
Abstract
The beneficial effect of interventions with chondroitinase ABC enzyme to reduce axon growth-inhibitory chondroitin sulphate side chains after central nervous system injuries has been mainly attributed to enhanced axonal sprouting. After traumatic brain injury (TBI), it is unknown whether newly sprouting axons that occur as a result of interventional strategies are able to functionally contribute to existing circuitry, and it is uncertain whether maladaptive sprouting occurs to increase the well-known risk for seizure activity after TBI. Here, we show that after a controlled cortical impact injury in rats, chondroitinase infusion into injured cortex at 30 min and 3 days reduced c-Fos+ cell staining resulting from the injury alone at 1 week postinjury, indicating that at baseline, abnormal spontaneous activity is likely to be reduced, not increased, with this type of intervention. c-Fos+ cell staining elicited by neural activity from stimulation of the affected forelimb 1 week after injury was significantly enhanced by chondroitinase, indicating a widespread effect on cortical map plasticity. Underlying this map plasticity was a larger contribution of neuronal, rather than glial cells and an absence of c-Fos+ cells surrounded by perineuronal nets that were normally present in stimulated naïve rats. After injury, chondroitin sulfate proteoglycan digestion produced the expected increase in growth-associated protein 43–positive axons and perikarya, of which a significantly greater number were double labeled for c-Fos after intervention with chondroitinase, compared to vehicle. These data indicate that chondroitinase produces significant gains in cortical map plasticity after TBI, and that either axonal sprouting and/or changes in perineuronal nets may underlie this effect. Chondroitinase dampens, rather than increases nonspecific c-Fos activity after brain injury, and induction of axonal sprouting is not maladaptive because greater numbers are functionally active and provide a significant contribution to forelimb circuitry after brain injury.
Introduction
In previous work, we have used cABC to further digest the CSPG side chains to prevent their eventual increase after injury. 23 However, although the resulting significant increase in cortical sprouting did significantly improve behavioral outcomes, it did not confer robust functional improvements. We therefore initiated a study to determine whether newly sprouting axons do, in fact, contribute to brain circuitry using accumulation of c-Fos protein as an indication of cortical cell activation. We have used a well-known controlled cortical impact (CCI) injury adult rat model of TBI to produce a cortical injury over the forelimb cortex, together with an improved method of cABC delivery to produce a greater sprouting response. The data show that cABC infusion promotes a significant enhancement of the forelimb cortical map, and that newly sprouting, growth-associated protein 43 (GAP43)+ axons and/or reductions in perineuronal nets contribute greatly to this.
Methods
Experimental protocol
An initial experiment was performed to determine the best method for cABC intervention. After left CCI injury, cABC was injected through an in-dwelling catheter (n=3) at 30 min and at 3 days (2 μL, 48 U/μL, and 100 nL/min) using a digital microinfusion pump (Stoelting, Wood Dale, IL) and compared to vehicle (artificial cerebrospinal fluid; aCSF) injections after injury (n=3), and to a single 30-min injection of cABC followed by chronic administration using an osmotic pump (n=3; 10 U/mL and 0.5 uL/h), according to our previously published methods. 23 Rats were perfusion fixated at 7 days postinjury and quantified for total cortical GAP43+ immunostaining (Fig. 1). This time point was chosen because previous studies showed maximal cortical sprouting in concert with CSPG-deficient regions by 7 days post-CCI, with growth-inhibitory CSPGs reappearing 14–21 days post-CCI. 1,3
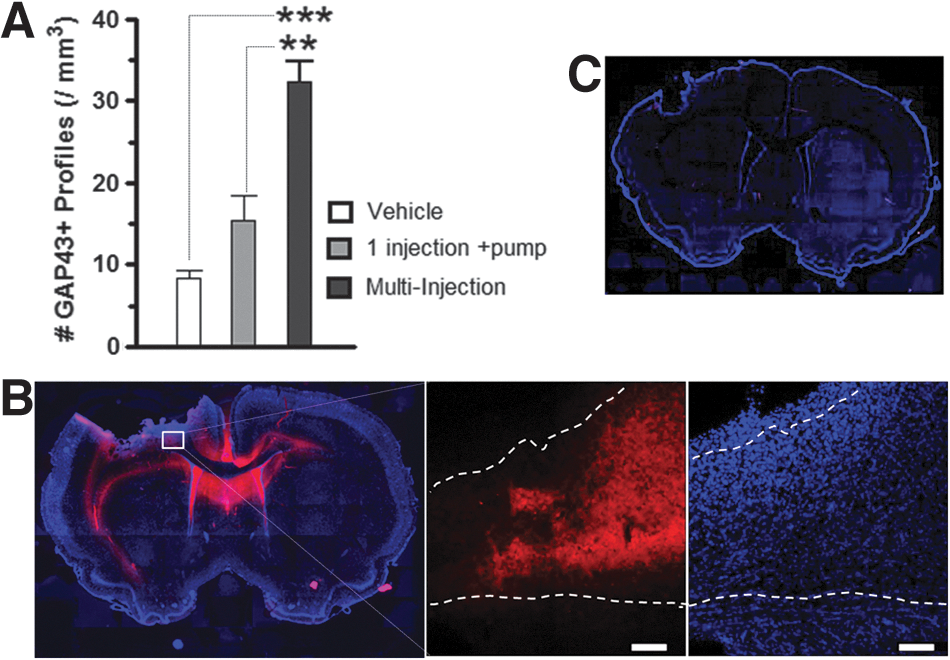
Multiple injections of chondroitinase (cABC) increase cortical axonal sprouting after brain injury. (
All other studies were performed after left CCI injury, and chondroitinase (cABC; n=6) or CSF vehicle or no vehicle (n=3 and 5, respectively) was administered at 30 min and 3 days later through an in-dwelling catheter. All rats, together with a naïve group (n=4), were perfusion fixated at 7 days postinjury after 1 h of right (affected) forelimb electrical stimulation. Additional groups of naïve, injured-vehicle and injured-cABC rats (n=3, 5, and 3, respectively) were not stimulated and served as controls. After immunostaining, cortical tissue was quantified for gray and white matter c-Fos+ profiles, GAP43+/c-Fos+ profiles, and c-Fos+ cell phenotype.
Surgical procedures
All study protocols were approved by the University of California, Los Angeles (Los Angeles, CA) Chancellor's Animal Research Committee and adhered to the Public Health Service Policy on Humane Care and Use of Laboratory Animals. The method for induction of moderate CCI injury was performed in the manner similar to that previously described. 1,23 –27 Briefly, male Sprague-Dawley rats (220–250 g in body weight) were anesthetized with 2% isofluorane vaporized in O2 flowing at 0.8 L/min and placed on a homeostatic temperature-controlled blanket while being maintained in a stereotactic frame. CCI was produced using a 4-mm-diameter impactor tip that was advanced through a 5-mm craniotomy (centered at 0.0 mm Bregma and 3 mm left lateral to the sagittal suture) onto the brain using a 20-psi pressure pulse and to a deformation depth of 2 mm below the dura. In rats receiving intraparenchymal cABC or CSF infusion, a small hole was made in the dura using a 30-G needle and a 2-mm-thick polyethylene plastic window was cemented to the bone around the craniotomy using gel superglue. A metal cannula (33 G in diameter) mounted on a pedestal (Plastics One, Roanoke, VA) was advanced through a predrilled hole in the window, through the dura to a depth 1.5 mm below the bone. The pedestal was cemented in place on the window with superglue and to the surrounding bone using dental acrylic cement, which also served to close the craniectomy site.
Forepaw stimulation paradigm
At 7 days after injury, rats were very briefly anesthetized with isofluorane (2% in oxygen) to enable an intravenous bolus injection of the sedative, dexmedetomidine hydrochloride (0.05 mg/kg; Pfizer, New York, NY) through the penile vein and for placement of two metal stimulating electrodes (30 G in diameter; Grass Technologies, Warwick, RI) through the palmar pads of the right (affected) limb, immediately adjacent to digits 1 and 4, subcutaneously (s.c.) to a position proximal to the wrist. An s.c. cannula was placed on the back, through which a continuous infusion of dexmedetomidine was begun (0.1 mg/kg/h). 28 After confirming correct placement of the electrodes by brief stimulation to elicit a forepaw twitch, isofluorane was immediately discontinued and visual input to the rat was reduced by fitting a box around the head, and laboratory noise was minimized to prevent auditory stimulation. Continuous electrical stimulation of the forepaw was begun 20 min later at 3.5 mA (10 Hz for a 60-minute period), following which the dexmedetomidine infusion was discontinued. This stimulus intensity was chosen because it gives robust regions of activation using functional magnetic resonance imaging in the same model. 29 Although it is disputable whether this is an innocuous or nociceptive stimulus, 30,31 at least within the sensorimotor cortex, there is little difference in the volume of activation, compared to an activation evoked with a lower-intensity 2-mA stimulus. 30 After stimulation, the rat was maintained in a quiet environment for another 2 h, 32 still under partial sedation before terminal pentobarbital anesthesia (100 mg/kg intraperitoneally) was administered for transcardial perfusion fixation with 0.1 M of phosphate-buffered saline (PBS) at approximately the mean arterial pressure for an adult rat, followed by 4% paraformaldehyde.
Immunohistology
Brains were removed, cryoprotected, and snap-frozen in liquid nitrogen-cooled isopentane. Coronal sections were cut on a cryostat at 50 μm, and all sections were stored in buffered ethylene glycol/glycerol. Immunohistology for bright-field microscopy was performed as described before. 1,23 Briefly, after antigen retrieval using citrate buffer and heat, sections were quenched and then blocked in TXTBS [Tri-buffered saline (TBS) containing 0.2% Triton X-100; Sigma-Aldrich, St. Louis, MO] with 10% normal horse serum (NHS; Vector Laboratories, Burlingame, CA) and 1% bovine serum albumin (BSA; Vector Laboratories) and placed in primary antibody (Ab) overnight at room temperature [c-Fos, 1:2000, #PC38 (Calbiochem, San Diego, CA), or GAP43, 1:1500, #MAB347 (Millipore, Billerica, MA)] in TBS containing 5% NHS. All steps were followed with three rinses in TBS after each incubation period. Signal amplification was achieved by incubating sections in species-specific, biotinylated secondary Abs (1:500; Vector Laboratories) in TBS with 5% NHS for 1 h, followed by the Vectastain Elite ABC kit (Vector Laboratories), and then visualized with diaminobenzidine (DAB). Sections were mounted on gel-coated slides, dehydrated, and cover-slipped with DPX mounting medium. Immunohistology for multi-label fluorescence microscopy was performed in a similar manner, as described before, 1,23 with direct detection of the primary Abs, c-Fos and GAP43, as described above, and NeuN (1:5000, MAB377; Millipore), glial fibrillary acidic protein (GFAP; 1:500 AB5804, Millipore), ionized calcium-binding adapter molecule 1 (Iba1; 1:500, Ab15690; Abcam, Cambridge, MA), and CC1 (1:5000, MAB1580; Millipore), 2B6 (1:200; Associates of Cape Cod, East Falmouth, MA), or biotin-conjugated lectin Wisteria floribunda (WFA; 1:500, No. L-1766; Sigma-Aldrich) using Alexa 555– or 488–conjugated, host-species–specific secondary Abs or conjugated streptavidin-Alexa (Molecular Probes, Carlsbad, CA). Cell bodies were stained with the nucleic acid marker, 4,6-diamidino-2-2-phenylindole (DAPI; Vectashield; Vector Laboratories), and sections were mounted on slides and cover-slipped with anti-bleaching medium (Vector Laboratories). Representative z-stack images were acquired from the full thickness of the sections at 3-μm intervals using structured illumination microscopy implemented on a Zeiss Apotome attached to an M2 microscope (Zeiss, Thornwood, NY) using high numerical aperture (NA) objectives ×20 (0.8 NA), ×40 (0.95 NA), and ×63 (1.4 NA) that govern the slice thickness used, and images were captured with an Axiocam MRm camera (Zeiss).
Cell counts and cortical atrophy measures
For the initial experiment to determine the best method for cABC delivery, cortical gray matter GAP43+ cell profile counts were made in predefined contours, as described before, 1,23 on three sections per brain at least 600 μm apart within the ipsilateral sensorimotor cortex (Bregma +1.2, +0.48, and −0.26m) at a ×20 objective magnification under bright-field illumination on a Zeiss M2 upright microscope and visualized using a camera (AxioCam MRm; Zeiss) interfaced to a computer running StereoInvestigator software (MicroBrightField, Inc., Williston, VT).
In subsequent experiments, c-Fos+ cells were quantified in a similar manner and digitally mapped under bright-field illumination, also on three sections at the same anteroposterior levels as described above, and bilaterally within the sensorimotor cortex, within predefined gray- and white-matter contours between the mid-line and the rhinal fissure, excluding heavy c-Fos+ cell labeling that is normally present irrespective of brain activation, in gray-matter regions medial to a line drawn from the apex of the cingulum to the most medial-dorsal point of the gray matter. GAP43+ profiles were mapped and counted under epifluorescence in the same regions, followed by the number of profiles double labeled for c-Fos. Data are expressed as cell densities using the contour areas and were averaged between all sections within each brain. Data were semi-quantitatively visualized by rigidly aligning the contours in three data sets representative of the group-level differences, as described before. 23 Cortical atrophy was determined by measuring the volume of ipsilesional cortical gray- and white-matter tissue remaining in at least six sections 600 um apart and normalizing by the volume of the contralesional cortex and expressing atrophy as percent tissue loss, as described before. 27
c-Fos+ cell phenotype
The phenotype of c-Fos+ cells was assessed semi-quantitatively on confocal image stacks acquired on three sections per brain at least 600 μm apart and in three regions per section placed either on the side or below the contusion in cABC- and aCSF-treated injured rats that were stimulated (nine fields total per brain). Image stacks were acquired on a Zeiss LSM 410 Pascal laser confocal microscope system (Zeiss) using a ×20 objective, 9–15 optical slices, 4 μm thick, and at 2.1-μm intervals in a total area of 369 μm2 per field. The number of c-Fos+ and c-Fos/NeuN+/GFAP+/CC1+/Iba1+ cells were counted by assessing the images in both the planar and projected orthogonal view to be certain of any double-labeled cell assignments. For c-Fos+/GFAP+ cells, only those c-Fos+ cell bodies surrounded by GFAP+ processes were considered double-labeled. Double-labeled cell data were expressed as a percentage of the total number of c-Fos+ cells in each image stack and averaged between all stacks in each section and then between all sections analyzed.
Statistical analysis
For all cell-count data, Kolmogorov-Smirnov's with Lillie's correction and Levene's median tests were used to describe distribution of data and determine equality of variances, respectively. Having passed these normality tests, group numerical data were expressed as the means±standard error of the mean (SEM) or as raw cell counts. Cell-density-count differences were explored using one or two-way analysis of variance (ANOVA), according to our a priori hypothesis that chondroitinase increases axon sprouting. Non-normally distributed data were tested for significance using Kruskal-Wallis' one-way ANOVA on ranks. Statistical significance (α) was set at p<0.05, one-tailed for all comparisons, consistent with our hypothesis that increases in neuronal activation was expected.
Results
In previous work, we have shown that a single intraparenchymal injection of cABC, together with osmotic pump infusion, results in significant increases in cortical GAP43+ axon sprouting. 23 We now show that in the same brain injury model, two cABC injections at 30 min and 3 days after injury result in significantly more cortical gray- and white-matter GAP43+ axon profiles at 7 days (p<0.01; Fig. 1A). Immunostaining for the CSPG glycosaminoglycan “stub removal” region on the core CSPG protein with 2b6 Ab revealed that multiple injections result in large regions of the brain with digested CSPG side chains, including the contralateral side surrounding the corpus callosum and cingulum, when compared to vehicle (aCSF) infusion at 7 days postinjury (Fig. 1B,C).
Cortical c-Fos+ cells increased after injury alone, but decreased after cABC infusion
The presence of increased axonal sprouting alone does not necessarily confer improved brain function, and so we used a forelimb stimulation paradigm of the affected limb to determine whether cABC also enhances activation of the ipsilesional cortex. We used immunostaining of the protein product of the immediate-early gene, c-Fos, as a readout of cell activation. We first determined whether the number of cortical c-Fos+ cells were merely altered by injury or cABC infusion alone in groups of rats not subjected to forelimb stimulation. An initial comparison of injured rats with no infusion and injured rats receiving vehicle infusion showed that there was no effect of vehicle (p>0.05, not shown), and so data were pooled and collectively referred to as injured + aCSF. The findings show that at 7 days after injury, there were very consistent 4- to 5-fold increases in the density of ipsilesional cortical gray- and white-matter c-Fos+ cells resulting from injury alone and compared to similar regions in the naïve group (p<0.05; Fig. 2A–C). Smaller, but significant, increases were also found in contralesional gray- and white-matter regions (p<0.05). In contrast to this, cABC infusion into the ipslesional injured cortex significantly decreased the density of c-Fos+ cells bilaterally in gray matter, compared to vehicle infusion (p<0.05), but had no effect in white matter, although a trend toward a reduction was readily apparent (Fig. 2A–C). We found no significant neuroprotective effect of cABC; tissue loss was −10.3±2.22 and −13.4±5.12% of the contralateral hemisphere in vehicle and cABC-treated injured rats, respectively (p>0.05).
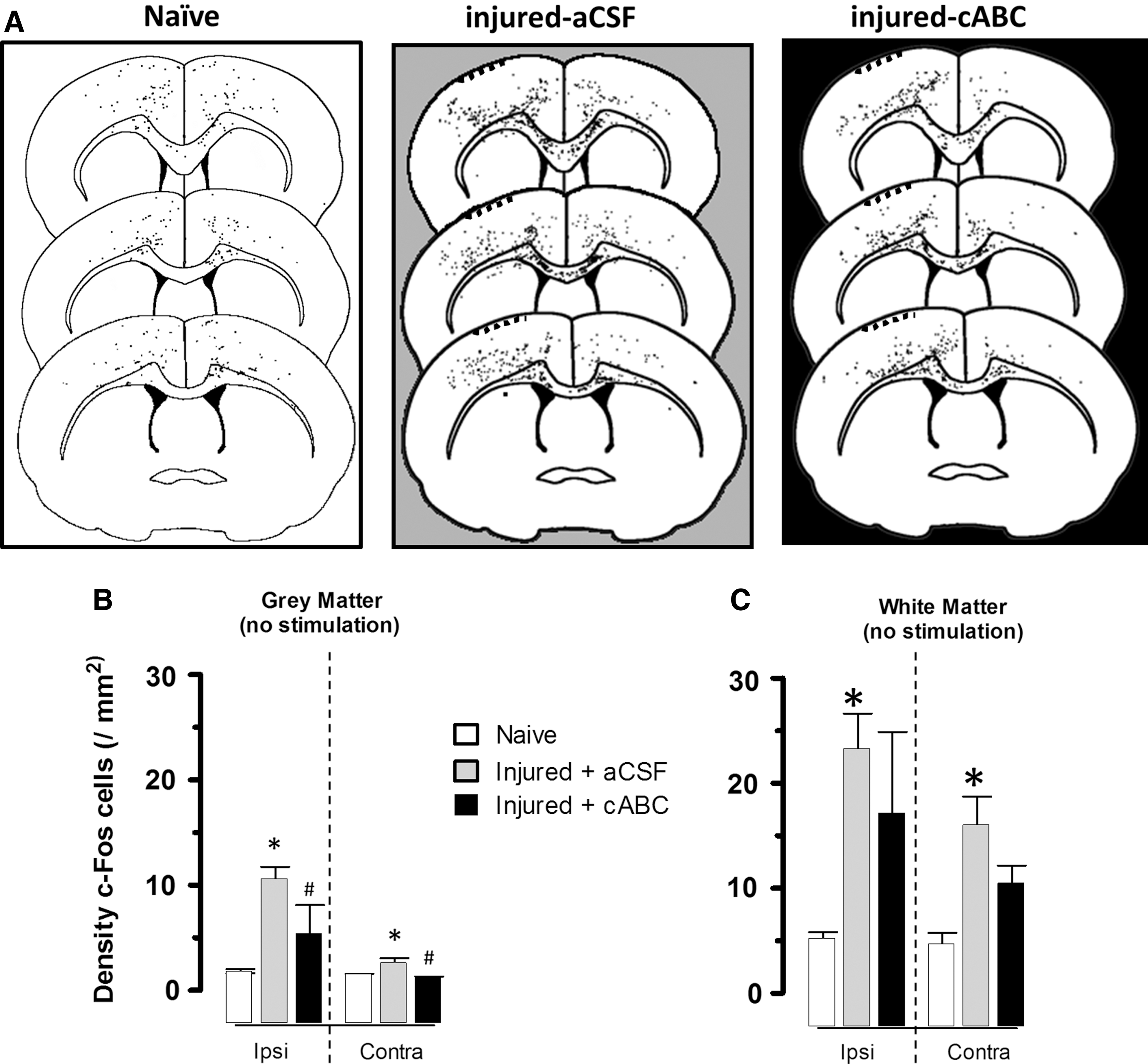
Injury alone without forelimb stimulation increases c-Fos expression. (
Forelimb-stimulated c-Fos+ cell activation increases with injury severity and with cABC infusion
Stimulation of the right forelimb in naïve rats increased left cortical c-Fos staining above baseline levels in only 2 of 4 rats, so that, overall, it was not significantly raised above naïve animals with no stimulation (p>0.05; Figs. 3A,D and 4). Likewise, c-Fos+ cell staining was raised in the majority of affected (right) forelimb-stimulated injured + aCSF treated rats (Fig. 3A), but the effect was variable between animals and in all regions examined, so that, overall, it was not significantly different from naïve rats (p>0.05; Fig. 3D,G) or nonstimulated injured rats (Fig. 4). However, further analysis of the severity of the injury revealed that after removing 1 injured + aCSF rat with no detectable tissue loss, the density of c-Fos+ cells was linearly and positively associated with the degree of tissue loss in ipsilesional gray and white matter (r=0.73 and 0.68, respectively; p<0.05; n=7), and this correlation also approached significance in the contralesional gray matter (r=0.65; p=0.058). cABC infusion after injury resulted in a profound increase in the number of c-Fos+ cells stained after affected forelimb stimulation, compared to other groups (Fig. 3A), so that even without normalizing to the degree of c-Fos staining in unstimulated cABC-infused rats, c-Fos+ cell density was significantly increased bilaterally in gray matter and in contralesional white matter, compared to naïve and injured+aCSF rats (p<0.001; Fig. 3). Normalizing c-Fos+ cell data in the stimulated group for baseline (unstimulated) c-Fos+ staining that was solely the result of injury + aCSF or injury + cABC infusion similarly revealed a significant effect of cABC after injury in all regions examined (p<0.05; Fig. 4). This effect was especially consistent within the ipsilesional cortex, where c-Fos+ cell density was increased to levels well above naïve and injured + vehicle groups.
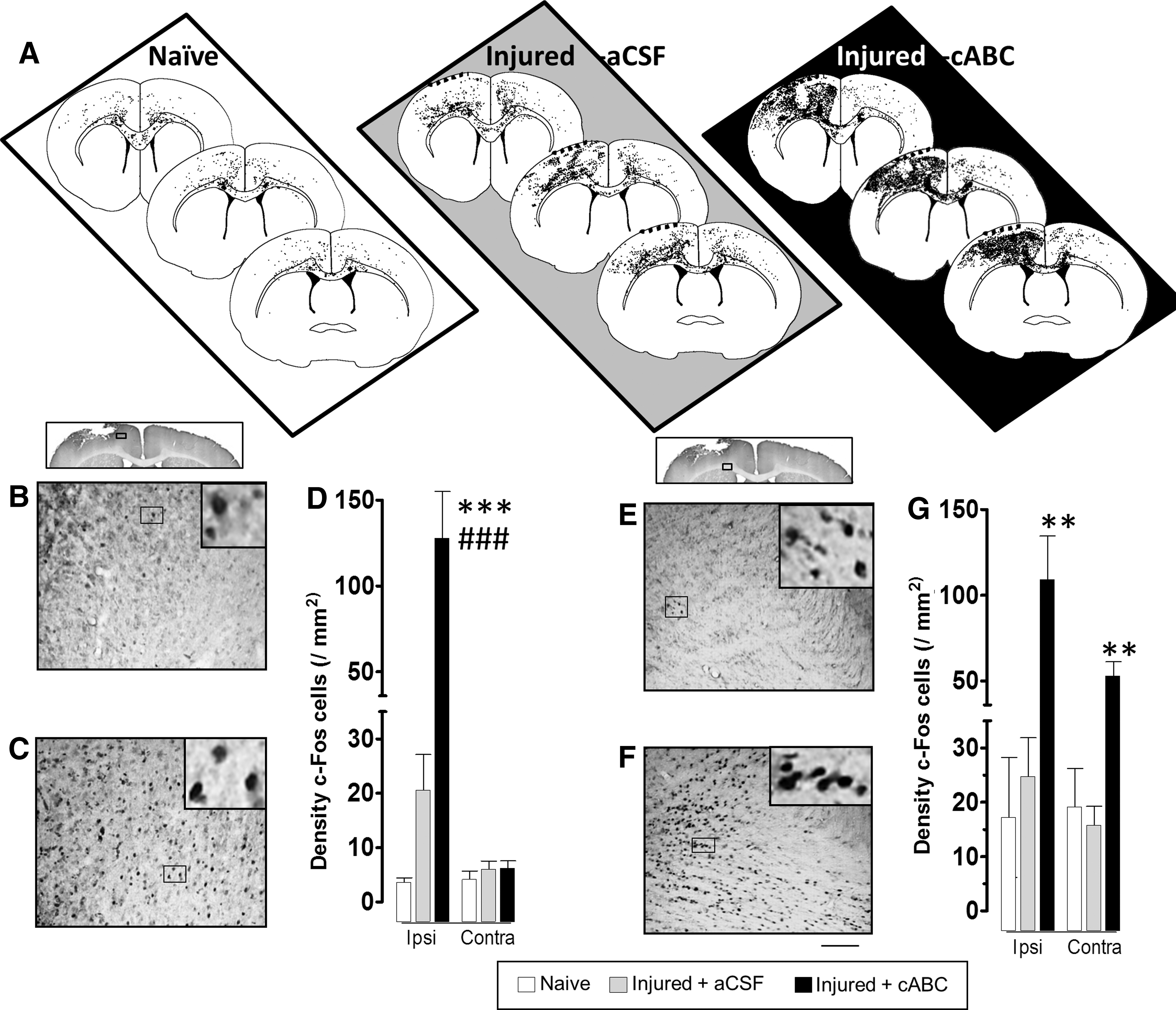
Chondroitinase (cABC) increases c-Fos+ cortical cellular staining in response to electrical stimulation of the affected (right) forelimb. (
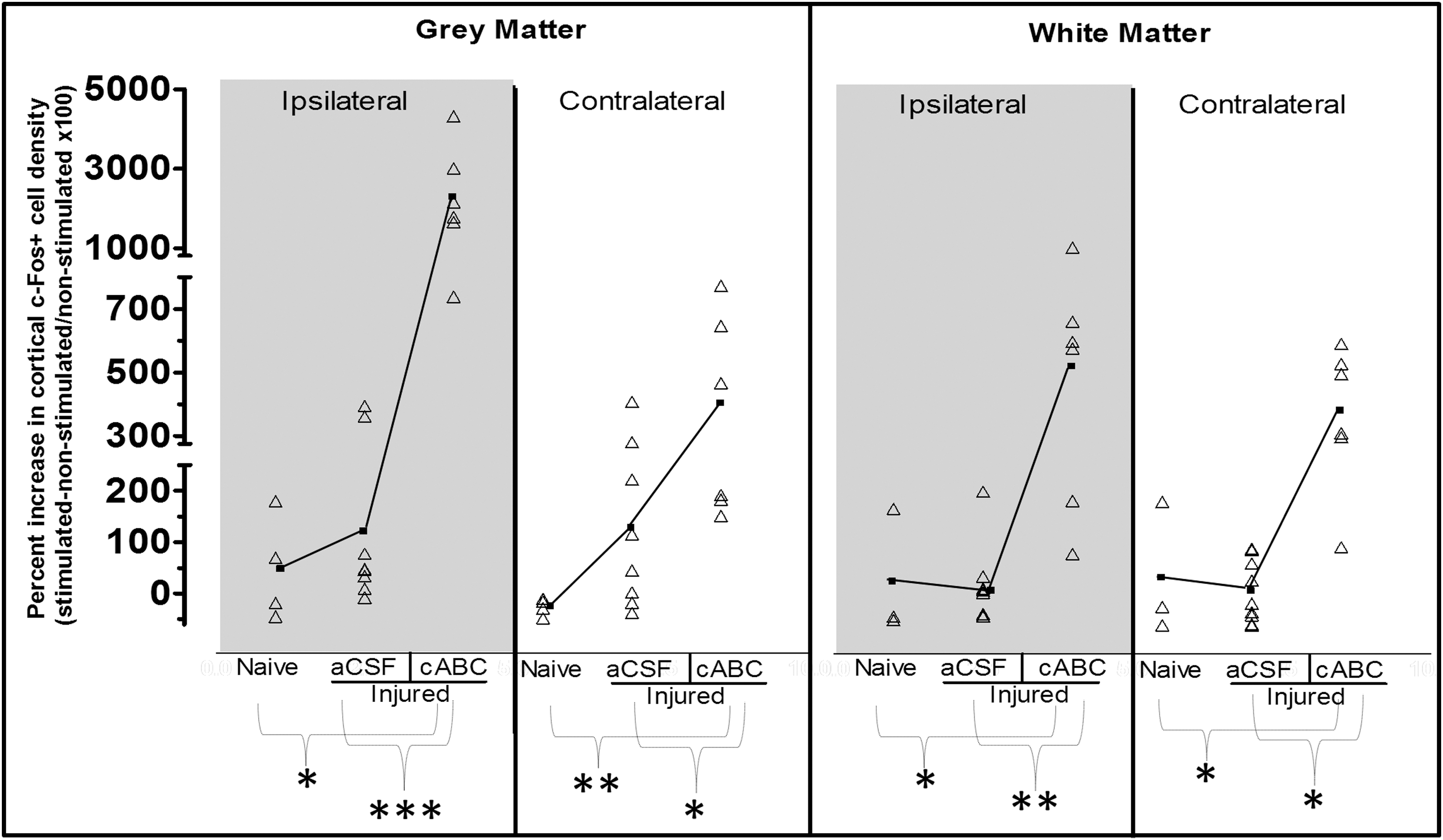
Number of cortical c-Fos+-activated cells during forelimb circuit activation normalized to the mean c-Fos+ cell counts in nonstimulated controls. Nonspecific c-Fos+ cell activation that was unrelated to right forelimb circuit activation was removed by normalization of c-Fos+ cell numbers obtained during forelimb stimulation (data from Fig. 3) to the mean c-Fos+ cell numbers in the corresponding nonstimulated control groups (data from Fig. 2). These computed data are plotted as raw values (open triangles) and group means (closed squares and solid connecting line) for all regions examined. Despite data normalization, significant increases in cortical c-Fos+ cell activation in injured + chondroitinase (cABC)-treated rats remained in all regions examined (p<0.05, compared to the nonstimulated injured + cABC group). This effect was consistent among rats in ipsilateral gray matter and is not merely related to the effect of injury, but to the effect of cABC infusion after injury. After normalization of c-Fos+ cell numbers to nonstimulated controls in naïve and injury + aCSF (vehicle) rats, large increases remained in many, but not all, rats and so, overall, was not significantly different (p>0.05, compared to nonstimulated). In injured + aCSF rats, this variability was related to the severity of injury (see text). Even after normalization in injured + cABC rats, c-Fos+ cell density was significantly increased versus naïve and injured + aCSF groups and in all regions examined (p<0.05). ***/**/* p<0.001/0.01/0.05. ns, not significant; aCSF, artificial cerebrospinal fluid.
cABC increases the number of forelimb-stimulated c-Fos+ cells of neuronal phenotype after injury
We next examined the cell phenotype of c-Fos+ cells in forelimb-stimulated rats to determine whether the overall increase in c-Fos+ cell density was attributable to any particular cell type (Fig. 5). Data indicate that cABC significantly increased the fraction of c-Fos+ cells that were neurons (p<0.05; Fig. 5E), but reduced the number that were astrocytes and microglia, although this was just outside significance, compared to injured + aCSF-treated rats (GFAP+/c-Fos+, p=0.068; iba1+/c-Fos+, p=0.081). There was no effect of cABC on the fraction of c-Fos+ cells that were gray-matter oligodendrocytes. However, white-matter c-Fos+ cell body staining immediately under the contusion (Fig. 5F) was almost entirely attributable to CC1+ oligodendrocytes (Fig. 5G), because no other cell types examined were double labeled in this region (data not shown).
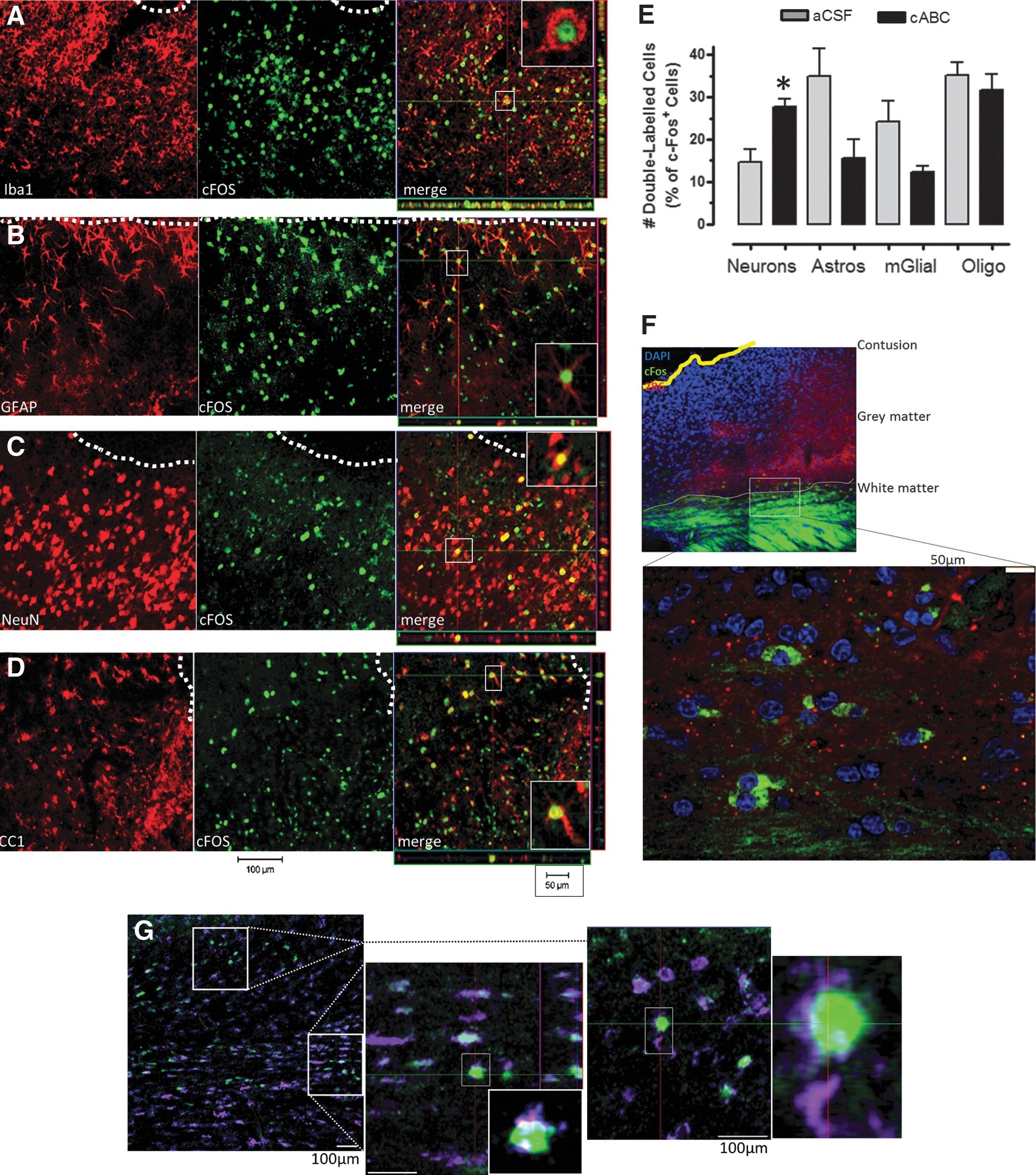
Chondroitinase (cABC) increased the percentage of forelimb-stimulated cortical neuronal/c-Fos+ cells and lowered astrocyte/c-Fos+ and microglial
Forelimb-activated c-Fos+ cells do not contain perineuronal nets after injury
cABC digests not only the extracellular arrangement of CSPGs, but also those condensed around neurons forming perineuronal nets (PNNs). Although the specific function of these PNNs is unknown, it is generally thought that they stabilize existing synapses and prevent new synapse formation, especially as indicated by their role during the developmental critical period preceding visual field formation. 33 Because there is evidence that cABC increases synapse formation in vitro, 34 we reasoned that the significant rise in forelimb-stimulated c-Fos+ cells after cABC infusion might preferentially occur in neurons with digested PNNs. This was indeed the case, and, in fact, we could find no ipsilesional cortical c-Fos+ cells that were double labeled for the PNN marker, WFA (WFA+/c-Fos+; five sections from three brains per group examined; Fig. 6A,B), even within the contralesional gray matter, where PNNs are relatively intact (Fig. 6C). This is in direct contrast to forelimb-stimulated naïve rats, in which, although c-Fos+/WFA− cells were present throughout the sensory-motor cortex, unlike in injured + cABC rats, c-Fos+/WFA+ cells were consistently present in all three examined (Fig. 6D).
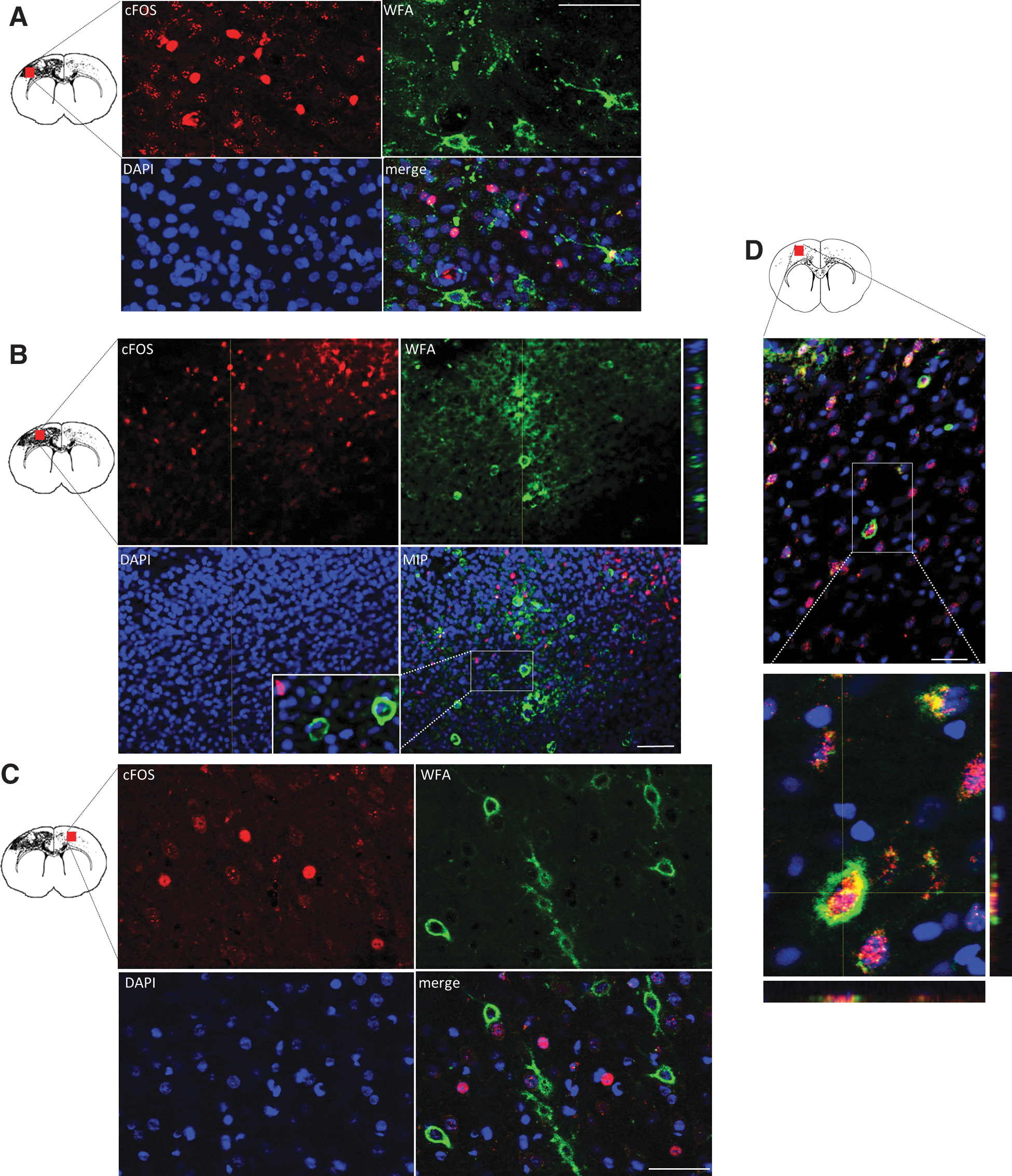
C-Fos+-activated cells do not contain perineuronal nets after injury in the sensory-motor cortex. Representative apotome-acquired images and z-stacks of c-Fos+ cells from forelimb-stimulated injured + cABC treated rats at 7 days after injury from an ipsilateral intact gray-matter region relatively remote from the contusion (
cABC increases the number of sprouting axons that are activated in the forelimb circuit
Next, we investigated whether digestion of CSPGs and CSPG-containing PNNs results in somatosensory circuit-appropriate cell activation consistent with newly sprouting axons or whether indiscriminate c-Fos+ occurs as a result of some other process that we had not anticipated. We double stained for GAP43 and c-Fos in brains from a subset of the forelimb-stimulated rats that were treated with either cABC or vehicle (n=3/group). We determined the extent to which newly sprouting axons and GAP43+ cell bodies were also c-Fos+, as an indication of their inclusion in the activated forelimb circuitry (Fig. 7A–C). Infusion of cABC significantly increased the number of sensory-motor GAP43+ profiles overall (p<0.05), representing a 1.7- to 2.5-fold increase that approached significance regionally within gray- and white-matter regions (p=0.08 and 0.07, respectively; Fig. 7D,F). The number of GAP43+ profiles that were also c-Fos+ significantly increased in cABC-infused rats, compared to vehicle, by 6- to 7-fold in both gray- and white-matter regions (overall group×region effect, p<0.05; Fig. 7E,G), and this represented a 2- to 3.5-fold increase in the fraction of GAP43+ sprouting axons that were activated after cABC, compared to vehicle (Fig. 7H).
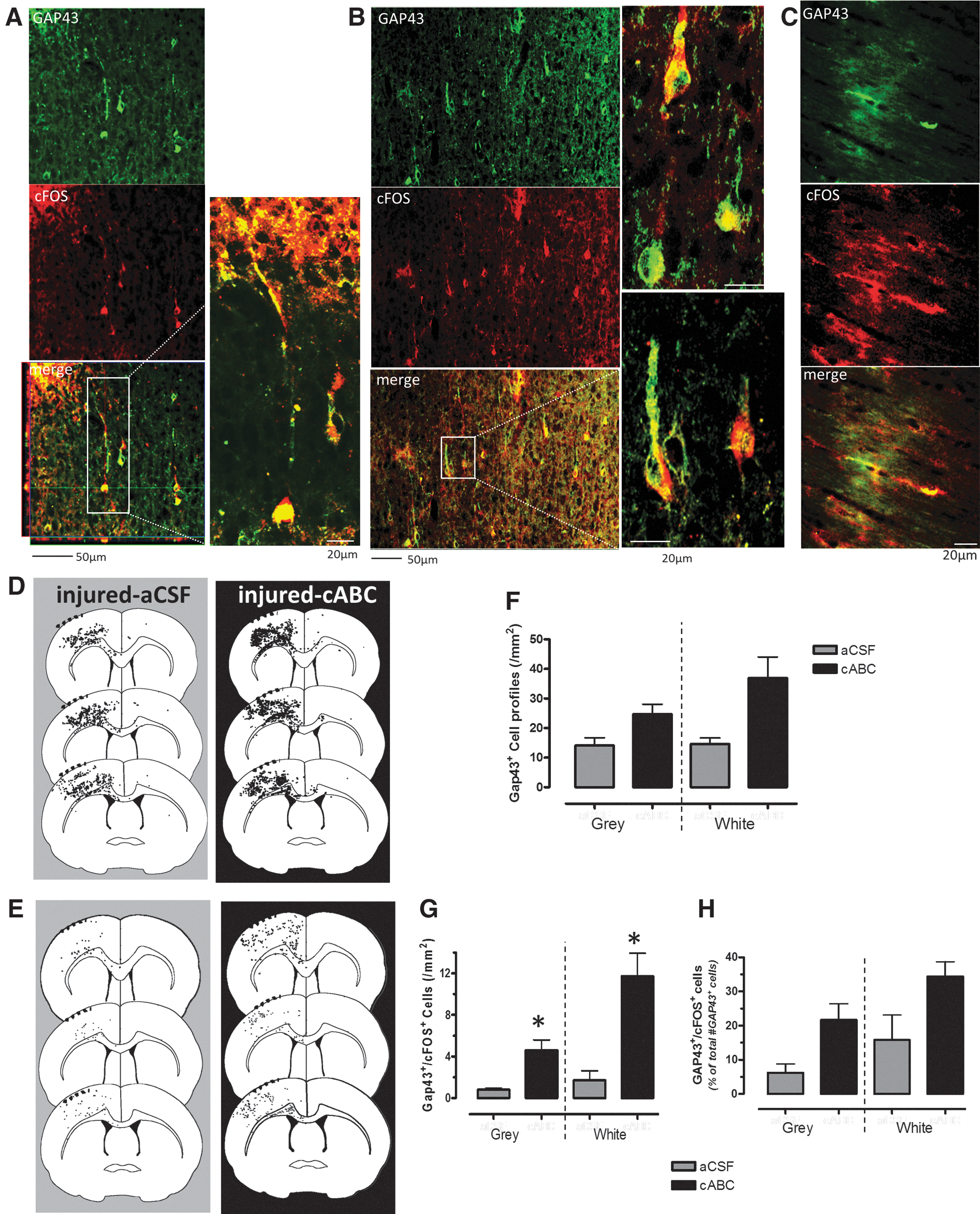
cABC increases the number of cortical sprouting axons that are activated during forelimb stimulation. Representative confocal immunohistochemical images from a forelimb-stimulated injured + aCSF (
Discussion
Chondroitinase reduces injury-related spontaneous c-Fos+ cell activity and potentiates the forelimb-evoked cortical c-Fos+ map
Numerous studies have used cABC enzyme infusion after CNS injury to reduce the growth-inhibitory influence of CSPGs that accumulate in the developing glial scar. 1,6,8,16,20,35 We now extend these data to show that cABC infusion significantly increases the number and extent of sensory-motor cortex, c-Fos+-activated neurones in response to forelimb stimulation after TBI, when compared to injured-vehicle treated. Though cABC infusion to reduce CSPGs does produce an environment that encourages vigorous increases in axonal sprouting, one potential problem is that these new connections may not be appropriate for reestablishing connectivity. In fact, they might present as a maladaptive response, so that taken to the extreme, seizure-type activity might occur. Clearly, an intervention that hastens the onset of TBI-induced epileptiform activity, a known risk for traumatically injured patients, 22 is not useful as a potential therapy. However, we report that in injured animals at baseline non-forelimb-stimulated conditions, cABC infusion does not produce a nonspecific increase in c-Fos+ cells. On the contrary, cABC significantly reduced the amount of abnormally high background c-Fos+ activation in the unstimulated injured brain. Although very acute increases in excitability may well have occurred during cABC infusion, as occurs in hippocampal brain slices when treated with cABC, 36 the observed dampening of c-Fos activity does agree with more-recent work showing that cABC affects L-glutamate receptors, leading to decreases in both their sensitivity and efficacy, as indicated by reductions in miniature excitatory postsynaptic currents. 34 We did not determine the major cellular phenotype that underlies this effect, but the increased layer V neuronal excitability and spontaneous epilieptiform activity that normally occurs at 1 week in pericontusional tissue in this model without cABC infusion 37 would suggest that decreases in spontaneous neuronal output are likely to be responsible for the lowered c-Fos+ cell density. Regardless of the underlying cause, the reduction in c-Fos+ cells after cABC infusion indicates that cABC does not appear to increase the risk of nonspecific cell activation 1 week after injury.
A mechanism for cABC map plasticity: Intracortical sprouting or PNN digestion?
The mechanism for functional gains after cABC infusion is generally assumed to result from increases in axonal sprouting, leading to restoration of map representation of a peripheral limb or target of a spinal pathway. cABC-induced enlargement of the cortical map responsive to afferents at 11–12 weeks after dorsal column lesions in the squirrel monkey was suggested, though not validated, to occur as a result of sprouting at three different sites along the CNS axis. 5 Increased axon sprouting may well underlie map plasticity after brain trauma, because the current c-Fos map enlargement coincides temporally with the peak of the sprouting response in this model after cABC intervention. 23 Spatially, the ipsi- and contralateral increases in gray- and white-matter c-Fos cell density observed were consistent with the contralateral spread of enzyme that we recorded. The increase in the number of c-Fos-activated GAP43+ profiles that occurred in cABC-infused animals, compared to vehicle-infused injured rats, where the c-Fos forelimb map was considerably smaller, also supports the idea that sprouting does underlie the newly activated regions. We report that the degree of injury was positively associated with greater forelimb-evoked c-Fos+ cortical cell density bilaterally after injury alone, which implies greater reorganization of the forelimb map with increasing injury severity. Because injury severity is also directly correlated to the amount of cortical sprouting in the same model, 23 this further underscores the idea that axonal sprouting does, at least partly, support changes in map plasticity after brain injury.
We observed that in addition to cABC increasing the number of sprouting axons, the percentage of these axons that contributed to the forelimb circuitry indicated by c-Fos+ profiles was also increased. On the one hand, this indicates that the intervention does not appear to lead to inappropriate connectivity, that functional gains measured by forelimb reaching after cABC infusion after TBI 1 may, at least partly, result from increased sprouting. However, this does not rule out that other mechanisms unrelated to axonal sprouting, and more directly related to reductions in growth-inhibitory CSPGs (e.g., increases in synaptic connectivity of the existing circuitry), may account for the increase in the c-Fos+ cortical map. This might occur by increased numbers of synapses and sprouting at the level of dendrites, as has been shown after neutralization of another growth-inhibitor molecule, NOGO-A, after experimental stroke. 38 The observation of a decrease in PNNs containing condensed CSPGs is a consistent finding both after cABC infusion and after injury alone, 1,23 and one hypothesis is that PNN reduction potentiates an increase in synaptic connections. Although the exact function of the PNN remains unknown, they are not required for neuronal survival 39 and appear to be compartmental barriers governing function and limiting plasticity because their removal increases AMPA-type glutamergic receptor trafficking between extrasynaptic and synaptic compartments, resulting in altered synaptic performance and more juvenile-like plasticity. 40 Indeed, in vitro data after cABC digestion of PNNs of cultured hippocampal neurones report an increase in synaptic puncta, 34 although earlier data using a similar preparation reported no change. 36 We were unable to find c-Fos+ cells with an intact PNN both after injury and after cABC infusion + injury, yet they persisted, although they did not predominate, in naïve stimulated rats. Previously, we have shown that the cortical density of PNNs are decreased by TBI 23 and are likely to decrease further coincident with the reduction in CSPGs after cABC infusion. 1 It therefore remains a possibility that increases in synaptic function and/or synaptic number occur as a result of further denuded PNNs and account for the remarkable increase in c-Fos+ cell activation after intervention with cABC. Enhanced AMPA receptor function secondary to PNN removal 40 may well be the downstream effect of cABC digestion of PNNs after injury, especially given the evidence that AMPA receptor agonist enhancement of glutaminergic function improves outcome after forebrain stroke. 41
PNNs are preferentially distributed around inhibitory neurones,
42
and disturbances or reductions in molecular components of PNNs result in persistent plasticity and increased perisomatic inhibition.
43,44
This evidence suggests that the recovery of the cortical c-Fos+ map after cABC might occur through reduced intracortical inhibition as a result of digestion of PNNs and altered inhibitory cell function. Recent work shows that alleviating increased tonic inhibition ipsilesionally after forebrain stroke can significantly enhance functional outcome.
41
Moreover, optogenic inhibition of somatostatin-expressing gamma-aminobutyric acid–mediated (GABAergic) neurones in mouse barrel cortex strongly increases the number and firing rate of nearby excitatory neurones,
45
consistent with the current work showing increased c-Fos+ cells after CSPG digestion. If cABC produced a similar reduction of inhibitory cells by PNN digestion, this might well provide one potential mechanism by which map plasticity occurs. However, at least acutely in cultured embryonic hippocampal cells, enzyme digestion of PNNs has no effect on postsynaptic inhibitory cell function,
34
which would argue against such a mechanism. However, more chronic effects on cell function were not determined, and it remains possible that alterations in the balance of excitation and inhibition conjointly regulate the cortical map in a particular spatial arrangement, in a manner similar to that observed after cortical plasticity induced by intracortical microstimulation.
46
In this interpretation, enhanced c-Fos cell activity that we observed after cABC infusion would occur in glutamatergic neurones, limited by inhibitory zones of increased cell numbers containing GABA. This would seem to concur with previous in vitro work showing increased
cABC reduces c-Fos+ glial activation
The coexistence of c-Fos among multiple cellular phenotypes in addition to neurons is not without precedent, because similar labeling occurs in spinal cord and brain stem after cell activation with tumor necrosis factor alpha, 47,48 in spinal cord glial cells after sympathetic nerve discharge, 49 in Schwann cells in response to axonal action potentials, 50 and in optic nerve oligodendrocytes in response to ischemia. 51 In the present study, the large, bilateral, white-matter c-Fos activation observed in cABC-treated rats occurs in both Gap43+ axons as well as in surrounding oligodendrocytes. We interpret this as increased axonal activity requiring significant metabolic support from oligodendrocytes for underpinning the bilateral increase in cortical gray-matter map plasticity that also occurs.
In addition to the increase in neuronal c-Fos+ cell activation underlying the enlargement of the c-Fos+ cortical map after cABC, there was a reduction in the fraction of c-Fos+ cells that were astrocytes or microglia. This is difficult to interpret, given the somewhat controversial nature of the mechanisms of glial involvement in synaptic transmission and the effect of gliotransmitters. 52 The more traditional idea of astrocytes as a K+ and glutamate sink within the tripartite synapse 53 would tend to suggest a mechanism whereby a cABC-mediated reduction in extracellular or PNN-associated CSPGs reduces astrocytic function at the synapse, resulting in reduced sequestration of neurotransmitters and excitotoxic substances as well as enhanced neuronal activation. The reduction in glial cell function indicated by the present data would suggest a decrease in neurotransmitter sequestration, so that combined with the known increase in extracellular calcium diffusion that occurs after digestion of CSPGs by cABC, 54 enhanced neural activation recorded in the present data would certainly be a plausible result. The cABC-induced reduction in number of c-Fos+ microglial cells is interesting because reduction of inflammation can be neuroprotective after TBI, 55 which might underlie the neuroprotective effects of cABC after SCI, 56 although not after TBI, in the current data. One mechanism by which microglial cell activation is reduced after cABC infusion is by the cABC cleavage of CSPGs to a disaccharidic degradation product that has previously been shown to markedly reduce T-cell infiltration and microglial activation after experimental autoimmune encephalomyelitis. 57 Clearly, cABC has a multitude of effects in the brain, in addition to the well-studied sprouting response, that have only just begun to be unraveled.
In summary, the data support a role for cABC in effecting recovery of the cortical map evoked by forelimb stimulation. Though newly sprouting axons may well underpin a large part of the map reorganization, a number of mechanisms downstream of disruption to the perineuronal net arrangement of CSPGs may also contribute to cortical map changes after brain injury.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH NINDS; award no.: NS055910) and the UCLA Brain Injury Research Center.
Author Disclosure Statement
No competing financial interests exist.