
Abstract
Mild traumatic brain injury (mTBI), particularly mild “blast type” injuries resulting from improvised exploding devices and many sport-caused injuries to the brain, result in long-term impairment of cognition and behavior. Our central hypothesis is that there are inflammatory consequences to mTBI that persist over time and, in part, are responsible for resultant pathogenesis and clinical outcomes. We used an adaptation (1 atmosphere pressure) of a well-characterized moderate-to-severe brain lateral fluid percussion (LFP) brain injury rat model. Our mild LFP injury resulted in acute increases in interleukin-1α/β and tumor necrosis factor alpha levels, macrophage/microglial and astrocytic activation, evidence of heightened cellular stress, and blood–brain barrier (BBB) dysfunction that were evident as early as 3-6 h postinjury. Both glial activation and BBB dysfunction persisted for 18 days postinjury.
Introduction
M
Although there is an ample body of literature on the role of brain ischemia, excitotoxicity, oxidative stress (OS), inflammation, and edema in moderate-to-severe TBI, less is known about its mild counterpart. 17 –21 Early events triggered by TBI include increases in the cytokines, IL-1β and TNF-α, known to contribute to monocyte cellular activation and infiltration, glial activation, neuronal and myelin loss, and further persistent inflammation. 1 –7
Mild traumatic brain injury (mTBI), particularly mild “blast type” injuries resulting from improvised exploding devices and many sport-caused injuries to the brain, result in long-term impairment of cognition and behavior. 22 –25 Our central hypothesis is that increases in IL-1α/β and TNF-α levels after mTBI may be responsible, in part, for triggering a cascade of inflammation with downstream effects on glial activation, neuronal function impairment, and blood–brain barrier (BBB) integrity. Behavioral and cognitive impairments are demonstrable for several rodent models of TBI by day 15 postinjury. 26 –28 Thus, we performed our assessments of inflammatory macrophage and microglial activation and BBB dysfunction at 3 and 6 h and 18 days postinjury. A corollary to our hypothesis is that the inflammatory consequences of mild lateral fluid percussion (mLFP) injury may persist and contribute to the development of long-term brain impairments associated with mTBI.
The rat lateral fluid percussion model of TBI 29,30 is widely accepted and causes a combination of focal cortical contusion and diffuse injury of subcortical brain areas, much like that present in human brain neuropathology after TBI. 31 We adapted the LFP injury model by decreasing the effect to 1 atmosphere (atm) pressure, which resulted in righting reflex response times of more than 4 min, but less than 10 min, as compared to righting reflex response times in excess of 15–30 min characteristic of moderate-to-severe LFP injury. We characterized the mLFP injury model at 3–6 h and 18 days postinjury in terms of acute inflammation biomarkers, such as increased IL-1α/β and TNF-α, macrophage/microglial and astrocytic activation, impaired BBB function, and activating transcription factor 3 (ATF-3) transcription factor activation, all known to affect brain function.
Methods
Surgical preparation: LFP injury
All animal manipulations were approved by the Department of Defense (DoD) and the University of Texas Medical Branch (Galveston, TX) institutional animal care committees, using guidelines developed by the DoD and National Institutes of Health (Bethesda, MD). Initial experiments were performed with LFP brain injury on rats at 1, 2, and 2.4 atm for comparison purposes, where the 2.0- and 2.4-atm levels of injury are known to result in what have been labeled in the literature as a moderate-to-severe TBI. 29,30 We defined mLFP based on righting reflex responses in the 4–10-min range after a 1-atm injury. Thus, all the immunohistochemical (IHC) assessments were performed on rats experiencing the 1-atm mLFP injury. Briefly, male Sprague-Dawley rats, weighing 350–400 g, were anesthetized with isoflurane in an anesthetic chamber, intubated, and mechanically ventilated with 1.5–2.0% isoflurane in O2/room air (30:70) using a volume ventilator (Edco Scientific, Inc., Chapel Hill, NC). Rats were then placed in a stereotaxic frame, and the scalp was sagittally incised. A 4.0-mm diameter hole was trephined into the skull 2.0 mm to the right of the sagittal suture and midway between the lambda and bregma. A modified Luerlok syringe hub was placed over the exposed dura, bonded in place with cyanoacrylic adhesive, and covered with dental acrylic. Isoflurane was discontinued; rats were connected to the trauma device and subjected to a mild 1.0-atm fluid-percussion injury, immediately after acquisition of a withdrawal reflex to paw pinch. The sham group was treated as described above, with the exception that no fluid-percussion injury was administered. After mLFP or sham injury, rats were disconnected from the fluid percussion device and righting reflex was assessed every 60 sec until there was a normal righting reflex. To test righting reflex, rats were placed on their backs and the time at which the animal righted itself 3 times consecutively was recorded as the righting reflex. Rats were then placed on 2% isoflurane, and wound sites were infused with bupivicaine and sutured with prolene. Isoflurane was discontinued, and rats were extubated and allowed to recover in a warm, humidified incubator.
Rats were allowed to recover for 3- or 6-h survival periods or 18 days, at which time rats were anesthetized before sacrifice with 150 mg/kg of Nembutal intraperitoneally and perfused transcardially with 4% paraformaldehyde (PFA) for IHC or, if earmarked for immunoassays, they were sacrificed and the hippocampus, parietal cortex, and thalamus were dissected and stored at −80°C until use.
Perfused brains were removed, postfixed overnight in 4% PFA, blocked into 3–4-mm coronal sections, and transferred to 30% sucrose until penetration was complete. Tissue was then embedded in optimal cutting temperature compound and frozen at −20°C. Tissue was mounted and then sectioned at 30 or 50 μm and collected both serially and free-floating. Once sectioned, every ninth section was collected for assessment with hematoxylin and eosin staining to provide a gross indicator of lesion severity. Sections were processed for widefield epifluorescent and confocal visualization as follows: A series of sections (every ninth section from the rostral to caudal extent) were rinsed in Tris-buffered saline (TBS), followed by incubation in TBS plus 0.025% Triton X-100 plus 5% serum of the appropriate species for 1 or 2 h. Parafin-embedded 3-mm-thick coronal blocks were sectioned at 5 um on a rotary microtome and immediately placed in dH2O at 46°C and placed on slides. Sections were deparaffinized at 56°C and rehydrated with xylene and a series of ethanols (100%, 95%, and 80%), followed by dH2O and phosphate-buffered saline (PBS). After the antigen retrieval step with 10 mM of Na citrate (pH 6.0 at 100°C for 30 min), tissue was washed in 0.18 M of PBS 4 times, blocked with 5% normal goat serum (NGS) plus 0.3% bovine serum albumin (BSA) in 0.18 M of PBS for 30 min. Sections were incubated with an antibody (Ab) in 1% NGS plus 0.3% BSA/0.025% Triton X-100 for 2 h at room temperature (RT), followed by washing 3 times with PBS plus Triton X-100 for 10 min, each at RT.
Except for sections incubated in secondary Ab in the absence of a primary Ab (negative control), sections were incubated in primary Ab or Abs for multi-antigen visualization overnight at 4°C on a rotating platform. Sections were then rinsed thrice in TBS and incubated in the appropriate secondary linked to an Alexa Fluor® dye (Molecular Probes, Eugene, OR) that excites at either 488 (green) or 568 (red) nm for 3 h. Sections were then mounted onto glass cover-slips, allowed to briefly dry, then mounted to glass slides using Fluoromount-G, after which slides were allowed to sit at RT for at least 20 min and were then put in a refrigerator for at least 24 h before storage in a freezer until viewing. For wide-field epifluorescent assessment and image acquisition, slides were viewed on an upright microscope with narrow band-pass filters to minimize fluorescence overlap and images were captured using a SPOT Flex digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI) before annotation using Adobe Photoshop CS4. For confocal acquisition, slides were first visualized under wide-field epifluorescent light on an Olympus BX51 upright microscope (Olympus, Tokyo, Japan). Suitable fields were imaged using a Bio-Rad Radiance 2000 confocal system (Bio-Rad, Hercules, CA). Resulting images represent projections from stacks of optical sections of no less than 25 and no greater than 30.
Abs used were as follows 1:20 IL-1β (goat polyclonal; R&D Systems, Minneapolis, MN); 1:5000 anti-NeuN (mouse monoclonal; Millipore, Billerica, MA);1:1000 anti-myelin basic protein (mouse monoclonal; Covance Inc., Princeton, NJ); 1:200 anti-RECA-1 (rat endothelium cell antigen 1; mouse monoclonal; AbD Serotec, Oxford, UK); 1:1000 anti-IBA-1 (ionized calcium-binding adapter molecule 1; rabbit polyclonal; Wako, Richmond, VA); 1:1000 anti-GFAP (glial fibrillary acidic protein; rabbit polyoclonal; Dako, Glostrup, Denmark); 1:1000 anti-GFAP (mouse monoclonal; Chemicon International, Temecula, CA); 1:400 anti-vimentin (mouse monoclonal; Sigma-Aldrich, St. Louis, MO); 1:1000 anti-nestin (chicken polyclonal; Millipore); 1:750 albumin (ALB; sheep polyclonal; Bethyl Laboratories, Inc., Montgomery, TX); 1:250 rat immunoglobulin G (IgG; Invitrogen, Carlsbad, CA); 1:20 IL-1β (goat polyclonal; R&D Systems), 1:1000 anti ATF-3 (rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA); and 1:3000 anti-SMI 71 (mouse monoclonal; Covance).
We incubated sections with 1:1000 Alexa Fluor 568 (red) or 488 (green) goat anti-rabbit IgG or goat anti-mouse IgG for 2 h at RT (shaking). We mounted sections on slides, dried for a few minutes, and added regular mounting medium and let slides sit at RT for at least 20 min, after which they were put in a refrigerator for at least 24 h before storage in a freezer until viewing.
Cytokine immunoassay
We measured cytokines using the Bio-Plex assay kit from Bio-Rad, a capture sandwich immunoassay that is very sensitive at detecting cytokines in the pg/mL range.
The Bio-Plex cytokine assay is a multi-plex bead-based assay designed to detect up to 100 cytokines in a single well of a 96-well plate. The assay uses up to 100 color-coded bead sets, which are conjugated with different specific reactants. Each reactant is specific for a different target molecule. Specific reaction is identified based on bead color. Samples in each well are drawn up into the flow-based Bio-Plex array reader. The reaction is measured by using fluorescently labeled reporter molecules that are also specific for each target protein. For our experiment, the Bio-Plex rat cytokine/chemokine assay was designed to detect and quantitate nine rat cytokines/chemokines: IL-1α; IL-1β; TNF-α; IL-2; IL-6; IL-4; IL-10; granulocyte macrophage colony-stimulating factor (GM-CSF), and interferon-gamma.
Image capture
Confocal images were acquired from immunofluorescently (IF) serial-labeled serial sections 40 um thick from at least three sections per animal, for 3 different animals for further sequential analysis. Each sample was initially compared by observing z-stack, optical selections 10 um thick, to verify planar continuity of sections. Conditions for scanning were 20×lens, no zoom, and an acquisition resolution of 1280×1024 pixels. Fixed iris (pinhole), laser intensity, gain, and offset were maintained throughout all samples of the same experiment. Images were taken at the same optical thickness (2 or 5 um) for all groups, based on countable cell numbers of biomarker-stained proteins present. Images on paraffin-embedded IHC sections 5 um thick, probed for endothelial barrier antigen (EBA) expression, were photographed by light microscopy at 20×. Counts were tallied and compared from unlabeled printed images taken on ipsilateral and contralateral sides of the same section.
Cell/intensity quantitation
Cell profiles for various markers were counted in specific anatomic regions (e.g., hippocampus CA1, dendate gyrus, and so forth) on histolgical sections, from at least three sections per animal, for at least 3 different animals, averaged, and calculated as means of profile counts±standard deviation within that specific region/animal/brain hemisphere. Thus, the final number was a mean number of profiles counted within a three-dimensional region (area of anatomical region×thickness of section). Thicknesses of sections were not significantly different between the ipsilateral and contralateral side for any given section; cell counts were stereological estimates based on profile counts. For each section, counts were performed by three different individuals on a blinded basis to eliminate bias. Though there were differences in resulting counts for any slice for the three different individuals counting, the relative differences between samples were consistent and there was a 10% or less variation for any sample count across individual counts from the three individuals performing counts.
For some analyses, it was not possible to count cell profiles (e.g., Iba-1, GFAP, and so forth). For these comparisons, after ensuring that staining was linearly related to secondary reporters, density of immunoreaction product was quantified utilizing optometric techniques. Analysis was performed using a Bio-Rad confocal laser system coupled to a Nikon light microscope using Bio-Rad 1.50 software. Sections (n=5/animal) from each group at each time point were viewed with a 10×objective, and images were captured using a SPOT Megapixel camera. The final projected image for analysis was 1000×that of the original section for cytological analyses. Thus, pixels were included in the analysis that were not readily detectable in the low-magnification photodocumentation shown in the results. Integrated optical density area was confined and measured in the regions listed above. Density levels and distribution were quantified both ipsilaterally and contralaterally and compared to data collected from shams. For all sections, nonstaining gray or white matter was empirically chosen as background and staining was normalized to this signal intensity.
Beam balance assay
At day 11 postinjury or post–sham treatment, rats were placed on a balance beam apparatus consisting of a beam 91.5 cm long (L)×1.7 cm wide (W) elevated 30 cm off the surface below and secured to a platform on either end. Rats were placed on the center of the beam and released (start time) and were allowed to walk to either end. Animals were returned to the center of the platform if they walked onto one of the end platforms. This was continued for 1 min or until rats fell off (stop time). For each rat, 10 trials were performed and average time duration on the beam was recorded. Naïve rats were also tested for purposes of establishing a baseline.
Foot fault assay
At day 11 postinjury or post–sham treatment, rats were placed on a wire mesh 69.5 cm W×45 cm L with 3-cm gaps stretched out over a wooded frame. The number of times from 10 attempts on the wire mesh that the forelimb or hindlimb fell through the gaps rats transversed the mesh were recorded separately as a percentage (% foot faults).
Statistical analyses
Analyses were performed with GraphPad Prism 5 software (GraphPad Software, San Diego, CA). For analyses involving two groups, a Student's t-test was performed and a p-value less than 0.05 was considered significant. If variances for two groups were deemed significantly different, a Welsh correction was added to the Student's t-test. For analyses of more than two groups, a one-way analysis of variance was performed with a Tukey-Kramer post-hoc test to compare groups. A p-value less than 0.05 was deemed significant.
Results
There were increased righting reflex response times for rats after mLFP injury
An accepted measure of the severity of a TBI incident in humans is the return of consciousness, which, in rodent models, is mimicked by measuring the righting reflex response time. In our LFP injury model, we observed a significant increase in righting reflex response times of approximately 75% after injury with a 1-atm LFP injury. Righting reflex response time after LFP increased as the impact force was increased to 2 atm, the latter being typically used in moderate-to-severe LFP injury models (Fig. 1). All subsequent experiments were performed using the 1-atm LFP injury as a model of mLFP with righting reflex response times greater than 4 min or lesser than 10 min.
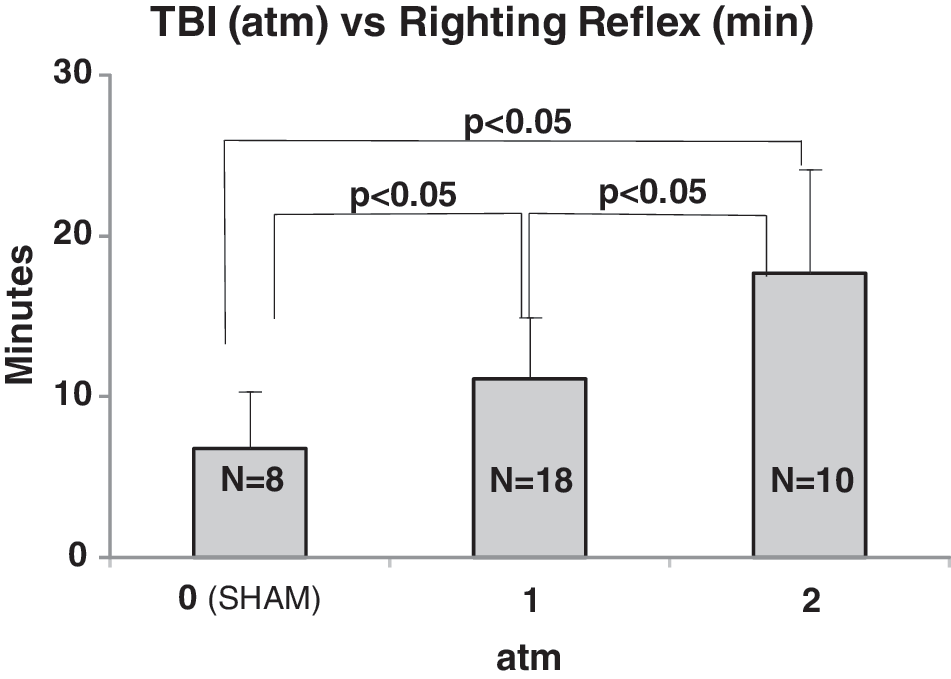
Time of righting reflex responses in minutes as a function of mLFP injury severity measured in atm. Responses for 1 and 2 atm mLFP injuries are significantly different from shams (0 atmospheres) and from each other (p<0.05).
Locomotor responses to mLFP injury after 11 days of show impaired function
One index of injurious outcome often used in assessments of TBI in rodent models is the measurement of vestibulomotor function, as assessed by measuring the time that a rat can balance on a beam with defined dimensions 32 and the number of foot faults incurred by a rat transversing a mesh with specific parameters. 33 Exposure to mLFP injury showed a significant impairment in both balance ability and locomotor coordination at 11 days postinjury (Fig. 2).

mLFP increases cytokine expression at 3–6 h
We focused on early time points after injury given the known prompt and robust inflammatory response to the more moderate and severe brain injury reported for rat TBI models, spinal cord injury, and ischemia. 13 –15 Injury to the brain is known to have an early component in which prompt cell-death–promoting signaling mechanisms are triggered, followed by the prompt activation of signaling pathways responsible for more-pervasive delayed chronic inflammation and persistent plastic changes. Thus, we chose the 3- and 6-h time points as useful for acute biomarker-based assessments to characterize this mild version of brain injury.
To test the hypothesis that mLFP injury triggers an early inflammatory response, we compared ipsilateral (right hemisphere, injured) to contralateral levels of nine cytokines in three brain regions (parietal cortex, hippocampus, and thalamus), using an array immunoassay that measures cytokines in the pg/g cytokine protein/total protein range (Fig. 3A–C). In the ipsilateral parietal cortices, compared to their contralateral counterparts, there were significant increases in levels of IL-1α, IL-1β, and TNF-α as early as 3 h after mLFP injury. By 6 h, there were also significant increases in IL-1α, IL-1β, TNF-α, IL-6, and IL-10 (a noninflammatory cytokine) as well. Increases in IL-1β were the most prominent, relative to changes in all the other inflammatory cytokines. In the ipsilateral injured hippocampi, there were significant increases in levels of IL-1α, IL-1β, TNF-α, and IL-6 as early as 3 h after mLFP injury, when comparing ipsilateral to contralateral hemispheres. By 6 h, there were also significant increases in IL-1α, IL-1β, TNF-α, IL-6, and the anti-inflammatory IL-10 as well. Again, increases in IL-1β were the most prominent, relative to the changes in all the other cytokines. In the ipsilateral injured thalami, ony IL-1β showed significant increases at 6 h after mLFP injury, when comparing ipsilateral to contralateral hemispheres. By 18 days postinjury, we could only detect, by IHC for IL-1β, its presence in a sparse population of IL-1β+ cells proximal to the site of injury (Fig. 3B).


mLFP injury increases the levels of inflammatory cellular markers after 3–6 h
Increases in parenchymal levels of IBA-1 are associated with an increased presence of activated macrophages and microglia, both indicators of an ongoing inflammatory response. Six hours after mLFP injury, there was a significant increase in IBA-1+ cells throughout the cortex, which colocalized with IL-1β and displayed a “reactive” morphological phenotype (Fig. 4), consistent with inflammation. This increased localized IBA-1 presence persisted for 18 days after injury (Fig. 4C).

Representative images of cortical inflammatory response.
mLFP increases astrocytic activation
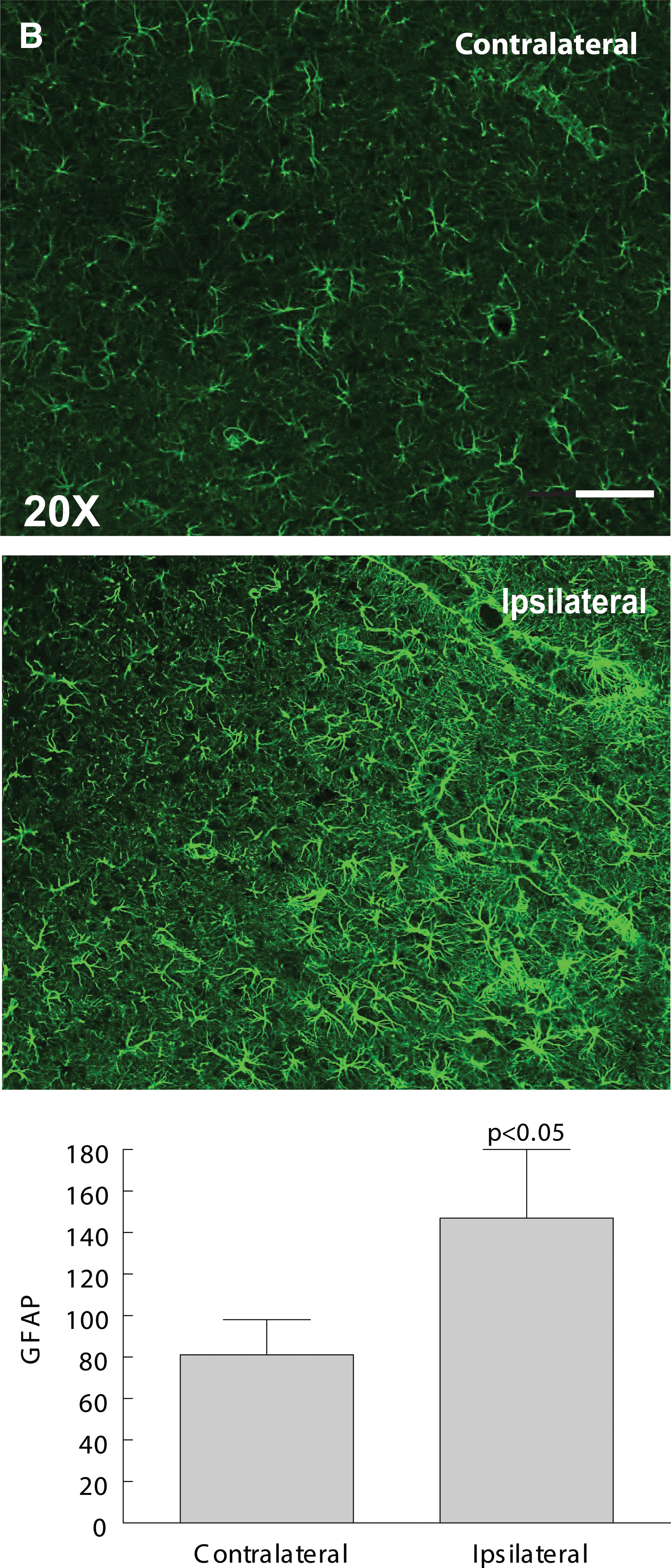
Given the increased levels of inflammatory cytokines and the increase in levels of the inflammatory marker, IBA-1, as early as 3–6 h after mLFP injury, we used IF to assess the state of activation of astrocytes, also known to play a role in inflammatory responses in the CNS. 34,35 As early as 6 h after mLFP injury, there was a significant increase in GFAP staining intensity (GFAP+), a marker for astrocytic activation, in the cingulate cortex ipsilateral to the injury site (Fig. 5). Figure 5 shows the classical star-like hypertrophied configuration of astrocytes responding to injury that, in part, defines the inflammatory cascade induced by CNS trauma and is a contributor to later pathology. We observed similar levels of activation in the thalamus and hippocampus (data not shown). The injury-induced increase in GFAP+ cell labeling persisted for 18 days. We also used the glial biomarkers, nestin and vimentin, and, as can be seen in Figure 5, the mFLP injury induced significant increases in nestin and vimentin labeling of activated astrocytes.
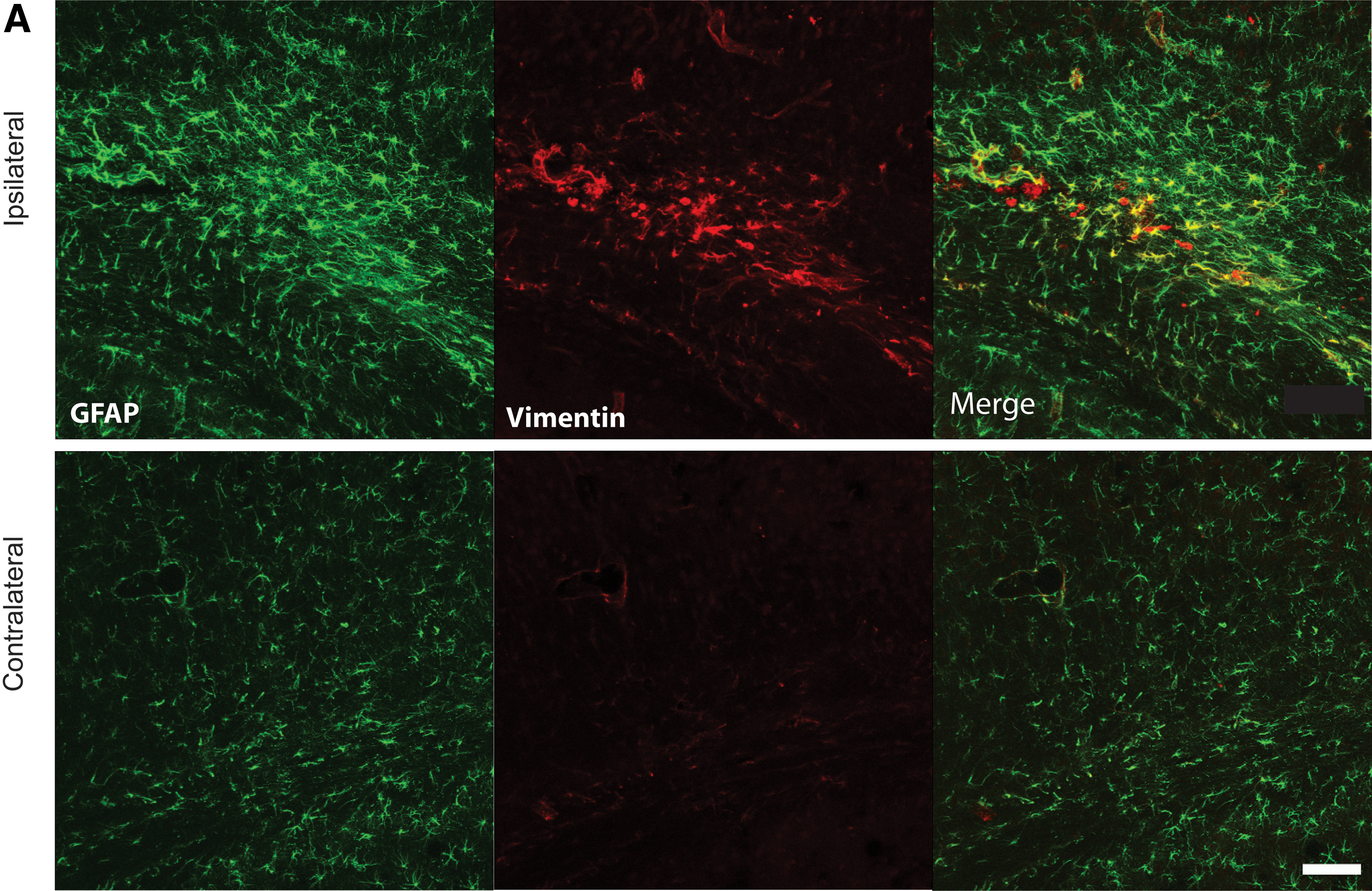
Representative immunostaining 6 h after 1-atm mLFP injury. Images of inflammatory activated astrocytes in ipsilateral parietal corticex, as indicated by GFAP+ cells and colocalized
mLFP perturbs the BBB
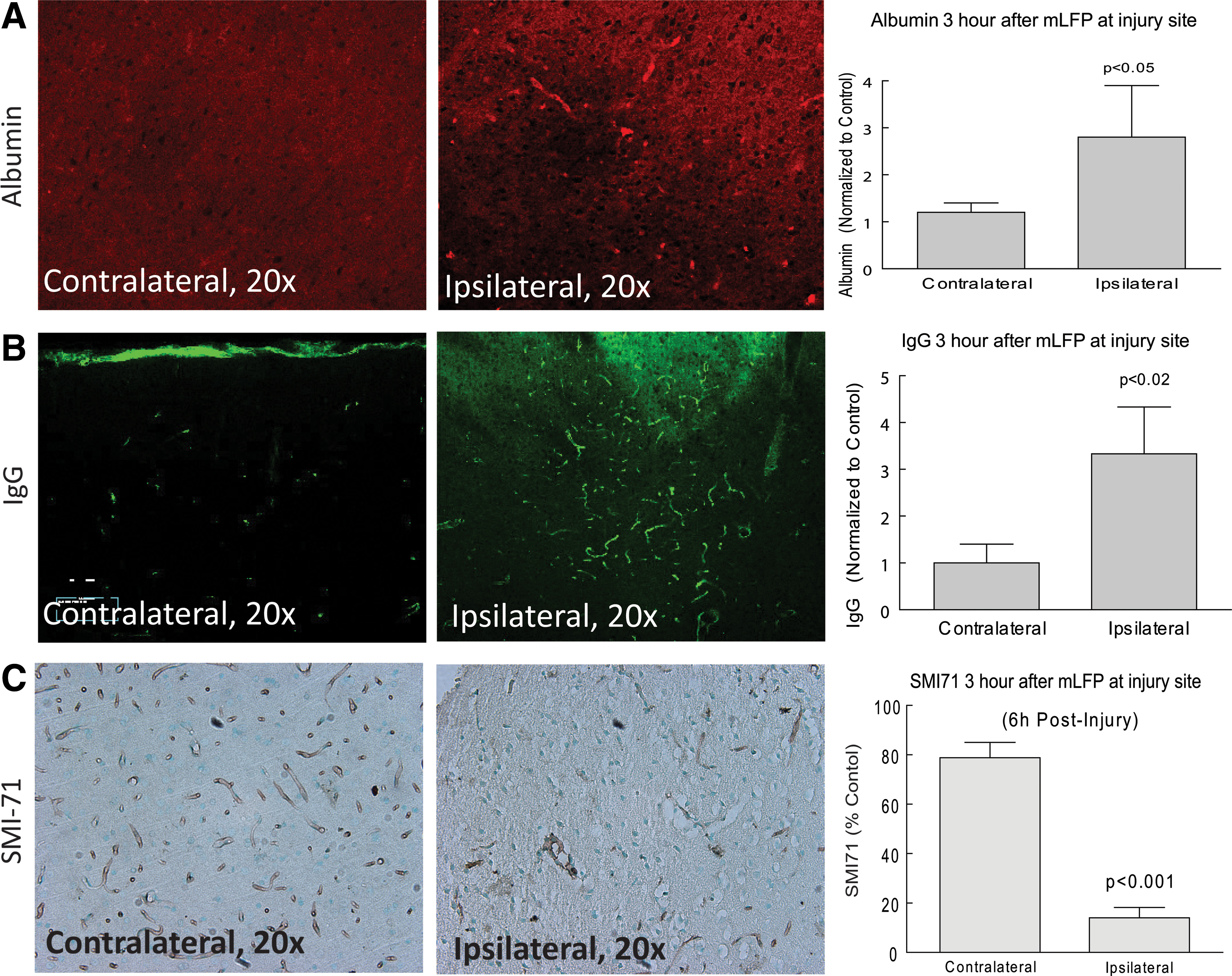
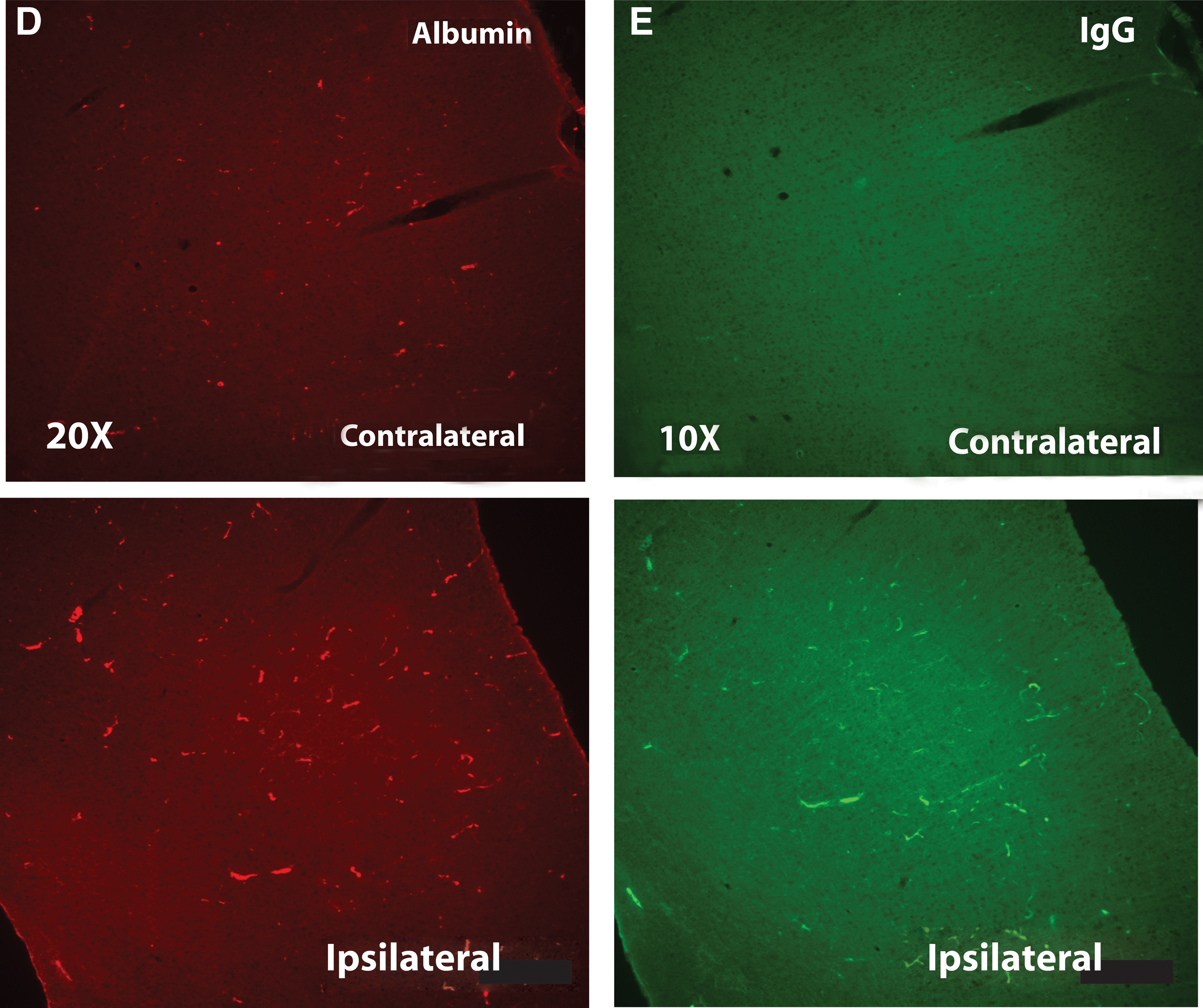
Moderate-to-severe TBI is known to cause significant perturbations in the integrity of the BBB with the resulting influx of blood-derived substances, such as ALB and IgG, contributing to a growing state of inflammation and OS. 20,36,37 It was unclear, however, whether an injury that can be described as mild would elicit a similar loss of BBB integrity and subsequent release of blood-derived proteins. We therefore asked whether there was evidence of an mLP injury-induced BBB breakdown that would allow, and perhaps stimulate, intrusion of injury-activated macrophages as well as the blood-derived proteins, ALB and IgG, into the parenchyma. Both were significantly increased at the site of injury in the parenchyma after mLFP injury, when compared to controls (Fig. 6). There was also a decreased appearance of EBA, as detected with the SMI-71 Ab, a marker for BBB integrity and more direct indicator of compromised BBB integrity (Fig. 6 A–C). These changes persisted up to 18 days after mLFP injury when they colocalized to the endothelial lining of blood vessels, as shown by colocalization with the RECA, an endothelial marker consistent with impaired BBB functionality (Fig. 6 D–E).
Immunostaining of

There was neuronal loss in the hippocampus of rats 18 days after mLFP injury
To determine the identity of the tissue loss in brains of rats exposed to mLFP injury after 18 days, cells were stained for NeuN, a marker for neuronal nuclei. Eighteen days after mLFP injury, there was significant neuronal loss of more than 50% of NeuN+ cells in the ipsilateral hippocampus, as compared to sham-treated rats. Although there was also significant neuronal loss in the contralateral hippocampus, compared to shams, contralateral hippocampi also showed more severe neuronal losses, compared to their ipsilateral conuterparts (Fig. 7).

Immunostaining of NeuN+ cells in hippocampus 18 days after mLFP injury. Representative hippocampal images from mLFP-injured rats after 18 days immunolabeled with NeuN (original magnification, 4×, 20×, and 60×, compared to sham-treated rats. Quantitation of NeuN+ cells: p<0.01. Color image is available online at
There was significant myelin loss 18 days after mLFP injury
A significant indicator of neuronal dysfunction is the extent of losses in neuronal myelination. We measured the relative loss of myelin in hippocampus by IHC using a myelin basic protein (MBP) Ab. Eighteen days after mLFP injury, we observed a better than 40% loss in MBP IF consistent with neural dysfunction (Fig. 8).
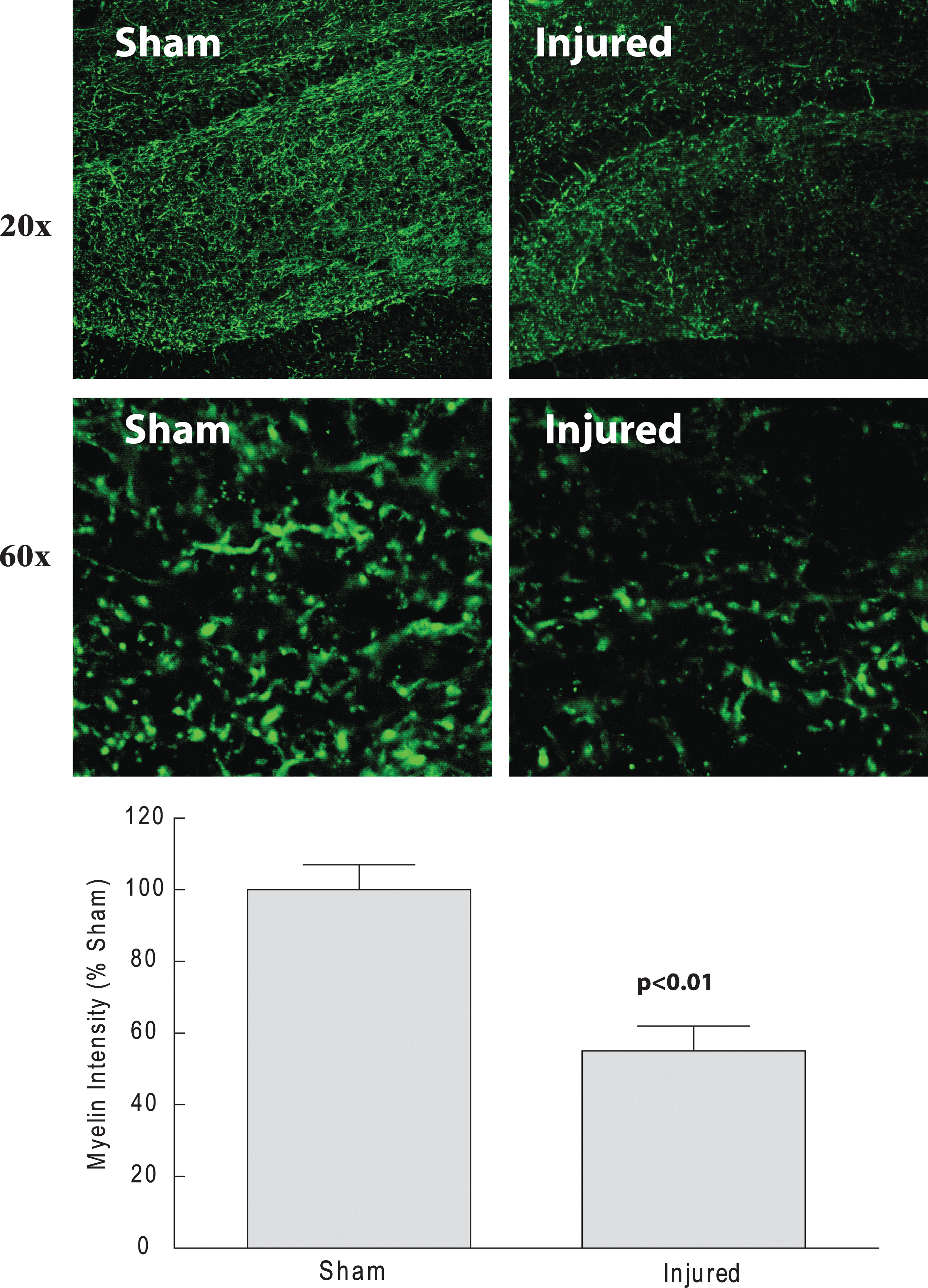
Immunostaining of MBP+ cells in hippocampus 18 days after mLFP injury. Representative hipppocampal images of mLFP-injured rats after 18 days immunolabeled with MBP (original magnification, 10×; p<0.01). Color image is available online at
Discussion
Mild “blast” injuries resulting from improvised exploding devices have long-term cognitive and behavioral deficits stemming from mTBI that seriously impair quality of life. Brain damage is a result of the initial mechanical trauma (or primary inury) and delayed secondary events. Secondary events include breakdown of the BBB, edema formation with concomitant swelling, infiltration of peripherally circulating blood cells, activation of resident glial cells, and increased parenchymal levels of stress-response proteins, including proinflammatory cytokines.
For moderate and severe TBI, it is known that the resultant triggered inflammatory cascades and vascular dysruption result in cell death and neuronal dysfunction 17 –20 ; however, less is known about the “mild” forms of TBI. The diversity of clinical outcomes associated with mTBI and the diffuse nature of the injuries classified as mTBI would suggest that no single animal model of mTBI is likely to match 100% of the spectrum of mechanisms and outcomes that have been documented for clinical mTBI. 38 We chose a rodent model that fits many of the clinical observations that characterize mTBI as a useful tool to increase our understanding that the rodent model of mTBI, relying on a 1-atm LFP, would also provide a platform for the development and assessment of intervention strategies for the rational design of clinical therapies.
As a first step in our characterization of a rat mLFP injury model, we chose to use the righting reflex response time as an analog of return of consciousness time interval in humans, an accepted index of TBI severity, because it reflects the time of return of consciousness in the rat. 39,40 Comparison with sham-treated rats allows extrapolating the effects of anesthesia and handling alone; sham-treated rats display righting reflex response times under 4 min, as compared to righting reflex response times between 4 and 10 min, and thus represent a normalizing measure that is useful as a validating tool given the broad range of interanimal variabilty in behavioral and cognitive responses to brain injury. These differences may also reflect the known variability in outcomes present in a clinical setting. We were able to show correlative increases in length of righting reflex response times as a function of mLFP impact severity when measured in atmospheres of pressure (Fig. 1). This allowed us to define a 1-atm pressure injury as our standard for the model.
We also relied on two standard outcome measures of severity of injury by measuring vestibulomotor function as assessed with the beam balance performance test over a defined time interval 32 and number of foot faults on a beam with defined parameters. 33 Exposure to mLFP injury resulted in a significant impairment in locomotor coordination and balance at 11 days postlesion for both these tests (Fig. 2). These results were consistent with the assessment that though delays in righting reflex response times and impairment of beam balance performance and foot fault evaluations indicated a significant impairment associated with mLFP injury (1 atm), they were not as robust as those witnessed by us after 2- and 2.4-atm LFP injuries.
There is evidence of significant increases in inflammatory cytokines in the parenchyma of patients after severe TBI 5,41,42 and in various brain regions in experimental LFP injury, 3 as well as other rodent models of CNS injury. 8 –16 The most consistent finding was an increase in IL-1β and TNF-α cytokines and the observation that manipulations that decrease levels of both these cytokines result in improved outcomes. 13,14,43 Here, we report that analyses of three brain regions at 3–6 h after mLFP injury resulted in a robust increase in both IL-1α and in IL-1β as well as TNF-α protein levels, when compared to other known cytokines. Though there were significant increases in IL-6 and GM-CSF in the hippocampus, their very low levels, compared to IL-1α/β and TNF-α levels, would suggest that their role may not be critical after mTBI. Interestingly, there was also an increase in IL-10 at 6 h, consistent with its demonstrated response in moderate or severe TBI. 3 IL-10 is an anti-inflammatory cytokine that has been shown to increase locally after both TBI and spinal cord injury (SCI). 44 –49 Attempts to increase IL-10 levels in SCI as a potential therapeutic agent have yielded mixed results. 50,51
The early increases in IL-1 and TNF-α in the ipsilateral parietal cortices, when compared to their contralateral counterparts, were significant as early as 3 h after mLFP injury. By 6 h, there were also significant increases in IL-1α, IL-1β, TNF-α, IL-6, and IL-10 as well. Increases in IL-1β were the most prominent, relative to the changes in all the other cytokines. In the ipsilateral injured hippocampi, there were significant increases in levels of IL-1α, IL-1β, TNF-α, and IL-6 as early as 3 h after mLFP injury, when comparing ipsilateral to contralateral hemispheres; by 6 h, there were also significant increases in IL-1α, IL-1β, TNF-α, IL-6, and IL-10 as well. Again, increases in IL-1β were the most prominent, relative to the changes for all the other cytokines. In the ipsilateral injured thalami, only IL-1β showed significant increases at 6 h after mLFP injury, when comparing ipsilateral to contralateral hemispheres. Thus, the highest levels were present in the hippocampus and cortex, with thalamic levels being approximately 25% of those in the ipsilateral hippocampi and cortices.
Local IL-1β increases in the CNS are common to most CNS experimental trauma and ischemic injury models. Both IL-1β and IL-1α bind and activate the IL-1 receptor (IL-1R) that, in turn, activate the nuclear factor kappa B transcription factor, followed by increased transcription of cyclooxegenase-2 and inducible nitric oxide (NO) synthase, resulting in increased levels of reactive oxygen species (ROS) and nitrogen oxygen species. The latter, in turn, stimulate lipid peroxidation, DNA damage, and mitochondrial- and endoplasmic reticulum–mediated cell death by apoptosis, necrosis, and autophagy. A number of experimental TBI studies have shown that the resultant damage to the site of injury and hippocampus area are likely to be a major determinant of cognitive impairments associated with TBI. 52 –55
When we determined the cellular origin of cytokines as early as 6 h after mLFP injury, we focused on IL-1β and used confocal IHC, together with a standard cell marker for inflammation. Against a background of very few IL-1β+-labeled cells in uninjured or sham-treated rat brain tissues, the mLFP injured rat brain displayed a significant increase in IBA1+ cells, consistent with reports of IL-1β and TNF-α production by microglia after TBI. 56 Although an increase in IL-1 β+-activated microglia is consistent with their having a microglial role in subsequent pathology in mTBI, it does not preclude that BBB perturbation may also result, in part, from astroglial activation, macrophage activation, monocyte infiltration, or perturbation of water flux regulation-mediated endothelial responses.
There are both clinical and experimental data that show increased levels of GFAP after severe TBI, reflecting the downstream activation of astrocytes by cytokine-induced inflammation after TBI. 57,58 Two other useful markers for astrocytic activation after TBI are nestin and vimentin. 59,60 Nestin and vimentin are intermediate filament proteins coexpressed by GFAP-positive astrocytes shown to promote scarring and glial proliferation, both of which have detrimental effects on neuronal recuperative and regenerative potential. 61,62 There were significant increases in GFAP immunoreactivity after 6 h and persisting up to 18 days after mLFP injury, reflecting increased astrocytic activation in the cortex, hippocampus, and thalamus ipsilateral to the injury site (Fig. 5). The GFAP+ astrocytes displayed the star-like hypertrophied configuration associated with astrocytes responding to injury that, in part, defines astrocytic activation. mLFP injury induced significant increases in nestin and vimentin labeling of activated astrocytes. These results suggest that mTBI acute injury triggers a cascade of inflammatory local signaling associated with known astrocytic post-trauma pathology that is likely to involve BBB functional impairment and neuronal recuperative processes.
Astrocytes are classically thought to play roles in the regulation of extracellular concentrations of water, potassium and other ions, and glutamate and other transmitters and provide for general homeostasis in the extracellular and synaptic spaces through energy-dependent uptake mechanisms. Specialized processes of astrocytes also play roles in the BBB by end-feet apposition to endothelium of the vasculature in the brain. Microglia are classically thought to be principally phagocytes that are able to be mobilized after injury, infection, disease, and in seizures. When activated, glial cells are known to hypertrophy, increase production of GFAP and proinflammatory cytokines, ROS, adenosine triphosphate, excitatory amino acids, and NO. 63 –66 These downstream increases are candidates that can elicit changes in hyperexcitability, affecting sensory function. Additionally, chronically activated astrocytes can lead to permanent blood/spinal cord barrier breakdowns that ensure continued immune cell infiltration and feed-forward continued inflammatory signaling. 67
In the mammalian system, we propose that normal glial function becomes abnormal and dysfunctional after CNS injury and that the dysfunctional glial state contributes to conditions that initiate and ensure persistence of neuronal dysfunction. Though the concept of glia-neuronal and neuronal-glial interactions were described in invertebrate systems several decades ago, 68,69 the conceptual basis of dysfunctional glial cells contributing to changes in neuronal intracellular signaling and membrane properties is relatively new. 67,70 –73 We propose the term “gliopathy” to describe the dysfunctional and maladaptive response of glial cells to neural injury. We hypothesize that the initiation of gliopathy after neural injury is the sudden increase in the extracellular concentration of glutamate after nerve or CNS injury 74 that, in some cases, is 37-fold higher than resting concentrations and results in excitotoxicity 75 and glutamate receptor-mediated sensitization of both neuronal and glial populations. 76,77
Given the robust response of inflammatory signals to mLFP injury, we determined acute effects on BBB integrity, whose role is to isolate CNS tissue from circulating cytokines and immune-like cells. Severe TBI has been shown to disrupt the BBB, as determined by a number of criteria. 37 Measuring the BBB breakdown-mediated intrusion of the blood-derived molecules, ALB and IgG, into the parenchyma as early as 6 h after mLFP injury also showed their significant increase, consistent with an acute impairment of the BBB. Whereas ALB and IgG IHC will indicate the presence of protein infiltration, it will not determine whether that leakage is active or whether it occurred at an earlier time point. The significant mLFP-induced decrease in EBA protein levels, measured using SMI-71, is evidence of deterioration in the BBB, a conclusion based on the known association of decreased EBA with CNS injury resulting in BBB impairment, and the observation that after treatment with Abs to EBA there is an induction of BBB impairment. 78,79 Impairment of the BBB after mLFP injury would suggest that there are likely to be further downstream inflammatory signaling elements that would account for the long-term functional impairments associated with mTBI in the clinical context.
One potential contributor to the persistent inflammatory responses to mLFP injury could be continued BBB disruption and resultant increased BBB dysfunction-associated increased permeability. To confirm the published literature on increased BBB permeability after mTBI (the bulk of that literature addresses moderate TBI and/or stroke), 20,37 we also assessed the presence of blood-borne proteins in the brain at 18 days post-trauma. When we stained for ALB and IgG and compared results between naïve and injured, there was a qualitatively very definitive presence of blood-borne proteins in the injured brains at 18 days postinjury (Fig. 6), clearly documenting BBB impairment up to 18 days postinjury. This is important from the perspective of being able to introduce therapeutic proteins in the injured brain, because agents thought to be impermeable in uninjured brains would have access to the postinjury brain parenchyma.
Whereas ALB IHC will indicate the presence of ALB at different time points, it will not determine whether that leakage is active or whether it occurred at an earlier time point. EBA passage into CNS tissue indicates an actively leaky barrier. Finally, we also showed a decreased appearance of EBA, a marker for BBB integrity. 78,79 The resulting mTBI-induced decrase in EBA expression is indicative of BBB dysfunction, a conclusion based on the known association of decreased EBA with CNS injury resulting in BBB impairment and induction of BBB impairment after treatment with Abs to EBA. 78,79
We characterized BBB function by measuring levels of serum ALB in the parietal cortex 18 days after mLFP injury. As shown in Figure 6, there was more than a 2-fold increase in ALB in the ipsilateral parietal cortex, as compared to control hemisphere, 18 days after mLFP injury. Not surprisingly, there was also a 3-fold increase in IgG at the same site, consistent with an impaired BBB (Fig. 6). An indicator of BBB function is SMI-71. When SMI-71 was measured 18 days after injury, there was a decrease in SMI-71 levels colocalized to the endothelial lining of blood vessels, as shown by colocalization with the RECA endothelial marker, consistent with impaired BBB functionality (Fig. 6). This provides good evidence for the involvement of impaired vascular function after mTBI, which would be consistent with chronic low-level inflammatory events and which, because of cross-talk with other systems, may explain some of the persistance of behavioral outcomes. Thus, treatments that intervene in inflammatory receptor-mediated downstream pathways would be predicted to preserve neuronal function and, consequently, improve behavioral function.
These results are consistent with mLFP injury triggering a cascade in which the inflammatory cytokines, IL-1α and IL-1β, play a major role in triggering downstream impairment of the BBB and, further, more persistant microglial activation (Fig. 9).

Proposed mTBI-triggered inflammatory cascade. mTBI stimulates increased levels of IL-1α/β and TNF-α that, in turn, activate early robust inflammatory signaling by monocyte and glial activation, BBB impairment, and eventual neuronal and glial loss that are likely to affect behavior and cognition.
Footnotes
Author Disclosure Statement
No competing financial interests exist.