
Abstract
Traumatic brain injury (TBI) often occurs in conjunction with additional trauma, resulting in secondary complications, such as hypotension as a result of blood loss. This study investigated the combined effects of penetrating ballistic-like brain injury (PBBI) and hemorrhagic shock (HS) on physiological parameters, including acute changes in regional cerebral blood flow (rCBF), brain tissue oxygen tension (PbtO2), and cortical spreading depolarizations (CSDs). All recordings were initiated before injury (PBBI/HS/both) and maintained for 2.5 h. Results showed that PBBI alone and combined PBBI and HS produced a sustained impairment of ipsilateral rCBF that decreased by 70% from baseline (p<0.05). Significant and sustained reductions in PbtO2 (50% baseline; p<0.05) were also observed in the injured hemisphere of the animals subjected to both PBBI and HS (PBBI+HS). In contrast, PBBI alone produced smaller, more transient reductions in PbtO2 levels. The lower limit of cerebral autoregulation was significantly higher in the PBBI+HS group (p<0.05, compared to HS alone). Critically, combined injury resulted in twice the number of spontaneous CSDs as in PBBI alone (p<0.05). It also lowered the propagation speed of CSD and the threshold of CSD occurrence [induced CSD at higher mean arterial pressure (MAP)]. However, rCBF and PbtO2 were not responsive to the depolarizations. Our data suggest that PBBI together with HS causes persistent impairment of CBF and brain tissue oxygen tension, increasing the probability of CSDs that likely contribute to secondary neuropathology and compromise neurological recovery.
Introduction
Spontaneous CSD has been demonstrated in TBI patients 9,10 and in experimental animal models of brain injury. 8,11 Under pathological conditions, CSD causes vasoconstriction as a result of disrupted neurovascular response, resulting in spreading ischemia. 2 The mismatch between energy demand and supply prolongs neuronal depolarization, which triggers intracellular signaling pathways leading to cell death. 12 Whether CSDs contribute to post-traumatic secondary brain damage remains open to debate. On one hand, a study that induced additional CSDs by potassium chloride (KCl) injections into the injured hemisphere of TBI-injured mice failed to show a significant relationship between the number of CSDs and lesion size. 13 In contrast, Rogatsky and colleagues suggested that an increasing number of spontaneous CSDs accompanied by high ICP (≥20 mmHg) exacerbated brain damage. 14
Previous work investigating CSD occurrence in a rat model of penetrating ballistic-like brain injury (PBBI) demonstrated evidence of CSD from 5 to 72 h postinjury. 15 The acute incidence of depolarization (within 2 h postinjury) and the changes in CBF and PbtO2 have not been examined. Moreover, the effect of hemorrhagic hypotension on these acute physiological changes after brain trauma is unclear. In the present study, we investigated the effect of hypotension on spreading depolarization, CBF, and PbtO2 after PBBI. In particular, these changes were examined in core lesion and perilesional areas simultaneously, thus providing a comprehensive physiological profile acutely subsequent to the injury.
Methods
A total of 60 male adult Sprague-Dawley rats (280–320 g; Charles River Labs, Raleigh, VA) were used in these experiments. Animals were randomized between the following four groups: sham (n=16); PBBI (n=17); hemorrhagic shock (HS; n=12) or PBBI+HS (n=15). All procedures involving animal use were reviewed and approved by the institutional animal care and use committee of Walter Reed Army Institute of Research (Silver Springs, MD). Research was conducted in compliance with the animal welfare act, Guide for the Care and Use of Laboratory Animals (National Research Council, Washington, DC), and other federal statutes and regulations. Animals were housed individually under a 12-h light/dark cycle in a facility accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care.
General procedures
Anesthesia was induced with 3.5% isoflurane delivered in air/oxygen mixture (FiO2=0.26) and maintained at 1.5% throughout the surgery and recording. Core body temperature was maintained at 37.0°C by a homeothermic heating system (Harvard Apparatus, Holliston, MA). In all animals, the right femoral artery and vein were cannulated for mean arterial blood pressure (MAP) monitoring and fluid resuscitation respectively. In addition, the tail artery was cannulated for inducing HS by withdrawing blood and for blood gas analysis. Partial pressures of oxygen and carbon dioxide as well as pH were measured with a blood gas analyzer (ABL5; Radiometer America Inc., Westlake, OH) before and after injury. To detect the cortical spreading depolarization and its associated changes of regional CBF (rCBF) and PbtO2, electrodes (epidural) and probes (dorsoventral 2.5 mm from dura) were placed at the frontal and parietal cortices representing the core lesion and perilesional areas, as shown in Figure 1A. Signals were allowed to stabilize (∼30 min). After 20 min of baseline recording, rats were subjected to either PBBI or HS. In the PBBI+HS group, HS started at 5 min after PBBI. Sham control animals received craniotomy only. Signals were recorded continuously for 150 min, at which time rats were euthanized. As a positive control of the experiment, CSD was triggered by topical application of 1 M of KCl solution at the right frontal cortex (PBBI location) in a separate group of animals (n=5). The waveform, velocity of the CSD propagation and its duration, as well as the changes in rCBF and PbtO2 were examined.

(
Electrodes and direct current (DC) recording
DC shifts were recorded with epidural silver-silver chloride (Ag/AgCl) electrodes, as described previously. 11 Electrodes were prepared from Ag wire (0.010 in diameter, coated with Teflon; AM Systems, Inc., Carlsborg, WA), which was flamed to produce a spherical tip (0.8–1.0 mm diameter) and then chloridized in sodium hypochlorite solution. The tips of the electrodes were placed epidurally in the burr holes through the skull and affixed using 3M™ Vetbond™ Tissue Adhesive. The other ends were connected to an amplifier (EX4-400 Quad Channel Differential Amplifier; Dagan Corporation, Minneapolis, MN) through shielded cables. A sintered Ag/AgCl pellet electrode was placed subcutaneously at the neck region for reference. Signals were filtered with a 0.1-Hz low-pass cutoff and were acquired using a 16-channel PowerLab system (ADInstruments Inc., Colorado Springs, CO) at a sampling rate of 1 kHz.
CBF and PbtO2
rCBF was monitored continuously using laser Doppler probes (Moor Instruments, Devon, UK). The fiber optic probes were placed in the parietal cortices bilaterally through the burr holes on the skull. PbtO2 was measured using the Licox® Brain Tissue Oxygen Monitoring System (Integra Neurosciences, Plainsboro, NJ). The oxygen sensor probes (Licox CC1.R animal probe; temperature was set at 37°C) were placed in the injured ipsilateral parietal cortex. Both recordings were acquired simultaneously with the DC recording using the PowerLab system.
PBBI
Unilateral (right) PBBI was induced using a simulated ballistic injury device (Mitre Corp., McLean, VA) with a specially designed stainless steel probe (Popper & Sons Inc., New Hyde Park, NY). 15 The probe was mounted to a stereotaxic arm at an angle of 50 degrees from the vertical axis and 25 degrees counterclockwise from the anterior-posterior axis. It was then manually inserted through the right frontal cortex of the anesthetized rat by a cranial window (+4.5 mm posterior to bregma, +2 mm midline from bregma) to a distance of 12 mm (from dura). The elastic tubing on the probe was inflated by a rapid (<40-ms) water-pressure pulse, forming an elliptical balloon calibrated to 10% of the total rat brain volume and causing an intracerebral temporary cavity. The probe was then gently retracted, and the cranial opening was sealed with sterile bone wax. Our previous studies have demonstrated that PBBI produced extensive intracerebral hemorrhage, necrotic tissues, and degenerating neurons in the frontal cortex and striatum. Within 2 h postinjury, the core lesion expanded from approximately 5 to 9.5% of the contralateral hemisphere. 15,16
Controlled HS
Transient HS was induced in anesthetized animals by withdrawing blood through the tail arterial catheter using a withdrawal pump (Harvard Apparatus) at a constant rate of 0.25 mL/100g/min to reduce MAP to 40–45 mmHg. This hypotensive state was maintained for 30 min before receiving fluid resuscitation with lactated Ringer's solution (B. Braun Medical, Bethlehem, PA) at an infusion rate of 0.3 mL/100g/min through the femoral vein catheter. The infusion volume was three times that of the blood volume withdrawn. 4
Data processing and analysis
MAP, rCBF, and PbtO2 data were analyzed using LabChart v7.0 (ADInstruments). Data were analyzed as 2-min averages taken at 5-min intervals across the 170-min recording period. rCBF and PbtO2 data were normalized to their baseline values and are expressed as percent baseline (% baseline). CSD was identified as negative deflections of the DC potential that occurred sequentially on the electrodes with or without depression of the electroencephalography (EEG) signal (high-frequency component). The propagation speed was calculated by dividing the distance between two electrodes (3 mm) by the time required for one CSD to pass the distance between two epicortical recording electrodes (labeled as “latency” in Fig. 1B). The number of DC shifts was scored by visual inspection of the recordings. The duration was calculated using custom scripts in MATLAB (The MathWorks, Natick, MA). The beginning and the end of the wave were defined as the points of 2% drop of the predepolarization baseline and the postdepolarization baseline, respectively, as illustrated in Figure 1B.
Statistical analysis
Statistical analysis was performed using SPSS statistical software (v19.0; IBM Corp., Somers, NY) and SigmaPlot 12.0 (Systat Software Inc., San Jose, CA). Parametric analyses (i.e., analysis of variance; ANOVA) were used to analyze data that showed a normal distribution, and nonparametric analyses (i.e., Kruskall-Wallis' ranked analysis) were used on data that did not meet the criteria for normal distribution as assessed by Shapiro-Wilk's test. Two-way repeated measures ANOVA with Fisher's least significant difference (LSD) post-hoc tests were used to identify potential differences in outcome metrics between groups. Piecewise linear regression (two segments) was applied to the CBF and MAP data obtained from HS and PBBI+HS groups to determine the breakpoint, slope A (the first segment), and slope B (the second segment), as previously described by Engelborghs and colleagues, 17 using SigmaPlot 12.0. Mann-Whitney's test was used to evaluate the differences in these three parameters between the two groups. CSD occurrence and duration were analyzed using Kruskal-Wallis' tests, followed by Mann-Whitney's tests with Bonferroni-Holm's correction. The significance criterion was set at p<0.05. Data are presented as the mean±standard error of the mean.
Results
Acute physiological changes were recorded continuously before and after injury. Figure 1B shows the representative tracings of rCBF, PbtO2, and DC shifts evoked by KCl, which serves as a positive control. Application of KCl for 6 min triggered a mean of 10 depolarizations per rat, with a propagation speed of approximately 4.08 mm/min on the contralateral cortex and 4.10 mm/min on the ipsilateral cortex. CSD was coupled with slight increases in CBF and PbtO2 (Fig. 1B).
Reduction in blood pressure and heart rate after PBBI+HS
MAP remained between 85 and 100 mmHg throughout the recording period in the sham control group (Fig. 2A). In animals subjected to PBBI, significant reductions in MAP were detected at the time of injury that recovered within 5 min. In contrast, HS-induced reductions in MAP did not resolve in either the HS or PBBI+HS groups, even after fluid resuscitation (p<0.05). Notably, PBBI also caused a significant, abrupt drop in heart rate that recovered within 30 min post-PBBI (Fig. 2B). During hemorrhagic hypotension, the heart rate decreased significantly from 380 to 260 beats per minute in both groups, showing an initial uncompensated response resulting from blood loss. It remained significantly lower in the PBBI+HS group after fluid resuscitation, compared to that in the PBBI group (p<0.05). Notably, MAP was slightly lower in the HS-alone group than that in the PBBI+HS group during hypotension and after resuscitation. Levels of pH, partial pressure oxygen, and carbon dioxide in arterial blood between groups (Table 1) remained within a normal range after injury.
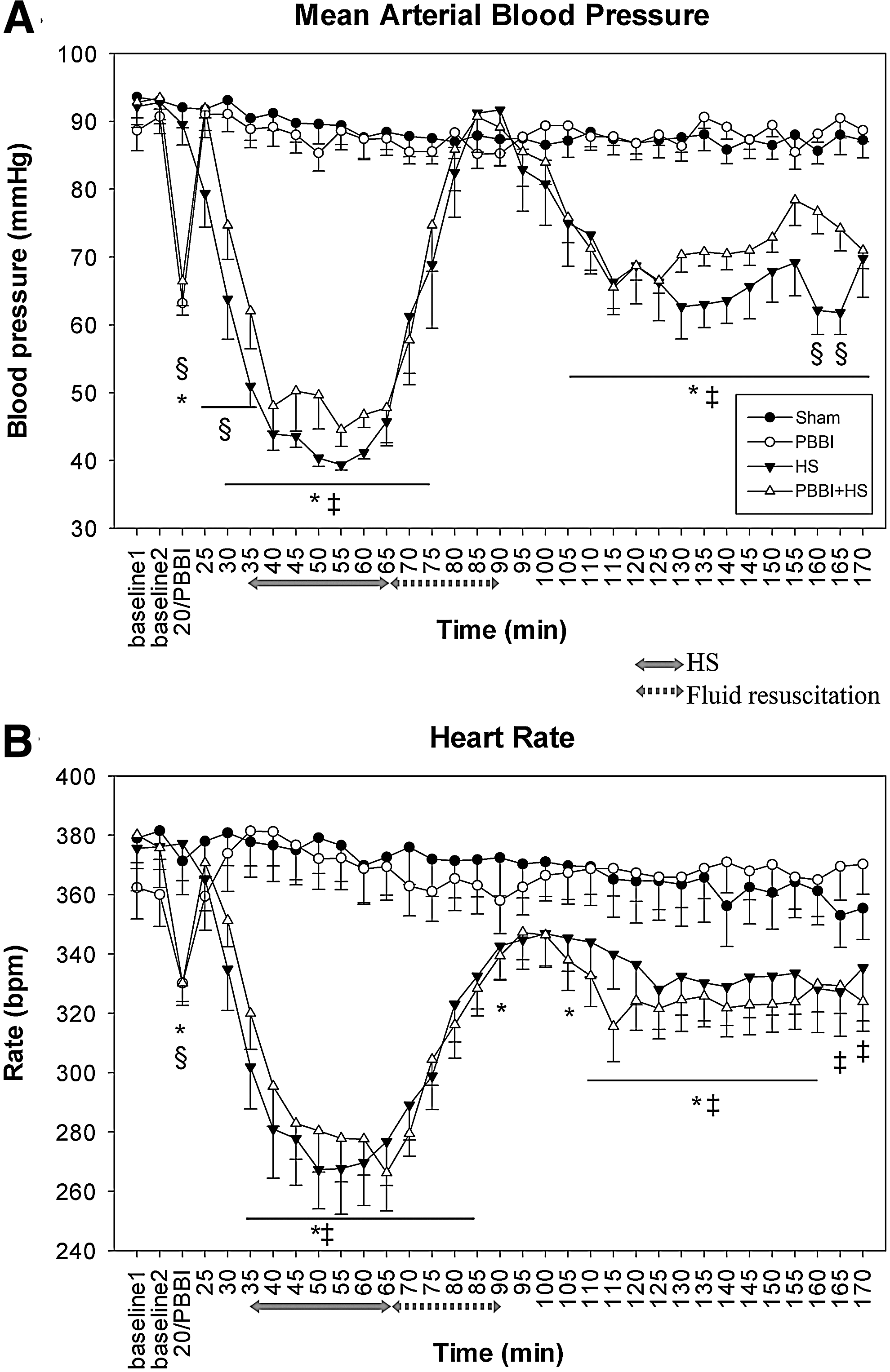
(
PBBI, penetrating ballistic-like brain injury; HS, hemorrhagic shock; PBBI+HS, combined PBBI and hemorrhagic shock.
Increased CSD occurrence, reduced propagation speed, and lowered threshold
Spontaneous CSD occurred in 84% (10 of 12) of the rats in the HS group, 79% (13 of 17) in the PBBI group, and 85% (11 of 13) in the PBBI+HS group. No CSD was detected in sham control animals. PBBI caused a massive nonpropagating depolarization at time of injury on both contralateral and ipsilateral cortices (Fig. 1B). However, this was not counted as a CSD event in our analysis. The number of DC shifts occurring in the ipsilateral cortex in the PBBI+HS group (5.92±1.34/rat) was almost six times greater than in the HS-alone (1.00±0.22/rat) and twice the number of occurrences in the PBBI-alone (2.29±0.63/rat) groups (p=0.024; Fig. 3A). The PBBI+HS group displayed a slight increase in CSD in the uninjured (contralateral) hemisphere, compared to the other groups, but the difference was not statistically significant. Patterns of DC shifts varied widely between rats, yet the durations consistently ranged from 18.06 sec to 19.65 min. CSD induced by transient hypotension had a longer duration than that induced by KCl, PBBI, or PBBI+HS on both contralateral and ipsilateral hemispheres. Though the duration appeared to be more prolonged in the PBBI+HS group (contralateral, 150.81±12.82 sec; ipsilateral, 194.71±14.94 sec), compared to the PBBI group (contralateral, 102.34±11.43 sec; ipsilateral, 123.19±11.88 sec), these difference failed to reach statistical significance (Fig. 3B). In some cases, depolarization was detected at only one recording electrode, showing no propagation. In addition, simultaneous or synchronous depolarizations were observed at the two epicortical electrodes (speed, ≥8 mm/min). As such, these cases were excluded from subsequent analysis of CSD propagation speed. HS after PBBI caused a significantly slower propagation (2.97±0.22 mm/min; p<0.01) in the injured cortex, compared to the other groups (KCl, 4.36±0.20; PBBI, 4.84±0.45; HS, 4.62±0.54 mm/min). In addition, CSD induced by HS alone occurred at a MAP level of ∼50 mmHg. Critically, after PBBI, the combined HS insult was able to induce CSD at a significantly higher MAP level (∼65 mmHg; p<0.05), as shown in Figure 3C.

Dot density plots of CSD parameters, including (
Persistent reduction of PbtO2 near core lesion
On the ipsilateral side near the core lesion, PbtO2 reduced significantly after PBBI (70% baseline) and gradually recovered to 80% baseline at the end of the recording. The addition of HS after PBBI further decreased oxygen tension to 40% baseline. After resuscitation with lactated Ringer's solution, oxygen level increased slightly, but was still significantly lower than that in the HS-alone and PBBI-alone groups (p<0.05). In contrast, PbtO2 in animals subjected to HS only returned to baseline level after resuscitation (Fig. 4A). In the perilesional region (Fig. 4B), PbtO2 decreased to 80% baseline after PBBI, gradually returning to baseline. HS caused a significant reduction (60% baseline) in PbtO2 during the hypotensive state that was restored to 80% of baseline in the HS group and 70% in the PBBI+HS group after fluid resuscitation. PbtO2 increased with time in sham control animals on both contralateral and ipsilateral sides.
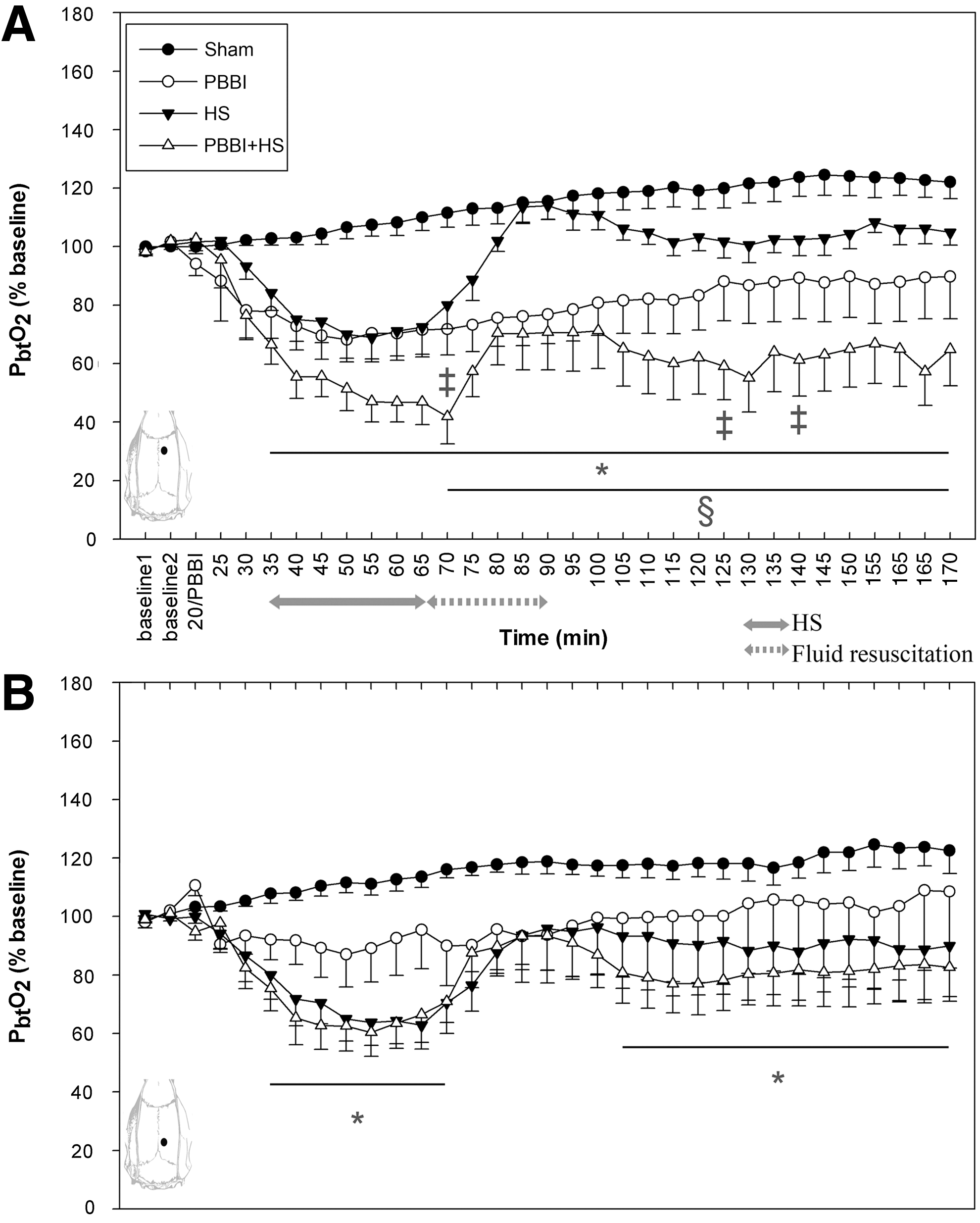
Temporal changes of PbtO2 in (
Significant decline of rCBF after PBBI in core and perilesion regions
Cerebral blood flow was measured at four locations: contralateral anterior parietal cortex; contralateral posterior parietal cortex; ipsilateral core lesion; and the ipsilateral perilesional area (Fig. 1A). In all regions, rCBF in the sham control group increased over time from 100% baseline to 120%, likely as a result of an isoflurane-induced vasodilation. PBBI caused a significant, persistent reduction of rCBF on the ipsilateral side at the both core (35% baseline; Fig. 5C) and perilesional areas (80% baseline; Fig. 5D), but a gradual increase on the contralateral side (Fig. 5A,B). During the hypotensive state, rCBF dropped to 60–70% of baseline, but was immediately recovered after fluid resuscitation, except in animals subjected to PBBI prior to HS. In this group (i.e., PBBI+HS), fluid resuscitation did not restore rCBF to normal levels in either the core lesion and/or perilesional region. On the uninjured side, the HS and PBBI+HS groups showed similar rCBF changes, in which the rCBF decreased significantly during the hypotensive state and increased immediately after resuscitation (Fig. 5A,B).

Temporal changes of CBF in the contralateral hemisphere (
PBBI and HS disrupted autoregulation
Examination of the relationship between rCBF and MAP (Figure 6A–D), which is reflective of cerebral autoregulation, showed that PBBI+HS significantly decreased the threshold for autoregulation (i.e., increased breakpoint), compared to HS alone (Fig. 6E; PBBI+HS, 67.41±1.51 mmHg; HS, 59.41±1.45 mmHg; Mann-Whitney's test, p<0.05). In addition, slope A in both groups were positive, indicating impaired autoregulation when MAP fell below the lower limit of autoregulation. On the contrary, slope B in both groups were negative, implying intact autoregulation when MAP was above the lower limit or the breakpoint. Both slope A and B were significantly greater in the HS-alone group than the PBBI+HS group (Fig. 6F,G), suggesting that after PBBI, the cerebrovascular system might be less responsive to changing MAP, possibly as a result of the severe damage of the vasculature especially on the side of injury (Fig. 6C,D).

Piecewise correlations (two segments) between MAP and CBF of representative rats from the HS and PBBI+HS groups at four locations: (
Discussion
In polytrauma patients, HS-induced hypotension accompanied by TBI is not uncommon. The secondary injury cascade in this combined event is complex, yet poorly understood. Acute physiological changes determine injury outcome and understanding these changes would facilitate the clinical design of neuromonitoring and early therapeutic interventions for polytrauma patients. Therefore, we sought to investigate how HS affects physiological parameters, such as CBF, PbtO2, and CSD, acutely following PBBI. The main finding of this study was that HS increased CSD incidence after PBBI. CBF and PbtO2 were significantly reduced, yet they did not change in response to CSD. Additionally, PBBI prior to HS did not adversely affect HS-induced cardiovascular responses.
Our previous study showed that ICP increased abruptly and dramatically in conjunction with the epoch of the PBBI injury. 18 It is likely that the dynamic pressure caused by the rapid expansion of the elastic tubing on the PBBI probe propagates through the brain parenchyma to the brainstem, causing abnormal cardiovascular and respiratory responses. Because the inflation only lasted for 40 ms, these abnormal responses resolved rapidly and blood pressure returned to normal after the pressure wave dissipated. In the present study, PBBI caused a similar sudden, profound decrease in arterial blood pressure and heart rate. However, the addition of HS led to a more persistent reduction in blood pressure and heart rate. Paradoxical bradycardia was evident in clinical and animal studies of severe blood loss 19 and was suggested to be a mechanism involving parasympathetic reflex to prevent cardiac collapse. 20 This can be reversed by infusing crystalloid solution, such as lactated Ringer's solution, to restore intravascular volume and thus blood pressure. The combined effect of PBBI and HS on this cardiac reflex was similar to that of HS alone. However, previous animal studies using fluid percussion injury (FPI) have reported negative effects of brain trauma on HS-induced cardiovascular response, where the response was delayed during the initial phase of HS in brain-injured animals, compared to that in animals subjected to HS alone. 21,22 The previous brain trauma also impeded the efficacy of fluid resuscitation. 22 One plausible explanation of the conflicting result is that the lesion produced by PBBI is mainly the frontal cortex and striatum, whereas FPI affected primarily the parietal cortex and brainstem. 23 Thus, the effect of brain injury on HS-induced cardiac reflex does not depend solely on the injury severity, as suggested by McMahon and colleagues, 21 but is also dependent on the brain regions affected in the TBI model. Interestingly, MAP in the PBBI+HS group was slightly higher than that in the HS-alone group during the hypotensive state and after fluid resuscitation, suggesting that systemic hemodynamics may be augmented by PBBI. Such hyperdynamic response was observed acutely in severe TBI patients and was related to sympathetic nervous hyperactivity. 24 Nevertheless, additional studies specifically designed for cardiovascular homeostasis is required to fully understand this physiological response.
During PBBI, ionic homeostasis was massively disrupted, leading to a near-complete depolarization on both contra- and ipsilateral cortices. A few or zero spontaneous CSDs occurred after this substantial depolarization; however, more recurrent CSDs occurred in animals subjected to HS after PBBI, suggesting that the additional insult worsened the ionic imbalance and energy depletion. In human TBI, a higher incidence of spontaneous CSD has been observed, compared to experimental TBI. 25 Indeed, spreading depolarization has been shown to be less readily elicited in the gyrencephalic brain than in the lissencephalic brain. 26 The high CSD incidence in humans may be explained by the observation that clinical TBI is often accompanied by secondary insults, such as hypoxia and hypotension, and its pathophysiology is more complicated and heterogeneous than occurs in a less-complex TBI animal model. 27
HS alone caused sustained depolarization (i.e., prolonged duration of DC shift). A previous study has demonstrated the prolongation of the CSD duration by reducing MAP to 50 mmHg in rats subjected to systemic hypotension. 28 Unlike KCl-induced CSD, CBF or PbtO2 were not responsive to HS- or PBBI+HS-induced CSDs in the present study, implicating that the normal hyperemic response was impaired. PBBI rats, however, lacked prolonged DC shift duration. This is consistent with other TBI rodent models, in which the CSDs were of short duration. 13 In contrast to the animal studies, a prolonged DC shift has been reported in TBI patients and suggested to be associated with poor prognosis. 29 A possible explanation for this difference between human versus experimental TBI is that the experimental models simply cannot reproduce the complex and heterogeneous nature of the clinical TBI. Notably, the additional HS insult after PBBI in our study appeared to extend CSD duration. Sustained depolarization results in intracellular calcium overload and increased free radical production, triggering apoptotic signals and proteases leading to cell death. 2 Apart from extending DC shift duration, combined PBBI and HS also reduced the propagation speed of CSD. Several studies suggested a positive correlation of CSD speed with cortical excitability 30 and MAP 28 as well as inverse correlation with cortical myelination. 31 PBBI causes ionic imbalance, damage to white matter, elevated ICP, inflammation, and necrotic and apoptotic cell death. 15 These might be worsened by the hypotension insult after PBBI, where, in the present study, the PBBI+HS group showed a significant reduction in MAP. The balance between TBI-induced cortical hyperexcitability and hypotension-induced slow propagation determines resultant CSD speed. Further study is required to examine this hypothesis.
PBBI lowered the threshold of spontaneous CSD. Our data indicated that CSD occurred when blood pressure dropped to ∼50 mmHg in rats subjected to HS only, whereas it occurred at higher blood pressure in animals subjected to PBBI prior to HS. Although MAP is an indirect factor, it is associated with cerebral perfusion. Cerebral hypoperfusion depresses electrical activity of the brain, 32 and it has been reported that in TBI patients, CSD incidence increased as a function of reduced MAP and cerebral perfusion pressure. 33 The threshold of CSD can be modulated by several factors, including reduced extracellular nitric oxide (NO) levels 34 and mutations in the Cacna1A gene that encodes calcium channels 35 and the ATP1A2 gene that encodes the astrocytic sodium pump. 36 Low NO levels as a result of reduced CBF 37 and mutated Cacna1A related to cerebral edema and coma after TBI 38 may account for the lowered threshold (higher susceptibility) to spontaneous CSD after PBBI.
Despite the emergence of CSD, the corresponding changes in rCBF or PbtO2 during the occurrence of CSD in the injury groups were not observed. Under normal physiological conditions, spreading depolarization is often associated with spreading hyperemia to meet the energy demand. 2 Brain tissue in the pathological condition, however, exhibits an inverse hemodynamic response, leading to spreading ischemia, 2 that was evident in patients with subarachnoid hemorrhage. 39 Spreading ischemia was not detected in the present study, probably because of the severely damaged cerebral vasculature and the extensive lesion where CBF in the injured hemisphere remained low throughout the recording period after PBBI or PBBI+HS. Interestingly, HS did not further reduce CBF after PBBI, possibly because of the vascular dysfunction and intracerebral hemorrhage. On the other hand, PbtO2 level in the PBBI+HS group was slightly lower than that in the PBBI group before and after fluid resuscitation, whereas in the PBBI group it decreased shortly after injury, followed by a gradual increase. This gradual increase in PbtO2 may be a result of the prolonged isoflurane anesthesia in spite of the presence of a lesion. 40 In addition to the anesthesia effect, the primary insult causes substantial necrotic cell death 16 and reduces oxygen demand, resulting in excess oxygen supply. Levels of CBF and PbtO2 may also have been affected by the fluid resuscitation strategy. In the present study, lactated Ringer's solution was infused intravenously to resuscitate the animal after HS. This is a standard resuscitation regimen for acute hemorrhage, according to The American College of Surgeons Advanced Trauma Life Support guidelines. However, it only temporarily restored the systemic volume and electrolytes, but not CBF or PbtO2, on the side of injury. The solution rapidly equilibrates with the interstitial space and is not confined to the vascular space. This explains the reduction in MAP shortly after fluid resuscitation, and the reduced MAP might account for the decreased PbtO2 level in both the HS and PBBI+HS groups after resuscitation. Although infusion of lactated Ringer's solution caused a decrease in MAP, it was capable of restoring CBF to normal levels in the HS group as well as in the contralateral side in the PBBI+HS group. In the core lesion and perilesional regions, CBF remained steadily at a low level. In addition to MAP (beyond the autoregulatory range), ICP and cerebrovascular resistance (CVR) are important determinants of CBF. 41 Data on ICP and CVR are required to elaborate on the effect of the fluid resuscitation strategy on CBF after combined PBBI and HS.
Autoregulation represents the ability to maintain a stable CBF in response to changes in systemic blood pressure. When blood pressure drops below the autoregulatory range (50–150 mmHg), global hypoperfusion occurs, making the brain more susceptible to injury. 42 In the present study, the threshold of cerebral autoregulation impairment was lowered (increased lower limit of autoregulation) after PBBI. Our finding is consistent with a previous study showing an increased lower limit of autoregulation (LLA) after TBI (weight-drop model). The loss of cerebral autoregulation was triggered when MAP was 62.2±20.8 mmHg at 24 h after closed head injury in rats, whereas without brain injury it was triggered at 46.9±12.7 mmHg. This increased LLA may be related to hypotension-induced sympathetic activation. 17 In addition, it appeared that the cerebral autoregulatory response was not lateralized after a unilateral brain injury. A recent study using a neonatal swine model of severe CCI reported similar findings. 43 In spite of the intact cerebrovascular structure, impaired cerebrovascular response was found in the contralateral hemisphere in animals subjected to PBBI+HS. However, the effect on the contralateral hemisphere may be transient and indeed resolved within hours after injury, which may explain the discrepancy between the clinical and pre-clinical observations where asymmetric cerebral autoregulation has been reported in TBI patients 3 days postinjury. 44
The present study was designed to examine the acute changes in CBF, PbtO2, and CSD as well as the general physiology, including MAP, heart rate, and blood gas. Detailed histopathological analysis was not performed in these animals because of the acute nature of the study and the additional trauma caused by the invasive recording technique and possible neuroprotective effects of prolonged isoflurane anesthesia. Another limitation of our study is the Ag/AgCl ball electrodes used for recording DC shift. These electrodes record the electrical activity on the surface of the cortex by an intact dura where the amplitude of depolarization cannot be accurately measured and was therefore not reported. Glass micropipette electrodes are capable of measuring amplitude in the cortex with minimal tissue disruption. However, this technique is not suitable in the PBBI model because the extremely fine tip of the glass electrode could be broken during the PBBI injury. Nevertheless, duration and number of CSDs are of clinical relevance. Along these lines, our results were consistent with the CSD findings from reported clinical studies in which prolonged duration and increased CSD incidence were observed in TBI patients. 29
In conclusion, our data show that HS after PBBI increased the occurrence of CSD that appeared to have prolonged duration and slower propagation speed. In addition, ipsilateral CBF and PbtO2 remained low and were not responsive to the depolarizations. These detrimental events, together with the lowered threshold of cerebral autoregulation, are likely to make compromised brain tissue more susceptible to secondary injury, resulting in more cell death and, very possibly, enhanced metabolic dysregulation.
Footnotes
Acknowledgments
The views of the authors do not purport or reflect the position of the Department of the Army or the Department of Defense (para 4-3, AR 360-5). This research is funded by the Combat Casualty Care Research Program, U.S. Army Medical Research and Materiel Command. The primary author is sponsored by the National Research Council Research Associateship Program (National Academy of Sciences, Washington, DC). The authors thank Dr. Jed A. Hartings (University of Cincinnati, Cincinnati, OH) for his valuable advice on cortical spreading depolarization and SPC Shawn McLoughlin and Ms. Xiaofang Yang for their technical support.
Author Disclosure Statement
No competing financial interests exist.