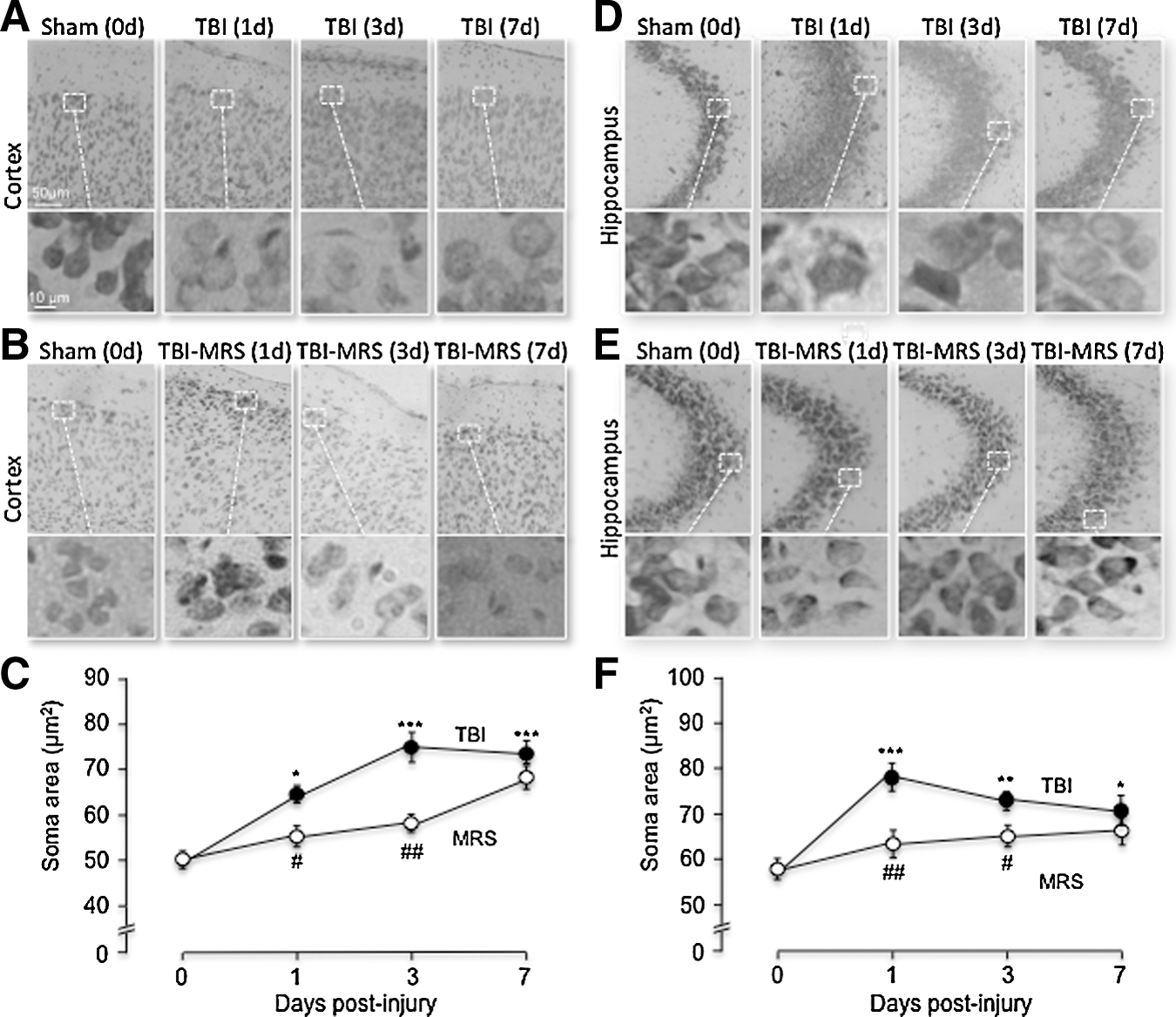
After TBI, Nissl staining of brain slices revealed significant increases in the soma size of neurons in both the ipsilateral cortex and hippocampus compared with sham mice, consistent with cytotoxic edema formation (Figs. 2 and 3). Twenty-four h post-TBI, the average soma size in the ipsilateral cortex increased to 64.2±3.3 (n=6) μm2 compared with the sham-treated soma size of 51.6±1.62 (n=6) μm2 (Fig. 2C). Neuronal soma size remained significantly elevated above sham mice at both days 3 and 7 post-injury in the cortex, with mean sizes of 75.5±7.5 (n=6) μm2 and 73.2±5.2 (n=6) μm2, respectively. Mice treated with 2MeSADP (100 μM tail-vein injection) 30 min after TBI demonstrated a significant reduction in the soma size compared with non-treated TBI mice at each time point. The average soma sizes at days 1, 3, and 7 post-injury were 54.1±3.5 (n=6) μm2; 62.1±4.3 (n=6) μm2; and 63.5±3.7 (n=6) μm2, respectively. Similarly, neurons found in the molecular layer of the CA3 region of the hippocampus exhibited significantly increased soma sizes after TBI compared with sham-treated mice (Figs. 2D–F). The average pre-trauma soma size of CA3 neurons was 59.31±1.62 (n=6) μm2. Twenty-four h post-TBI, there was a significant increase to 78.65±2.8 μm2 (n=6). This decreased to 73.81±3.2 (n=6) μm2 by day 3 and to 70.15±5.7 (n=6) μm2 at day 7 post-injury. Mice treated with 2MeSADP had significantly decreased hippocampal soma size compared with TBI mice at 1 and 3 days post-injury with mean soma sizes of 68.58±3.4 (n=5) μm2, 61.78±2.4 (n=6) μm2, and 66.68±3.8 (n=6) μm2 at 1, 3, and 7 days, respectively (Fig. 2F).

P2Y1R stimulation (2MeSADP) reduces cytotoxic edema formation in neurons of the somatosensory cortex and CA3 region of the hippocampus demonstrated by Nissl staining. Mice underwent sham or traumatic brain injury (TBI) surgery under isoflurane anesthetic (n=6 for each group). A subset of TBI mice received 2MeSADP 30 min post-TBI. Mice were sacrificed (1, 3, or 7 days post-injury; n=5 (day 1), n=6 (day 3) and n=6 (day 7)), perfused, sectioned at 25 μm, and Nissl staining was performed. Cytotoxic edema was evaluated by measuring the soma area of 20 cells per field.
P2Y1R stimulation (MRS2365) reduces cytotoxic edema formation in neurons of the somatosensory cortex and CA3 region of the hippocampus demonstrated by Nissl staining. Mice underwent sham or traumatic brain injury (TBI) surgery under isoflurane anesthesia. A subset of TBI mice received MRS2365 30 min post-TBI by tail vein injection (n=5 (day 1), n=6 (day 3) and n=5 (day 7)). Mice were sacrificed (1, 3, or 7 days post-surgery), perfused, sectioned at 25 μm, and Nissl staining was performed. Cytotoxic edema was evaluated by measuring the soma area of 20 cells per field. (
The purinergic receptor agonist 2MeSADP primarily stimulates P2Y1Rs, but also has some affinity for P2Y12 and P2Y13 receptors. To further delineate the role of the P2Y1R in reducing brain injury, we administered the highly specific P2Y1R ligand, MRS2365, 30 min post-TBI using the same experimental protocol as for 2MeSADP treated mice. Mice treated with MRS2365 (100 μM, 100 μl) after TBI demonstrated a significant reduction in the soma size compared with untreated mice on days 1 and 3 in the somatosensory cortex and in the CA3 region of the hippocampus (Figs. 3C, F). The average soma sizes in mice treated with MRS2365 in the somatosensory cortex at days 1, 3, and 7 post-injury were 55.86±2.25 (n=5) μm2, 59.64±1.8 (n=6) μm2, 69.11±3.27 (n=5) μm2, respectively (Fig. 3C). In the CA3 region of the hippocampus, the average soma sizes at days 1, 3, and 7 post-injury were 62.048±4.74 (n=5) μm2, 63.85±1.53 (n=6) μm2, 66.96±1.26 (n=5) μm2, respectively (Fig. 3F). At day 7 post-injury, there was no statistical difference in the cortex or CA3 region of the hippocampus between mice treated with MRS compared with non-treated TBI mice.
IP3R type2 deficient (IP3R2-/-) mice were used to determine whether the neuroprotective effects mediated by 2MeSADP and MRS2365 were abolished when the IP3 receptor expressed in astrocytes was knocked out. We found that the neuroprotective effects of 2MeSADP on TBI injured IP3R2-/- mice were lost in both the cortex and hippocampus. Within the cortex of IP3R2-/- mice that underwent TBI, the soma size averaged 62.27±1.8 (n=3) μm2 24 h post-injury, while mice treated with 2MeSADP averaged 61.03±2.3 (n=3) μm2, both of which were significant compared with sham-treated mice (Figs. 4A, B). The average soma size in the CA3 region of the hippocampus of IP3R2-/- mice treated with 2MeSADP increased slightly post-injury compared with the untreated TBI group. The average soma sizes were 64.48±2.2 (n=3) μm2 and 66.04±2.1 (n=3) μm2, respectively (Figs. 4C, D). Both treated and untreated TBI knockout groups were significantly increased compared with sham knockout, 55.4±1.77 (n=3) μm2.
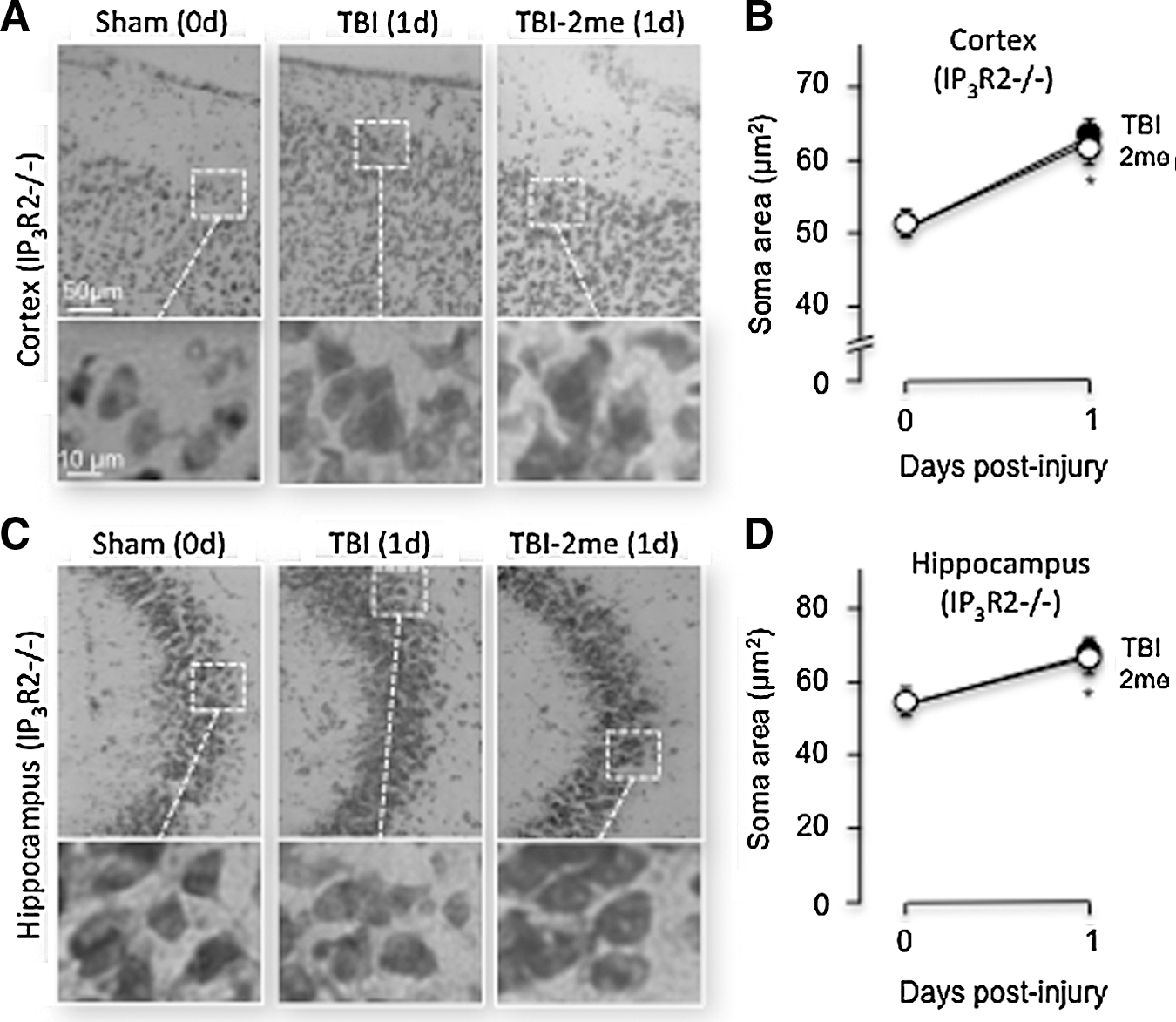
P2Y1R stimulation (2MeSADP) does not reduce cytotoxic edema formation in neurons of the somatosensory cortex or CA3 region of the hippocampus in IP3R2 -/- mice demonstrated by Nissl staining. Mice underwent sham or traumatic brain injury (TBI) surgery under isoflurane anesthesia. A subset of TBI mice received 2MeSADP 30 min post-TBI by tail vein injection. Mice were sacrificed (1 day post-surgery), perfused, and sectioned at 25μm, and Nissl staining was performed. Cytotoxic edema was evaluated by measuring the area of 20 cells per field.
AQ4 upregulation is reversed by purinergic stimulation
As stated earlier, AQ4 is the primary water channel in the brain, and it is assumed to be intimately associated with the regulation of water homeostasis. 43 –45 To determine the effects of TBI on AQ4 expression, Western blot analysis was used. We found significant increases in AQ4 protein expression in both the cortex and hippocampus of mice sacrificed at days 1 and 3 post-TBI (Figs. 5A, B). Twenty-four h post-injury, there was a 7%±6.08 (n=6) increase compared with sham-treated mice, which increased by 11.57%±4.89 (n=5) and 19.47%±8.93 (n=5) at days 3 and 7 post-injury within the ipsilateral cortex (Fig. 5A). There was a comparable increase in AQ4 expression within the hippocampus at 1 and 3 days post-injury with a 10.8%±6.42 (n=6) increase at 1 day and a 5%±5.18 (n=5) increase at 3 days compared with Sham mice (Fig. 5B). To determine the effects of P2Y1R stimulation on AQ4 expression after TBI, 2MeSADP was tail-vein injected 30 min post-injury. We found that AQ4 expression was not significantly changed at any of the time points examined, consistent with a reduction in brain edema post-trauma (Fig. 5).
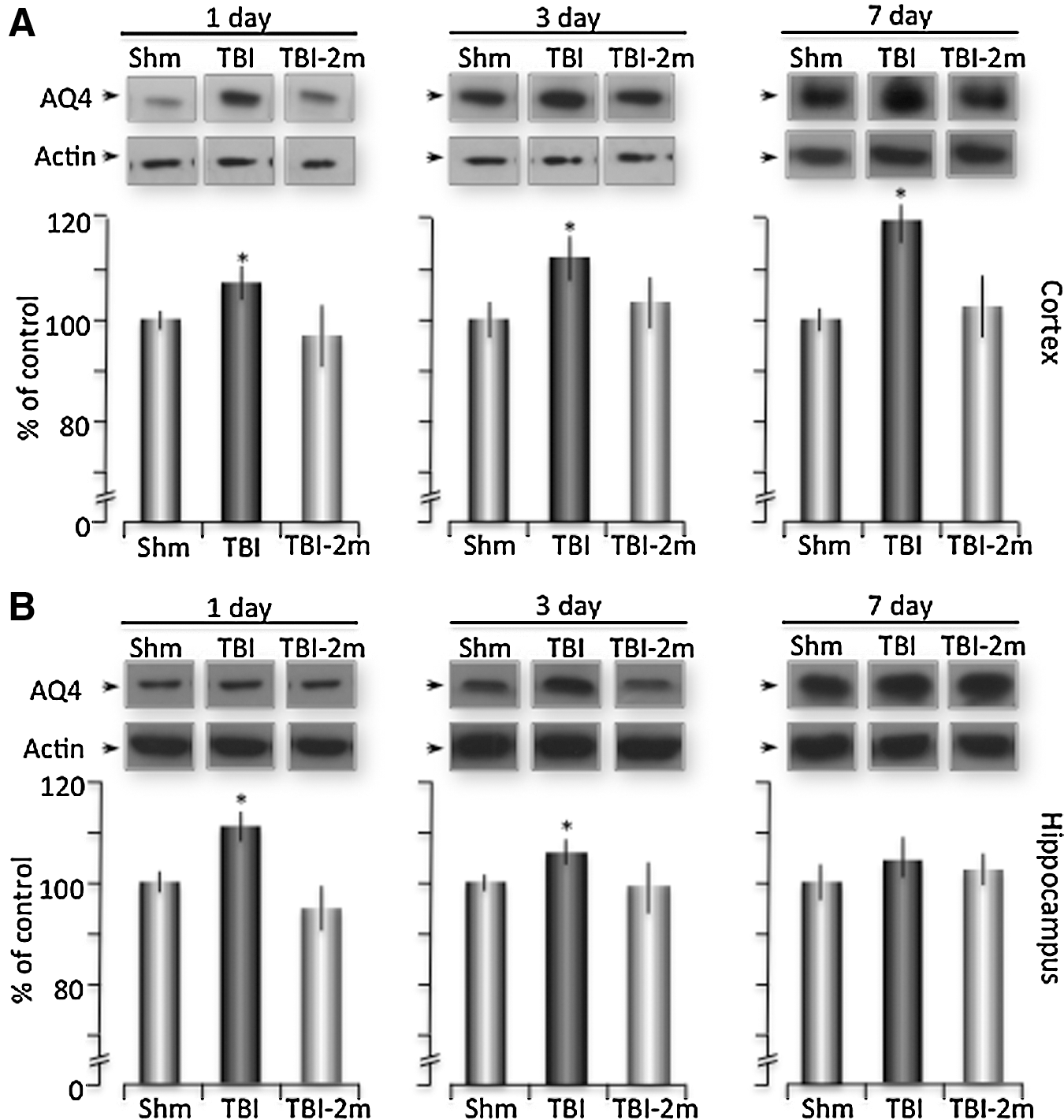
Increases expression of Aquaporin 4 is reversed with P2Y1R stimulation. Mice were prepared for sham or traumatic brain injury (TBI) under isoflurane anesthesia, with a subset of TBI mice receiving 2MeSADP 30 min post-TBI. The mice were sacrificed at 1, 3, and 7 h, and the cortex and hippocampus were isolated from ipsilateral and contralateral hemispheres. Western blot analysis was performed against AQ4 (1:500) and normalized to actin. Representative samples from sham, TBI, and 2MeSADP treated TBI mice are shown for both the cortex
Purinergic stimulation reduces GFAP expression in the cortex and hippocampus after TBI
GFAP is an intermediate filament protein involved in the structure and function of the cytoskeleton of astrocytes. Increased GFAP expression is also frequently used as a marker of reactive gliosis that occurs after brain injuries. 46 –48 To test our model of TBI-induced reactive gliosis, we performed Western blot analysis and immunofluorescent staining for GFAP expression in sham, TBI, or TBI and 2MeSADP mice sacrificed at 1, 3, and 7 days post-injury. First, Western blot analysis confirmed that TBI induced a significant increase in GFAP expression in both the cortex and hippocampus at 3 and 7 days post-injury (Fig. 6). Expression increased to 116.92%±6.19 (n=6) and 145.64%±12.15 (n=6) within the cortex and to 130.41%±5.49 (n=6) and 196.34%±18.28 (n=7) within the hippocampus, compared with sham mice at days 3 and 7, respectively. P2Y1R stimulation by 2MeSADP maintained these levels close to control values at each of these time points. (Figs. 6A, B).
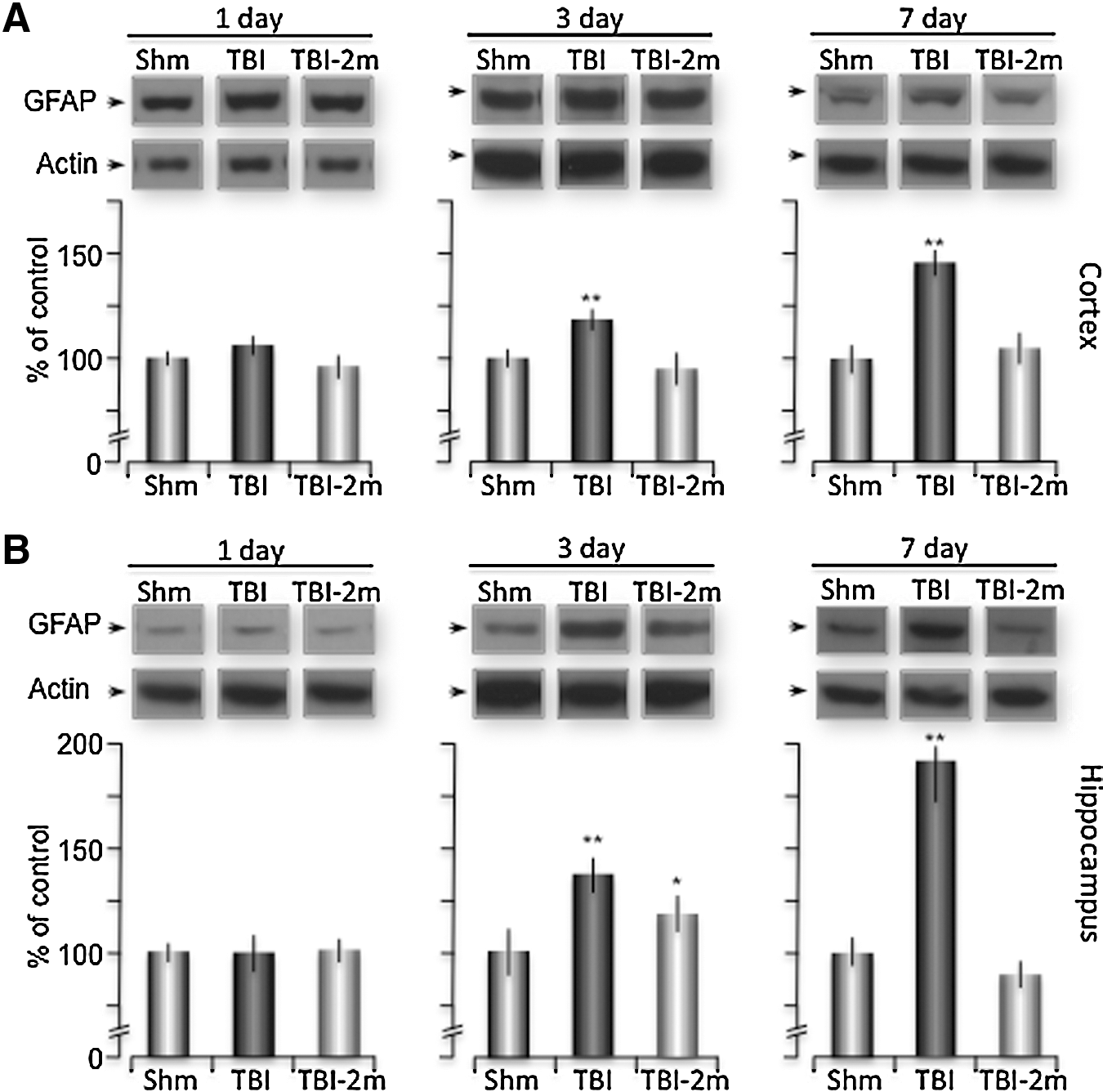
Increased GFAP expression is reduced by 2MeSADP treatment. Mice underwent sham or traumatic brain injury (TBI) with a group of TBI mice receiving 2MeSADP 30 min post-TBI. The mice were sacrificed at 1, 3, or 7 days post-injury, and the cortex and hippocampus were isolated from ipsilateral and contralateral hemispheres. Western blot analysis was performed against GFAP (1:1000) and normalized to actin. Representative samples from sham, TBI, and TBI + 2MeSADP (100 μM) are shown for the cortex
Second, we also observed a significant increase in the number of GFAP positive cells in both the cortex and hippocampus of TBI mice at all times tested (Fig. 7). The mean number of GFAP positive astrocytes in the cortex was initially 39.4±7.2 (per 1.59 mm2), which significantly increased to 82±5.8 (per 1.59 mm2), 65±5.7 (per 1.59 mm2) and 61.8±3.6 (per 1.59 mm2) at days 1, 3, and 7 post-injury. In mice treated with 2MeSADP, the number of GFAP positive cells in the cortex were significantly reduced to 49±3.4 (per 1.59 mm2), 52.4±3.3 (per 1.59 mm2) and 41±2.77 (per 1.59 mm2) at 1, 3, and 7 days post-injury. The same pattern of reactive astrocytes was observed in the hippocampus of mice after TBI (Fig. 7). Sham mice contained 432.6±19 (per 1.59 mm2) GFAP positive astrocytes within the hippocampus at day 0, while TBI mice sacrificed at days 1, 3, and 7 post-injury (n=6 per group) contained significantly higher numbers of GFAP positive astrocytes of 677±17.45 (per 1.59 mm2), 649.6±32.2 (per 1.59 mm2) and 625.4±12.3 (per 1.59 mm2), respectively. In addition, mice treated with 2MeSADP after TBI (n=6 per group) exhibited significantly lower numbers of GFAP positive astrocytes of 444.6±39 (per 1.59 mm2), 554.4±16.8 (per 1.59 mm2), 559.2±18.8 (per 1.59 mm2) (Fig. 7).
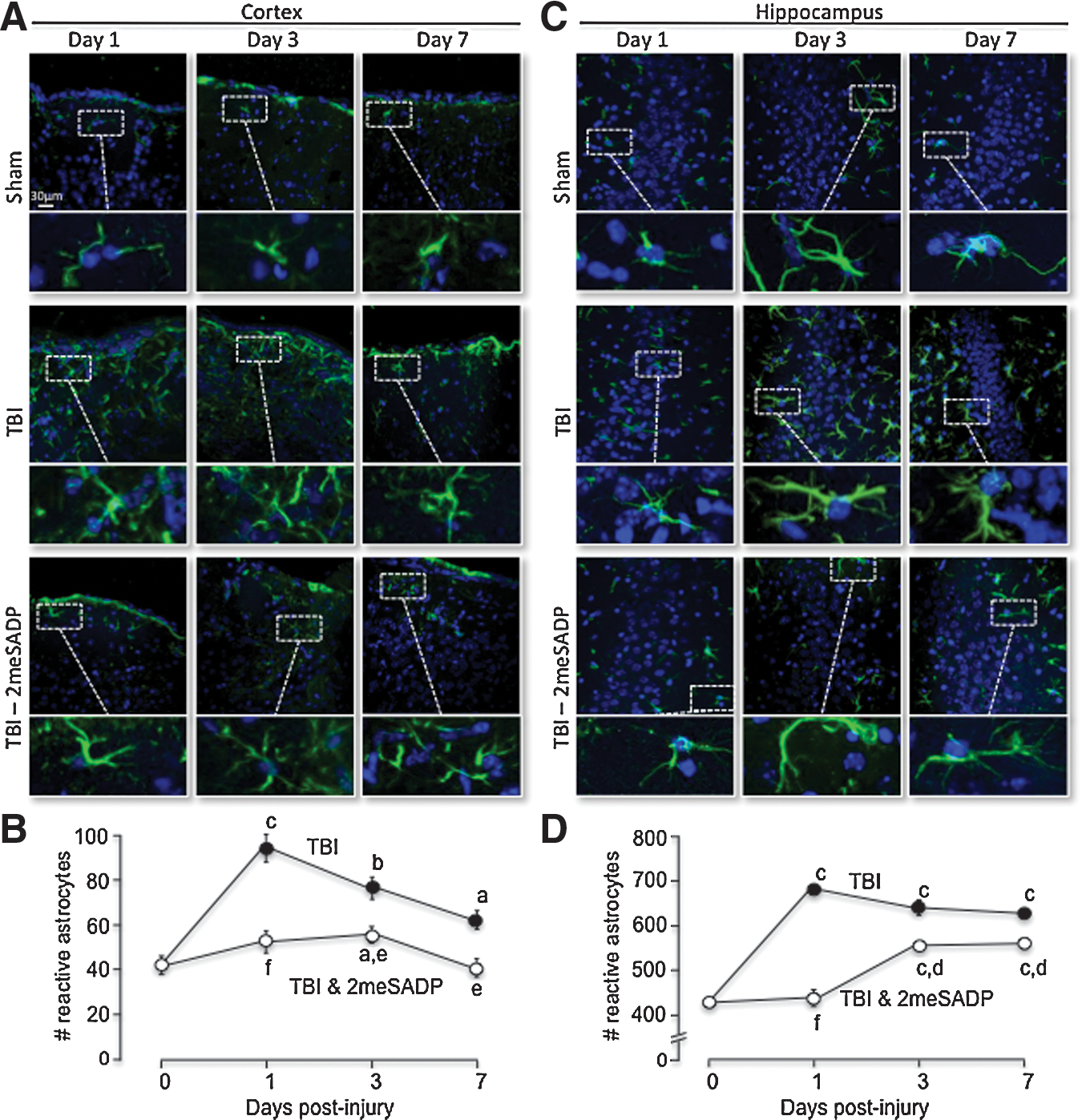
Reactive gliosis increases with traumatic brain injury (TBI) and is reduced with P2Y1R stimulation in the somatosensory cortex and the CA3 region of the hippocampus. Mice were prepared for sham or TBI under isoflurane anesthesia. After the mice were sacrificed at 1, 3, or 7 days post-injury and perfused with 4% PFA in 5% sucrose, brains were sectioned on a cryostat at 25 μm, and GFAP (green) immunofluorescence was performed using an anti-GFAP antibody. Nuclei are stained with DAPI (blue). The number of GFAP positive cells was evaluated using the Image J Cell Counter Pluggin.
IP3R2-/- mice were again used to test whether 2MeSADP-enhanced protection was dependent on IP3 signaling. We found that the number of cortical GFAP positive astrocytes in sham IP3R2-/- mice was 51.2±4.3 reactive astrocytes per field (1.26 mm 1.26 mm), which was comparable to wild type mice (Figs. 7 and 8). When these mice were injured, both 2MeSADP treated and untreated IP3R2-/- mice exhibited increased GFAP positive astrocytes with comparable values of 81.8±4.1 (per 1.59 mm2) and 83.2±3.9 (per 1.59 mm2) at 24 h, respectively (Fig. 8). In addition, the number of GFAP positive astrocytes in the hippocampus of sham IP3R2-/- mice was 447.9±36.3 per field (1.26 mm×1.26 mm). TBI increased these numbers to 589.3±66.2 (per 1.59 mm2) and 612.4±57.4 (per 1.59 mm2) at 24 h, respectively (Fig. 8). We concluded that P2Y1R-stimulated reductions of GFAP positive cell in TBI mice is dependent on IP3 signaling.
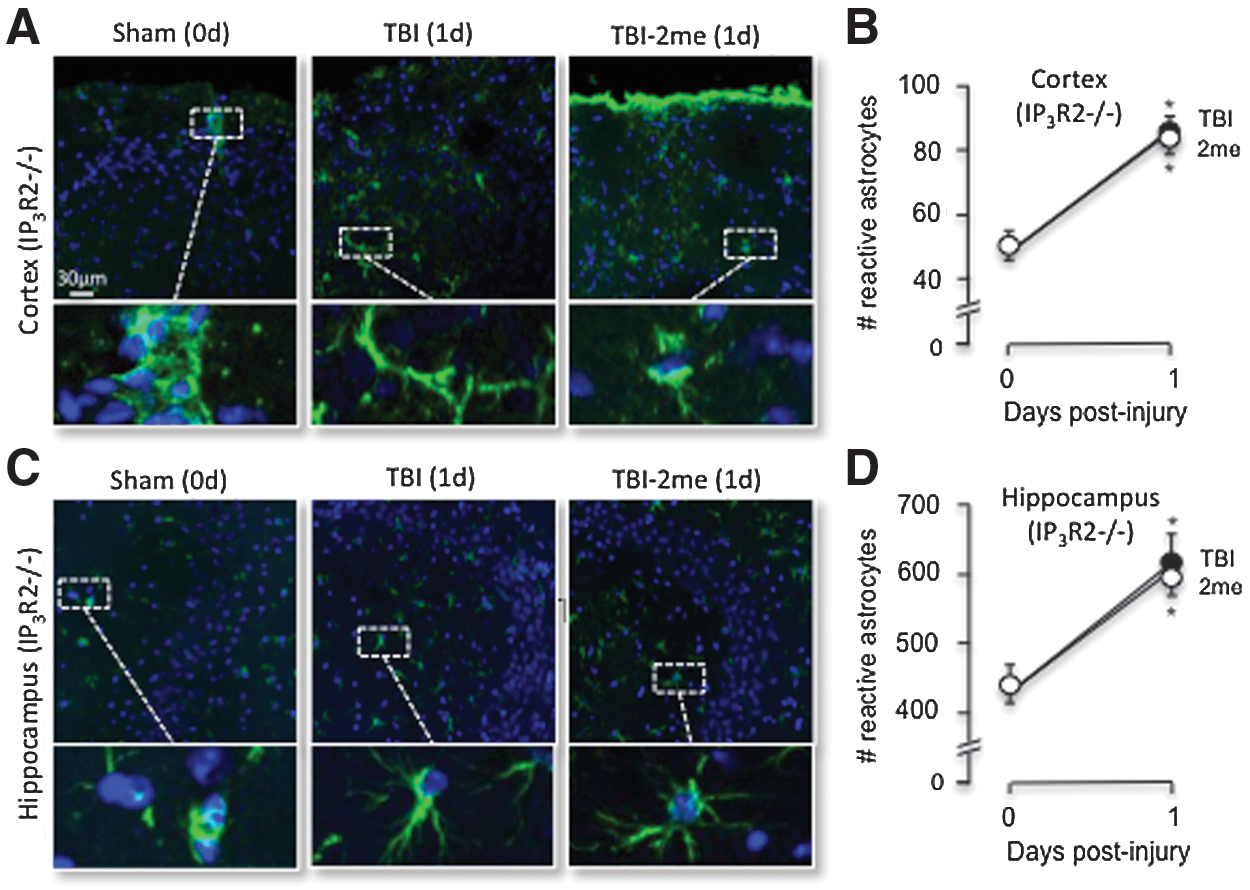
P2Y1R stimulation is not protective against increased reactive astrocytes in IP3R2-/- mice. IP3R2 -/- mice were prepared for sham or TBI with a subset of mice receiving 2MeSADP 30 min post-TBI. The mice were sacrificed 24 h post-TBI and were perfused with 4% PFA. Brains were sectioned on a cryostat at 25 μm, and GFAP immunofluorescence was performed as described in Methods. The number of GFAP positive cells was evaluated using Image J Cell Counter Pluggin.
Discussion
Treatment options to minimize and/or reverse brain damage after TBI are clearly needed. Our studies support a new treatment paradigm that significantly reduces severe injury in the primary injury phase of TBI, thereby also diminishing the elevated neurotoxic effects observed during the secondary injury phase.
3
–6
In brief, we enhanced the endogenous neuroprotective effects of astrocytes by stimulating their mitochondrial metabolism. Our working model is that stimulation of P2Y1Rs, which are primarily expressed on astrocytes (Supplementary Fig. 2; see online supplementary material at
An early critical need for energy utilization after TBI is the maintenance of ion homeostasis. TBI-induced ischemia decreases oxygen dependent ATP production. 17,49,50 This disrupts ion homeostasis, which increases osmotic stress and leads to water uptake, swelling, and, if left unchecked, cell lysis. The damaging role of edema during the early phase of TBI is well recognized. 12,36,51 Edema increases intracranial pressure, which further decreases cerebral blood flow. 9 –11 The precise mechanisms underlying edema formation following TBI, however, have not been fully elucidated. 7,8 Edema formation is separated into cytotoxic and vasogenic components. 13 Cell swelling from loss of ATP-maintained ion homeostasis is referred to as cytotoxic edema. We demonstrated that neuronal cytotoxic edema formation was significantly increased by our pneumatic model of TBI, in both the ipsilateral cortex and hippocampus. The ability of 2MeSADP and MRS2365 treatments to significantly reduce cell swelling is consistent with our working hypothesis that P2Y1R stimulation increases intracellular ATP levels, which in turn permits cells to better maintain ion homeostasis post-injury.
The second component of edema formation is vasogenic, which occurs when water moves from the vasculature into the extracellular space of brain tissue. Vascular edema is maximum during the height of TBI-induced BBB breakdown, which generally occurs between 3 and 6 h post-injury. During the hours and days after the initial insult, the processes associated with the secondary injury continue to induce both cytotoxic and vasogenic edema. 52 –54 Our pneumatic model of TBI clearly exhibited an expected progression of vasogenic edema in the days post-injury, as indicated by significant increases in whole brain water content and size. Tail-vein injections of 2MeSADP were very effective at reducing vasogenic edema, which presumably is because of decreased cytotoxic edema or potentially, energy-dependent regulation of the BBB integrity by astrocytes.
AQ4 has been implicated as the primary water channel within astrocytic end feet surrounding the brain vasculature. Both its distribution and expression play a major role in the severity of edema formation under normal and pathological conditions. 55 Mice deficient in AQ4 exhibit reduced water content and edema formation after acute water intoxication and ischemic stroke. 56,57 Overexpression of AQ4 accelerates the accumulation of water content after acute water intoxication, which induces rapid swelling of the brain. 58 Sun and associates 59 demonstrated that AQ4 mRNA is upregulated in male rats after a focal cortical contusion in the areas within the injured cortex. Ribeiro and colleagues 60 reported an increase in the expression of AQ4 on the endfeet of astrocytes within 1 h of stroke induction. In addition, Tomura and coworkers 61 demonstrated that AQ4 expression is increased after TBI using lateral fluid percussion injury in rats, and Rao and colleagues 62 demonstrated that in vitro trauma upregulates AQ4 expression after fluid percussion injury. Our pneumatic model of TBI is consistent with these observations, because we also found that AQ4 expression was significantly increased after injury.
For the mice treated with 2MeSADP, AQ4 expression was significantly diminished post-injury, which again was likely because of enhanced energization of astrocyte mitochondria. 34 An energy dependence of AQ4 expression is consistent with other reports in the literature where injuries are associated with edema formation. These injuries include intracerebral hemorrhage, 63 stroke, 64 and acute water intoxication. 44
Astrocytes are intimately involved in the maintenance of the brain under normal conditions. Their functional role after brain trauma, however, is less well understood. Astrocytes clearly play an important role in the restoration of ion homeostasis, neurotransmitter clearance, and they secrete a number of neurotrophic factors and amino acids that are crucial to neuronal survival. 65 –67 Because of the close association of astrocytes and the BBB as well as the presence of crucial metabolic substrate transporters, astrocytes are thought to play a prominent role in regulating the flow of essential nutrients to neurons that promote survival post-injury.
GFAP, a well-known protein that is primarily expressed in astrocytes, increases its expression after brain injury, and its protein levels are frequently used as a biological marker of TBI as well as a prognosis indicator after an injury to the brain. 68 –70 Consistent with a neuroprotective role, GFAP-null mice are more sensitive to both TBI and cerebral ischemia. 71,72 The production of neurotrophins, which promote neuronal survival and function, has been associated with reactive (high GFAP expression) astrocytes. GFAP is also associated with increased inflammation, the production of glial cytokines, and an increase in astrocyte numbers (astrogliosis). 73 –77
Finally, GFAP is highly associated with the structural integrity of the astrocyte membrane, which affords it greater control over the extracellular environment, a role crucial to neuronal survival. In our model of TBI, GFAP expression was significantly increased in astrocytes. We also found that treatment of injured mice with 2MeSADP significantly diminished GFAP expression, similar to its effect on AQ4 levels. The reduction in both GFAP and AQ4 expression could be a direct result on expression occurring during the secondary injury phase. Because all indications of injury (swelling, GFAP and AQ4 expression) were diminished by 2MeSADP treatment, however, we cannot attribute enhanced neuroprotection to any one specific process or injury phase, aside from the increased availability of ATP for energy dependent functions. In fact, enhanced protection during the primary phase of injury is beneficial simply because of the consequential decrease in the second phase of injury. In this light, the ability of P2Y1R stimulation to minimize edema may be particularly important, because the maintenance of cell integrity is clearly necessary to support other neuroprotective functions.
Because of the increasing prevalence of persons with TBI as well as the lack of treatment options, it is imperative that new treatment strategies are developed. We have established a new treatment paradigm that enhances the natural neuroprotective functions of astrocytes. Specifically, we demonstrated that enhanced astrocyte mitochondrial metabolism via stimulation of P2Y1Rs, provides a much needed energy boost that significantly decreases damage from TBI. The strength of this approach is that specific underlying neuroprotective mechanisms do not need to be identified and individually stimulated. Rather, stimulation of mitochondrial ATP production benefits all of the energy-dependent neuroprotective functions of astrocytes.
Footnotes
Acknowledgments
LTW, JDL, SS, and MD formulated the hypotheses, organized and designed the studies. LTW, SS, WZ, and RJG performed experiments and analyzed the data. DJ, MD, and JDL contributed reagents and supplies necessary for the study. LTW, RJG, and JDL wrote and edited the article and figures.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.