
Abstract
The incidence of blast-induced traumatic brain injury (bTBI) has increased substantially in recent military conflicts. However, the consequences of bTBI on the blood-brain barrier (BBB), a specialized cerebrovascular structure essential for brain homeostasis, remain unknown. In this study, we utilized a shock tube driven by compressed gas to generate operationally relevant, ideal pressure profiles consistent with improvised explosive devices (IEDs). By multiple measures, the barrier function of an in vitro BBB model was disrupted following exposure to a range of controlled blast loading conditions. Trans-endothelial electrical resistance (TEER) decreased acutely in a dose-dependent manner that was most strongly correlated with impulse, as opposed to peak overpressure or duration. Significantly increased hydraulic conductivity and solute permeability post-injury further confirmed acute alterations in barrier function. Compromised ZO-1 immunostaining identified a structural basis for BBB breakdown. After blast exposure, TEER remained significantly depressed 2 days post-injury, followed by spontaneous recovery to pre-injury control levels at day 3. This study is the first to report immediate disruption of an in vitro BBB model following primary blast exposure, which may be important for the development of novel helmet designs to help mitigate the effects of blast on the BBB.
Introduction
The BBB is a heterogeneous unit consisting of brain endothelial cells expressing tight junctions that are essential to ensure the barrier's integrity and low permeability. 8 –11 Tight junctions are made up of claudins, occludin, and the zonula occludens accessory proteins (ZO-1 and ZO-2), which form the restrictive paracellular diffusion barrier characterized by high electrical resistance. 10,12 Because the BBB plays a central role in preventing the transport of potentially neurotoxic blood-borne molecules into the neural parenchyma, damage to the barrier has been known to exacerbate several central nervous system (CNS) pathologies such as multiple sclerosis and stroke, and may be a critical mediator in bTBI. 6,13 –17 However, further investigation is warranted to understand the physical mechanism responsible for potential BBB breakdown resulting from militarily relevant levels of blast exposure.
BBB disruption associated with TBI results in brain edema and increased cerebrovascular permeability, both of which affect morbidity and mortality in head-injured patients. 14,18 In-theatre clinical observations of casualties resulting from explosive blast further report that edema, intracranial hemorrhage, and vasospasm are characteristic pathophysiological outcomes of bTBI. 2,19 Brain edema after TBI is thought to be initiated by BBB rupture, permitting the influx of protein-rich exudate through compromised endothelial tight junctions that may lead to delayed neuronal dysfunction and degeneration. 18,20,21 The net expansion of brain volume elicits increased intracranial pressure, impaired cerebral perfusion, and oxygenation, all of which impact patient outcome. 18 Although the role of vascular pathology in acute neurological dysfunction has only recently been explored, such neuropathologies directly linked to BBB breakdown have become a matter of major clinical importance. 20 –22
Breakdown of the BBB, lasting from several days to years after the initial injury event, has been observed in animal models of experimental TBI and in clinical studies. 23,24 Several mechanisms have been proposed to operate during the secondary wave of brain damage following BBB disruption, including altered interactions among the cellular components of the neurovascular unit and deficits in the structural integrity of BBB tight junction complexes. 8,21,25 Restoration of low permeability and BBB function has been shown to occur within days to weeks following concussive injury in vivo; however, there is a need for greater quantitative understanding of the relationship between cerebrovascular compromise and the physical initiators of TBI. 26,27
Several studies have demonstrated brain microvasculature and BBB breakdown resulting from blast exposure in rodent and small animal models of bTBI. 1,13,15 However, there is incomplete understanding of the mechanism(s) governing BBB damage following primary blast, distinct from a combination of primary and tertiary (acceleration-driven) blast injuries, which are typically difficult to separate in animal models. To explore the pathophysiological effects of primary blast exposure alone on the barrier, we subjected an in vitro model of the BBB (a brain endothelial monolayer mimicking cell-specific phenotype and functional properties of the BBB), to controlled blast loading at militarily relevant exposure levels. 10,28,29 We report isolated primary blast-induced barrier damage, quantify associated effects on BBB permeability and tight junction breakdown, and describe a time-course for spontaneous barrier recovery. We conclude that primary blast injury alone is capable of acutely disrupting the integrity of an in vitro BBB model at realistic blast loading conditions, suggesting a possible physical mechanism for vascular and subsequent neuronal pathology after blast.
Methods
In vitro BBB model
Brain endothelial monolayers representing the BBB were cultured using the bEnd.3 mouse brain microvascular cell line (American Type Culture Collection [ATCC], Manassas, VA). This cell line has been widely used in other in vitro BBB cell culture models for its rapid growth properties and ability to maintain BBB features and functions over multiple passages.
10,28,30
–32
For all experiments, a total of 60,000 bEnd.3 cells were seeded on Transwell inserts (1.12-cm
2
membrane growth area) pre-coated with poly-
Shock wave-induced injury of the BBB
A recently described in vitro bTBI model consisting of a shock tube and fluid-filled sample receiver was used to expose endothelial monolayers to controlled primary blast injury. 29 Prior to blast exposure, cultures were enclosed within sterile bags (Whirl Pak, Fort Atkinson, WI) filled with culture medium pre-warmed to 37°C. Any entrapment of air bubbles was meticulously prevented during sample preparation. For every experiment, each culture and bag was submerged in the fluid-filled sample receiver to a pre-determined depth, and oriented perpendicular to the direction of shock wave propagation.
Ideal Friedlander-like shock waves, mimicking overpressure/duration profiles similar to those observed in free-field blasts, were generated using a 76 mm-diameter shock tube with an adjustable-length driver section (25 or 50 mm used for the current study) pressurized with helium or nitrogen gas, and a 1240 mm-long driven section (Fig. 1A). 29,33 Pressure transducers (8530B; Endevco, San Juan Capistrano, CA) flush-mounted at the tube outlet recorded the incident pressure of the shock wave (Fig. 1B). A range of loading conditions was tested for the parameters of peak incident overpressure, duration, and impulse (Table 1). These blast parameters were determined in the open-tube configuration in the absence of interacting structures downstream, since this arrangement represented an independent and consistent measure of the injury input. Unless otherwise indicated, all blast parameters reported in this study refer to open-tube measurements. A transducer (8530B; Endevco) flush-mounted to the interior of the sample receiver test column measured the fluid pressure history directly beneath the submerged BBB sample (Fig. 1C). Blast parameters in the receiver fluid were measured for each corresponding open-tube configuration (Table 1). A previously published characterization of our in vitro bTBI model confirmed that the pressure wave was not attenuated as it passed through the BBB sample. 29 Sham controls were also enclosed in sterile bags and submerged in the receiver for a duration matching the injured cultures, but were not exposed to blast. 29
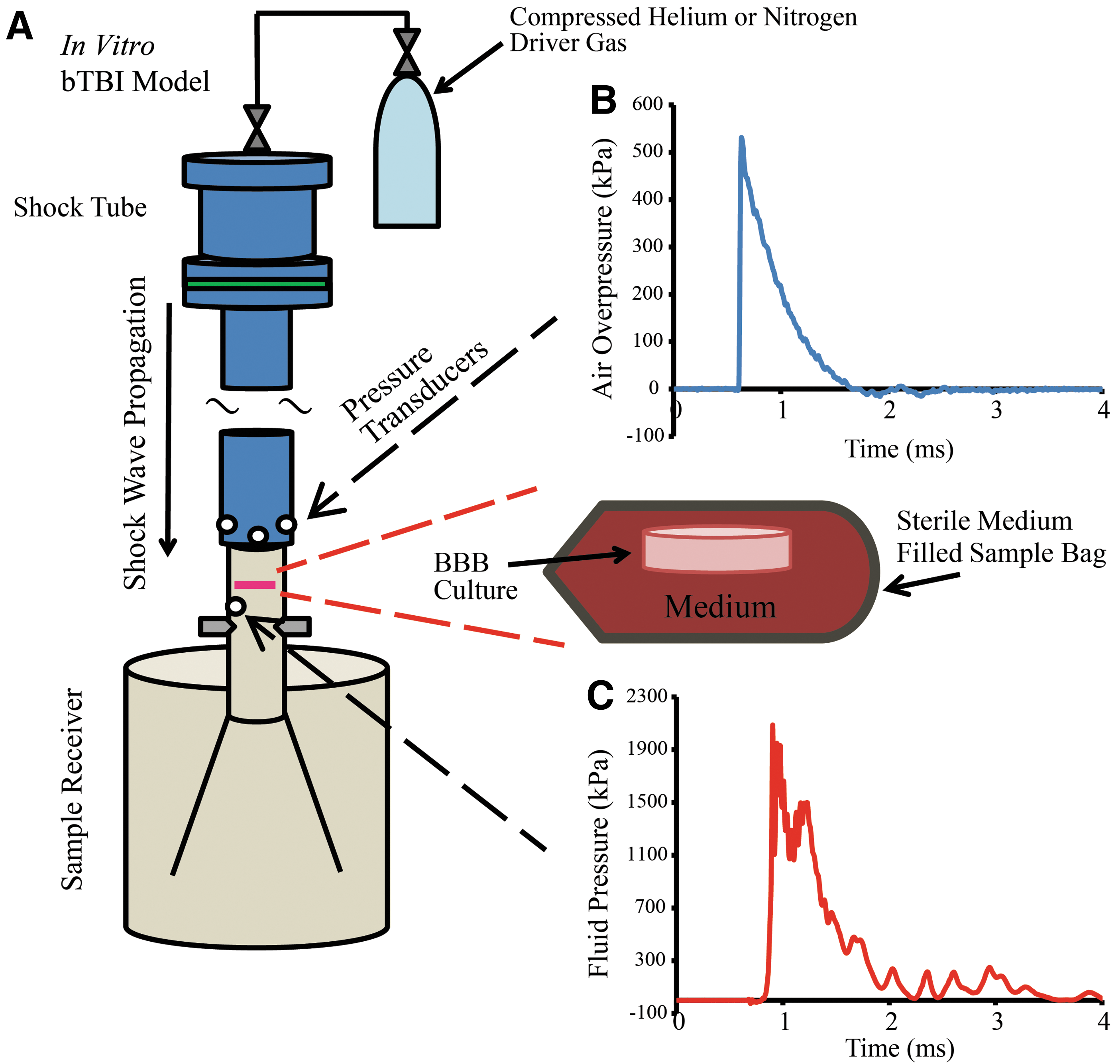
In vitro bTBI model consisting of shock tube and fluid-filled sample receiver as previously described.
29
Experimental loading conditions tested in this study were characterized by peak incident overpressure, duration, and impulse, measured in the open-tube configuration and in the fluid-filled sample receiver (mean±SEM; n≥3 for open-tube parameters; n≥2 for receiver parameters).
Quantification of TEER
TEER is a resistance measure dependent on ion flux through the cell monolayer, and thus provides a way to functionally characterize the integrity of the monolayer as well as tight junctions. 34,35 TEER was recorded immediately before and 2–10 min after blast exposure using an Endohm-12 electrode chamber connected to an EVOMX Epithelial Voltohmmeter (World Precision Instruments, Sarasota, FL). Consistent with TEER calculations for in vitro BBB models reported in the literature, 10,28,31 all TEER values were corrected for the TEER of a cell-free insert and then normalized for the insert surface area. Measurements reported in this study represent the TEER of each culture normalized to its pre-injury group average. For the time-course evaluation, the TEER of injured cultures was further normalized to age-matched and time point-matched sham controls. Sham control cultures were processed identically to injured cultures, but were not exposed to blast.
Quantification of BBB hydraulic conductivity
Hydraulic conductivity serves as a functional indicator of monolayer tightness, as the BBB restricts water flux from the systemic circulatory system to the brain in vivo.
10,36
To measure hydraulic conductivity between 30 min to 2 h post-injury, cultures were placed in a custom-built polycarbonate chamber.
10,28,37
Each BBB model was then subjected to a known hydrostatic pressure across the monolayer, giving rise to fluid flow through the culture. Water flux was quantified by tracking the displacement of an air bubble through a calibrated glass tube connected to the chamber. Hydraulic conductivity (Lp
, cm/s/cmH2O) was calculated using equation (1)
10,28
where
Quantification of BBB solute permeability
Solute permeability serves as a quantitative measure of the diffusive permeability of a blast-injured BBB to a range of molecular weights. To measure solute permeability between 30 min and 2 h post-injury, fluorescent solutes of specific molecular weights, including fluorescein-labeled 3 and 10 kDa dextrans (excitation 494 nm, emission 521 nm; Life Technologies), and Texas Red®-labeled 40 and 70 kDa dextrans (excitation 595 nm, emission 615 nm; Life Technologies), were added to the upper Transwell compartment above the BBB culture. Every 30 min for 4 h, 100 μL of medium was collected from the lower chamber, and the fluorescence was quantified using a SpectraMax M2 Microplate Reader (Molecular Devices, Sunnyvale, CA). The 100 μL sample was replaced with 100 μL of fresh medium, and this change in volume was accounted for when calculating the change in concentration over time. The diffusive solute permeability (P, cm/s) was calculated using equation (2)
10,28
where
Evaluation of ZO-1 morphology
To assess disruption of the structural integrity of tight junctions at 1–2 h following blast exposure, the BBB endothelial cultures were fixed and incubated with anti-ZO-1 rabbit polyclonal antibody (Life Technologies). The presence of ZO-1 tight junction proteins, a standard marker for BBB integrity, 10,28,31,38 was detected using the Alexa Fluor 488 anti-rabbit secondary antibody (Life Technologies). Cultures were also incubated with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies) to detect individual cell nuclei and to count the number of cells in each monolayer culture. Using antibodies manufactured from the same lot and applied at the same post-injury time-point, all samples were imaged with an Olympus IX81 (Olympus America, Center Valley, PA) fluorescence microscope and MetaMorph software (Molecular Devices, Sunnyvale, CA). The degree of ZO-1 immunostaining was quantified by applying an identical threshold for ZO-1 immunofluorescence to each of five randomly selected images acquired from every sham and injured culture. ZO-1 immunostaining was measured as the area-percentage exhibiting fluorescence above the threshold, normalized to the total number of endothelial cells in each image. This quantification method is similar to published methods on evaluating bEnd.3 barrier integrity by calculating the percentage of pixels with intensities above a threshold value. 10
Statistical analysis
Hydraulic conductivity, ZO-1 immunostaining, and cell-count comparison data were analyzed statistically by one-way analysis of variance (ANOVA) followed by Dunnett post hoc tests to sham controls. Dose-dependent TEER response data were analyzed by repeated-measures to determine the effect of blast on TEER in the acute phase post-injury, followed by one-way ANOVA and Dunnett post hoc tests to sham controls. Repeated-measures analysis was used to determine the effect of blast on the temporal recovery of TEER, followed by one-way ANOVA and Dunnett post hoc tests to the pre-injury group. Independent samples t-tests between sham and injured cultures were used to analyze solute permeability data (significance *p<0.05; SPSS v. 20; IBM, Armonk, NY).
Results
Decreased TEER after blast
An initial investigation was conducted to identify a threshold for this disruption as determined by a significant decrease in TEER measured acutely at 2–10 min after blast exposure. Using helium as the driver gas, TEER acutely decreased in a dose-dependent manner at increasing severities of blast from 231–571 kPa peak incident overpressure, 0.60–1.06 ms duration, and 40–186 kPa*ms impulse (Fig. 2A). Following the 469 kPa peak overpressure blast, TEER significantly (p<0.05) decreased to 78±8% of pre-exposure levels compared to 102±3% in sham controls (Fig. 2A). TEER continued to decrease at greater blast levels, while exposure to lower severity blasts with peak overpressure of 231–377 kPa, duration of 0.60–0.89 ms, and impulse of 40–96 kPa*ms did not significantly reduce TEER.
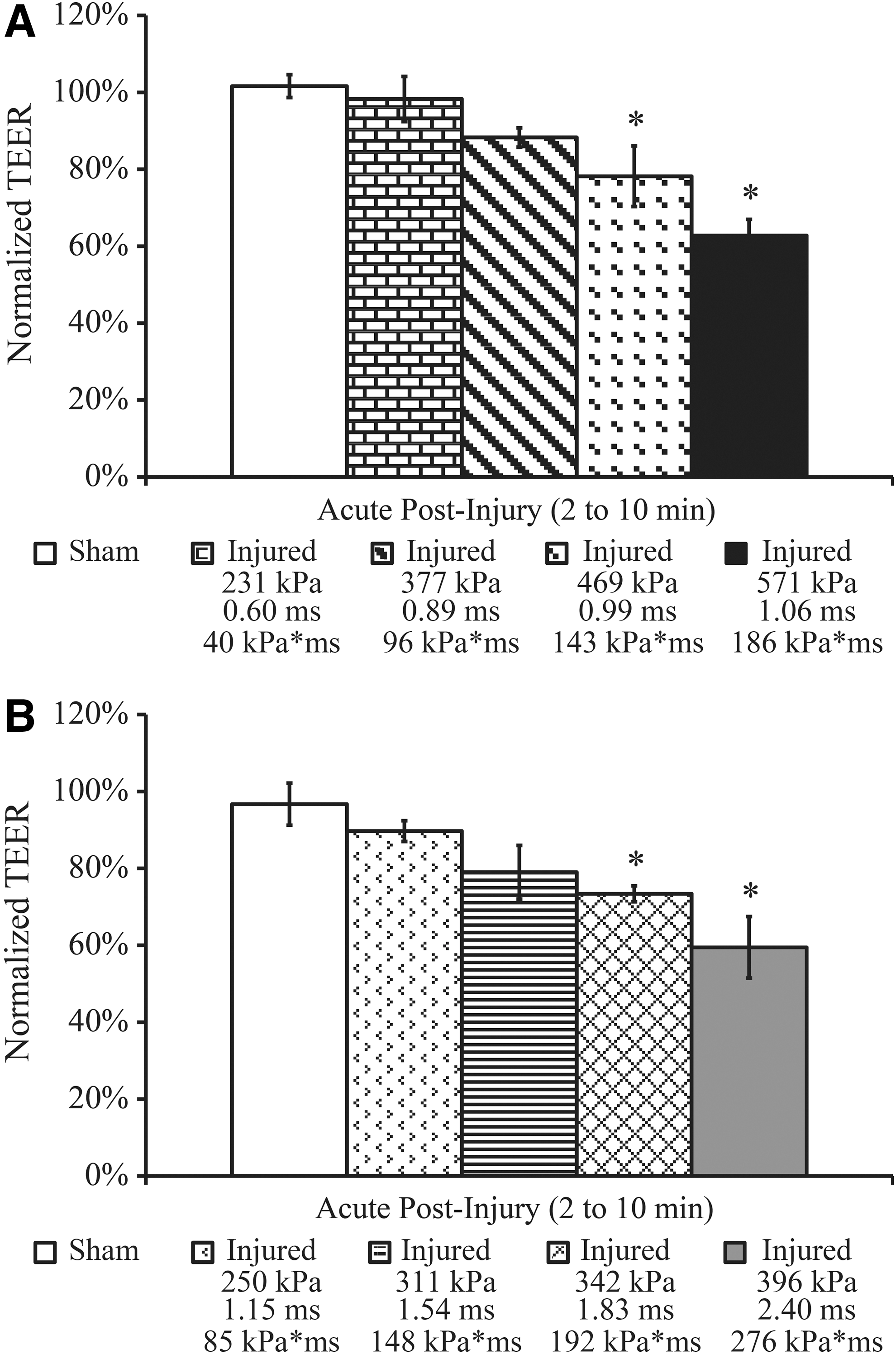
Dose-dependent TEER response in endothelial monolayers following exposure to a range of blast loading conditions.
We next sought to identify the most influential biomechanical blast parameter driving damage to the barrier. Changing the driver gas to nitrogen enabled generation of a different combination of parameters that included generally lower peak overpressures at longer durations and higher impulses compared to helium as the driver gas. TEER acutely decreased in a dose-dependent manner as the blast severity increased from 250–396 kPa peak incident overpressure, 1.15–2.40 ms duration, and 85–276 kPa*ms impulse (Fig. 2B). Following the 342 kPa peak overpressure blast, TEER significantly decreased to 73±2% of pre-exposure levels compared to 97±5% in sham controls (Fig. 2B). TEER continued to decrease as the blast severity increased, while exposure to milder blasts with peak overpressure of 250–311 kPa, duration of 1.15–1.54 ms, and impulse of 85–148 kPa*ms did not significantly reduce TEER.
To determine which blast parameter was most critical to BBB disruption, the correlation between changes in TEER and each of the three biomechanical injury parameters—peak overpressure, duration, and impulse—measured in the open-tube configuration (air) and in the sample receiver (fluid) was determined. As evidenced by the high R2 values of 0.92 (Fig. 3A) and 0.88 (Fig. 3D) for measurements in both the air and fluid, changes in TEER were overall most strongly correlated with impulse. A significant decrease in TEER after blast was observed at an air impulse as low as 143 kPa*ms, indicating functional deficits at TEER levels approximately 80% of pre-injury values (Fig. 2A,B). TEER was also strongly correlated with peak overpressure measured in the fluid, as indicated by a high R2 value of 0.93 (Fig. 3E). Weaker associations were evident between TEER and air peak overpressure, air duration, and fluid duration, which were all associated with comparatively lower R2 values of 0.52, 0.52, and 0.34, respectively (Fig. 3B,C,F).
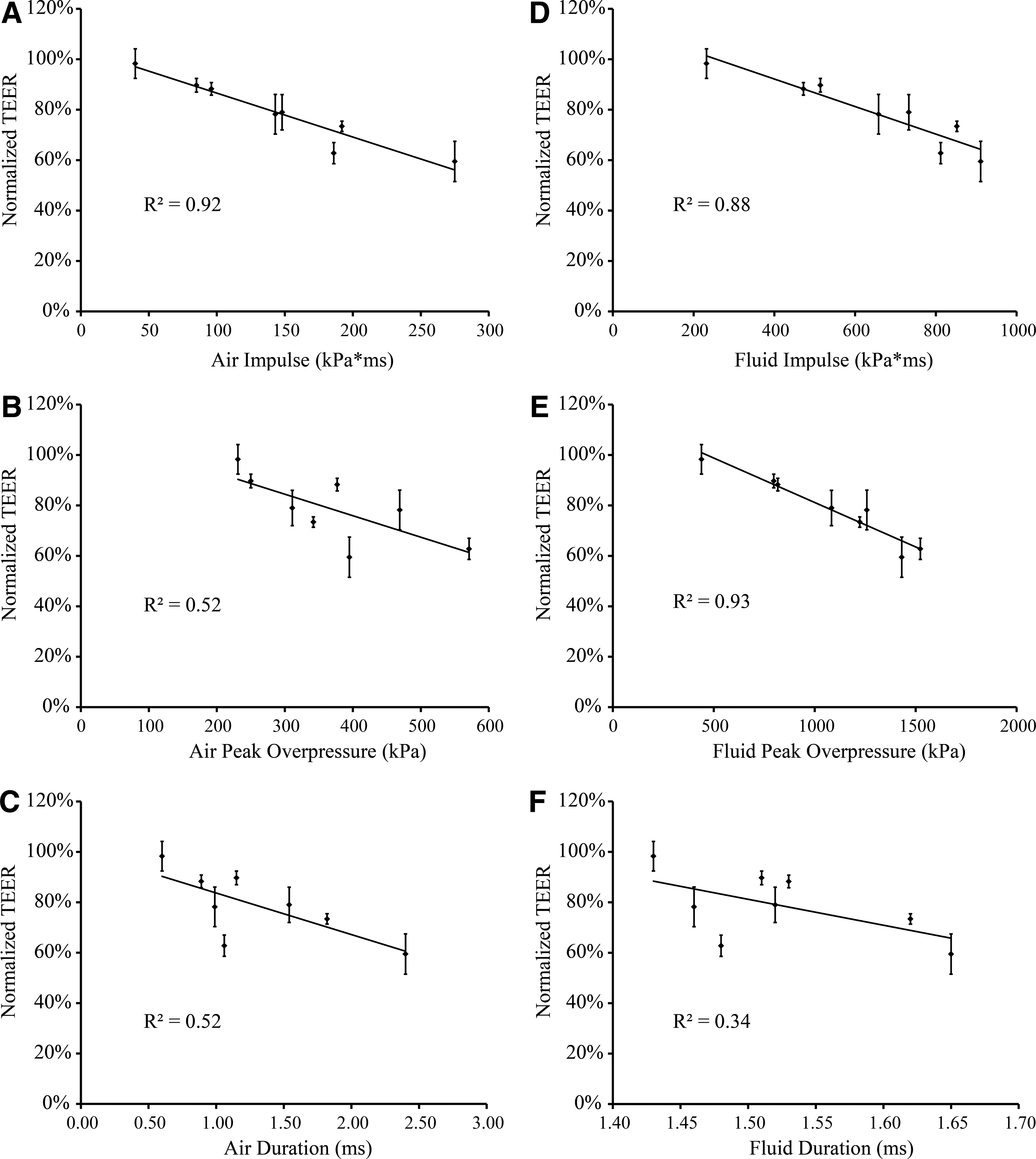
Correlations established for changes in TEER with different blast injury parameters measured in the open-tube configuration (air) and in the sample receiver (fluid). Linear functions were fit to the data for each association.
Increased hydraulic conductivity after blast
Hydraulic conductivity, Lp , was measured between 30 min and 2 h post-injury to quantify water flux through the BBB model. After exposure to blast with a peak overpressure of 571 kPa, duration of 1.06 ms, and impulse of 186 kPa*ms (helium driver gas), hydraulic conductivity was significantly increased to 11.4±3.5×10−7 cm/s/cmH2O compared to 2.9±0.4×10−7 cm/s/cmH2O in sham controls (Fig. 4). Similarly, blast with a peak overpressure of 396 kPa, duration of 2.40 ms, and impulse of 276 kPa*ms (nitrogen driver gas) significantly increased hydraulic conductivity to 7.5±1.9×10−7 cm/s/cmH2O (Fig. 4). Blasts with peak overpressures of 469 or 342 kPa, durations of 0.99 or 1.83 ms, and impulses of 143 or 192 kPa*ms did not cause significant increases in hydraulic conductivity.

Hydraulic conductivity of blast-exposed endothelial monolayers. Between 30 min to 2 h after injury, cultures exposed to the 571 and 396 kPa blasts exhibited a significant increase in hydraulic conductivity to 11.4±3.5×10−7 cm/s/cmH2O and 7.5±1.9×10−7 cm/s/cmH2O, respectively, compared to 2.9±0.4×10−7 cm/s/cmH2O in sham controls. (*p<0.05;±SEM; sham n=23; injured n≥6 per blast condition.)
Increased solute permeability after blast
Between 30 min and 2 h of exposure to blast with a peak overpressure of 571 kPa, duration of 1.06 ms, and impulse of 186 kPa*ms (helium driver gas), a trend of increased solute permeability was observed. The permeability of exposed cultures at the 10 kDa group significantly increased to 2.5±0.3×10−6 cm/s compared to 1.2±0.2×10−6 cm/s in shams (Fig. 5).

Increased solute permeability of 3, 10, 40, and 70 kDa dextran molecules after blast exposure. Injured cultures exhibited a significant increase in solute permeability to 2.5±0.3×10−6 cm/s compared to 1.2±0.2×10−6 cm/s in shams for the 10 kDa molecular weight tracer. (*p<0.05;±SEM; sham n=6; injured n=6.)
Disruption of ZO-1 after blast
In sham cultures, ZO-1 immunostaining revealed high levels of ZO-1 protein on the surface of cells and at the junctions between cells, indicating widespread expression of tight junction proteins (Fig. 6A). Visual examination of immunofluorescence images of the bEnd.3 cultures confirmed confluent cell monolayers exhibiting characteristic, elongated spindle-shape morphology for sham controls (Fig. 6A), bearing very close resemblance to the cell architecture of confluent bEnd.3 monolayers shown by light microscopy and tight junction immunostaining in previous studies. 10,28,29,35,39 Between 1 and 2 h of exposure, a blast with peak overpressure of 571 kPa, duration of 1.06 ms, and impulse of 186 kPa*ms (helium driver gas) substantially decreased ZO-1 staining and altered tight junction morphology (Fig. 6B). Similarly, a blast with peak overpressure of 396 kPa, duration of 2.40 ms, and impulse of 276 kPa*ms (nitrogen driver gas) substantially compromised ZO-1 staining (Fig. 6C). The morphology of tight junctions in both injury groups was more punctate and discontinuous, leaving regions between cells devoid of well-formed tight junctions. Consistent with the qualitative imaging data, the area-percentage of ZO-1 immunostaining per cell was significantly decreased in the 571 kPa injured cultures to 0.25±0.02% and in the 396 kPa injured cultures to 0.18±0.01%, compared to 0.31±0.02% in sham controls (Fig. 6D). Previous characterization of our bTBI model has confirmed cell viability following similar exposure levels, showing the absence of any significant cell death post-injury. 29
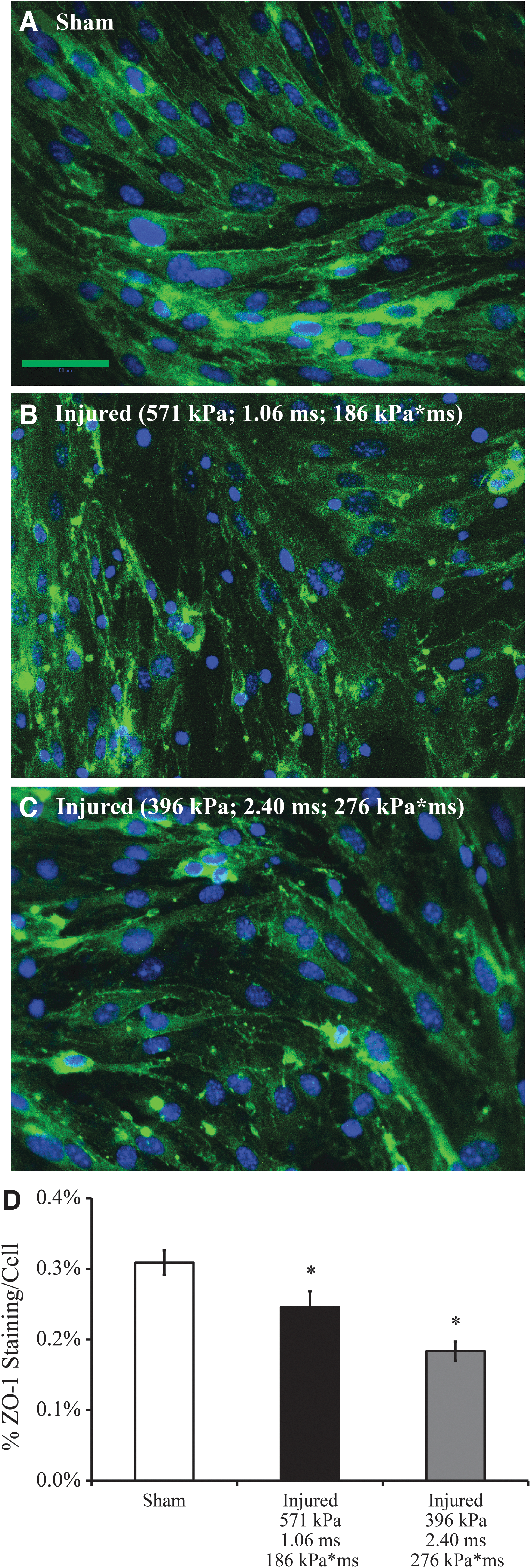
Immunostaining of ZO-1, a marker for tight junctions.
The average number of cells in each immunofluorescence image was quantified to determine the degree of cell detachment between 1 and 2 h of exposure to the 571 and 396 kPa peak overpressure blasts. In sham controls, the average cell count per field of view was 90±2, while corresponding cell counts for cultures injured at the 571 and 396 kPa overpressure levels were 87±3 and 79±2, respectively (Fig. 7). Significant cell detachment compared to sham was only observed for cultures exposed to the highest level blast (396 kPa overpressure, 2.40 ms duration, and 276 kPa*ms impulse, nitrogen driver gas).

Average cell count per immunostained image to quantify cell detachment between 1 and 2 h of blast exposure. The cell count was significantly reduced by 12% in cultures exposed to a blast with a 396 kPa peak overpressure, 2.40 ms duration, and 276 kPa*ms impulse (nitrogen driver gas). (*p<0.05;±SEM; sham n=50; injured n≥20 per blast condition.)
Temporal recovery of TEER after blast
The temporal recovery of TEER was measured after a blast with a peak overpressure of 571 kPa, duration of 1.06 ms, and impulse of 186 kPa*ms (helium driver gas). This level was selected because it was shown to cause a robust decrease in TEER based on the dose-dependent response in the in vitro BBB model without significant loss of cells (Fig. 2A). TEER of the injured cultures, normalized to age-matched and time point-matched sham controls, decreased in the acute phase 30 min after injury (Fig. 8). TEER remained significantly depressed for 2 days after exposure as compared to pre-injury levels (Fig. 8). Injured cultures recovered over time and exhibited full-recovery to pre-injury TEER levels at 3 days post-blast (Fig. 8).
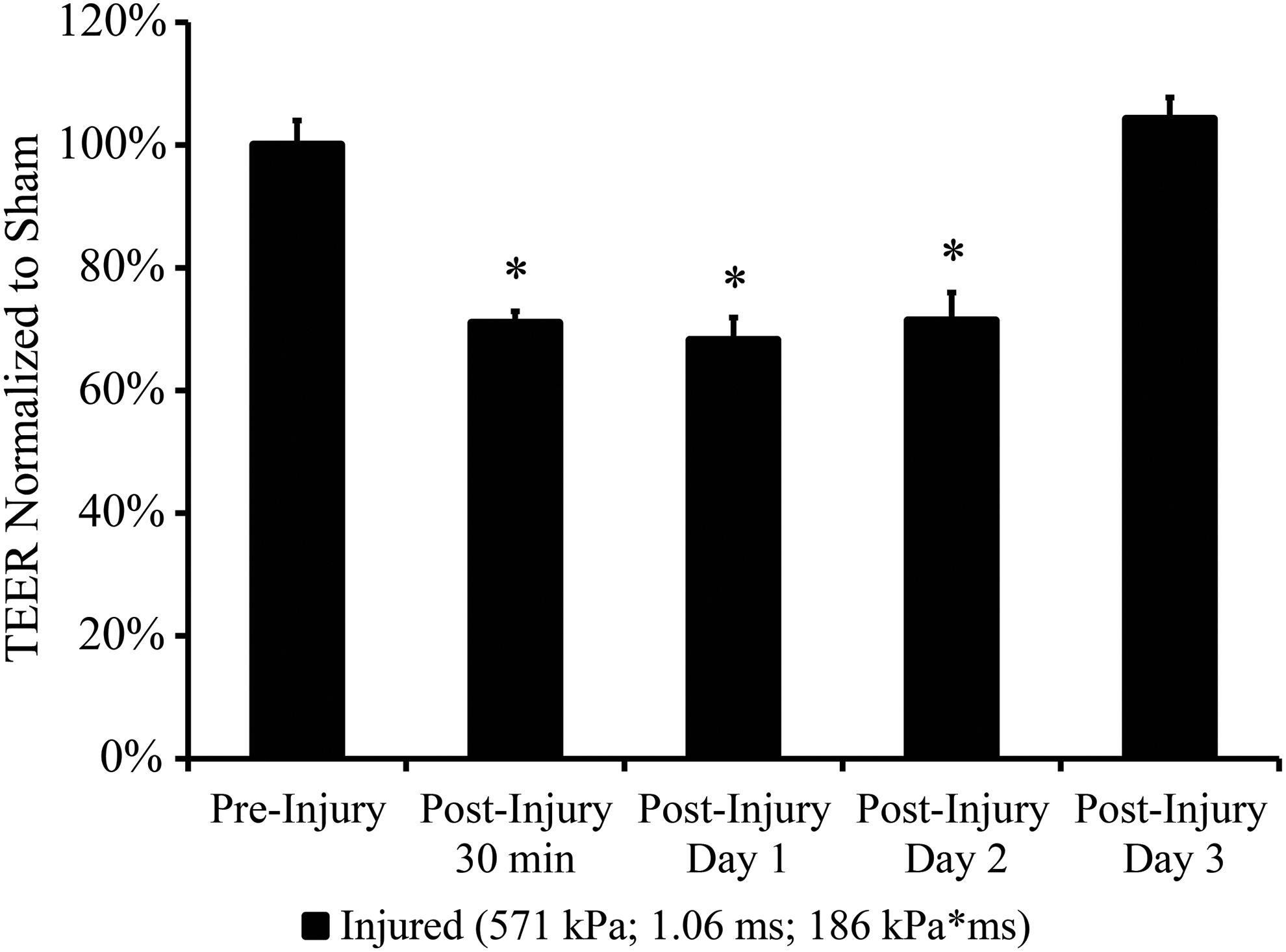
TEER time-course of blast-exposed endothelial monolayers normalized to age-matched sham controls. Depressed TEER of injured cultures fully recovered to pre-injury levels at 3 days post-blast. (*p<0.05;±SEM; n=3.)
Discussion
There exists a need to quantitatively assess changes in BBB integrity following blast loading at militarily relevant exposures. Several experimental bTBI models in rodents have demonstrated blast-induced BBB disruption through heightened permeability to endogenous circulating IgG. Elevated IgG immunohistochemical staining has been observed in the rat cortex after exposure to a 120 kPa peak overpressure blast, but with unreported duration and impulse. 13 Increased IgG immunoreactivity was observed in the contralateral cortex of rats 24 h after exposure to a 241 kPa overpressure blast with approximately a 4 ms duration. 15 Following a sub-lethal complex blast of 427–517 kPa peak overpressure delivered to the head of rats, abnormal IgG immunolabeling in the cerebellum and thalamus signified microvascular dysfunction. 1 Collectively, such evidence provides valuable insight into the vulnerability of the BBB to blast; however, the challenge remains of harmonizing differences between injury parameters used in the models. Analysis of the outcomes is further complicated by the biomechanical complexity of in vivo models, making it difficult to separate the specific contributions of shearing and stretching forces due to inertial loading from interaction of a primary blast pressure transient with the brain parenchyma and specialized structures like the BBB. 29,33,40 Exposure conditions of the aforementioned studies are also quite different from those used in this study. For instance, blast exposure to the unprotected torso of rats may potentially lead to injury cascades in the thoracic cavity that may affect the BBB and evoke transient effects within the brain. 13 The apparatus used in delivery of a complex blast potentially reflected a high-pressure wave off of the closed cranial surface of the rats, 1 while the integrated impulse of longer duration shocks (4–5 ms) described by others are substantially larger but may not be representative of operationally relevant blast conditions. 13,15,41
A definitive link between mild TBI (mTBI) associated with blast exposure and primary blast injury parameters (peak overpressure, duration, and impulse) has not been established. A mouse model of blast neurotrauma reported in a recent study demonstrated chronic traumatic encephalopathy (CTE)–related neuropathologies in blast-exposed mice, including tau protein hyperphosphorylation, vascular damage, and neurodegeneration, among others. 42 Functional deficits, including hippocampal-dependent learning and memory impairments, were also observed after delivery of a single sub-lethal shock wave with 77 kPa peak incident overpressure. 42 The investigators demonstrated that blast winds associated with the shock wave-induced oscillating head accelerations sufficient to cause brain injury, as their model permitted free movement of the animal head to mimic real-world blast loading conditions. 42,43 It is important to note that neurological deficits initially observed after exposure were reduced with head immobilization, suggesting that blast-induced inertial loading on the head (in addition to primary blast effects of the shock wave) was a major mechanism responsible for the ensuing neuropathology.
Capabilities provided by the in vitro blast injury model used in our study, however, enable separation of the effects of primary blast from secondary or tertiary (inertial loading) phases of blast injury. Using high-speed video, we have observed minimal to no gross deformation or movement of the sample during blast exposure. Our ability to isolate the shock wave component from confounding influences of the inertial component enables precise investigation of injury thresholds for primary blast, as opposed to the combination of primary and tertiary blast mechanisms. Furthermore, as previously described, our test methodology reproduces the intracranial loading whereby an external shock wave is translated to a fast-rising pressure wave. 29 Therefore, our in vitro model best represents the dynamic pressures that occur within the head/brain complex during a blast event.
Our results indicate that integrity of the BBB decreases in a dose-dependent manner with increasing severities of primary blast. Correlations established between TEER and each of the three blast parameters measured in the air and fluid confirmed impulse to be the overall most influential injury parameter responsible for disruption of the in vitro BBB model. We observed significant functional disruption of our BBB model after blast with an air impulse of 143 kPa*ms, with minimal alterations to barrier integrity evident at impulse levels as low as 40 kPa*ms. Other reports have suggested that the impulse component of the blast wave is a key parameter influencing severity of the damage and neurological deficits after blast exposure. 44,45 Interestingly, changes in TEER were much more strongly correlated with peak overpressure in the receiver fluid rather than in air, further implying the importance of measuring intracranial pressure history in addition to the external shock wave for accurately predicting the effects of primary blast injury. The weakest association was found between TEER and fluid duration, likely because material compliance in the sample receiver limited the range of durations that could be achieved with our specific device configuration. Future studies will examine if using more rigid materials of construction for the receiver will enable a broader range of fluid durations to be generated that could potentially be more closely correlated with changes in TEER. Nevertheless, the fact that injury outcome determined by TEER was so highly correlated with fluid impulse and fluid peak overpressure is encouraging, as such insight will critically inform the development of detailed injury tolerance criteria for brain tissue exposed to primary blast.
It is also encouraging that the threshold values we determined are above the sub-lethal shock wave input of 77 kPa overpressure reported in a previous study that resulted in no functional deficits when the mouse head was immobilized, 42 which is the approximate condition we are modeling in the current study. We also note that our impulse-driven functional disruption may be specific to the ideal Friedlander curve generated by our in vitro bTBI model, 29 and is potentially sensitive to more complex loading scenarios with altered pressure histories and frequency content that will be the subject of future studies. Previous characterization of our in vitro bTBI model has also demonstrated the absence of significant endothelial cell death following exposure to similar blast levels causing significant decreases in TEER. 29
Real-world blast exposures in the military setting, predicted by the Conventional Weapons Effects Program, span a range of 50–1000 kPa peak incident overpressure with 2 to over 8 ms duration. 33,46,47 Consistent with this range, loading conditions in this study were sufficient to cause functional changes similar to realistic blast threats. For instance, a blast of 571 kPa peak overpressure with 1.06 ms duration is similar to exposure to a 105 mm artillery round at a standoff distance of 5–10 m, which represents a common IED scenario. 47,48 Our in vitro bTBI model was capable of achieving single overpressure pulses of 0.60–2.40 ms in duration, comparable to loading conditions predicted by computational blast studies. 19,33,47
A major function of the BBB in vivo is to exclude water flux by the presence of tight junctions, 49 and measuring blast-induced modulation of hydraulic conductivity is important for linking exposure to the neuropathological conditions associated with vessel hyper-permeability. 50 –55 This study is the first to quantify a blast-induced increase in water flux across an in vitro BBB model. Hydraulic conductivity of our sham control cultures was in strong agreement with published values on the order of 10−7 cm/s/cmH2O for similar bEnd.3 culture systems. 10,28 We observed changes in hydraulic conductivity for our in vitro BBB cultures between 30 min and 2 h of blast exposure at increasing magnitudes of impulse. While there is a lack of published literature quantifying water flux across the BBB after primary blast, it is possible that this relatively long time-window—coupled with potential differences in the time-course of blast-induced BBB opening—accounts for why our hydraulic conductivity results did not mirror the acute TEER dose-dependent response observed (2–10 min after exposure) with increasing impulse levels. A previous study comparing the progression of BBB opening in different rat models of TBI demonstrated that impact acceleration resulted in immediate BBB opening followed by rapid closing of the barrier, whereas lateral cortical impact was associated with prolonged BBB opening for up to 4 h. 56 Therefore, we anticipate that more precisely defining the time point at which hydraulic conductivity is measured after injury will allow for improved mechanistic understanding of the high incidence of brain edema following bTBI. 2,57
A normally functioning BBB in vivo is capable of impeding the diffusion of molecules greater than 500 Da between the systemic and brain compartments. 58 Others have demonstrated shock wave–induced membrane permeabilization of in vitro cell culture systems by increased uptake of molecules of varying weights, including calcein (622 Da), propidium iodide (668 Da), trypan blue (873 Da), and fluorescein isothiocyanate-dextran (72 kDa). 59 –62 Importantly, it was found that permeability of solutes did not depend on peak pressure of the shock waves, but was critically dependent on impulse. 59 It is also vital to note that shock waves tested in these studies were of peak overpressures exceeding 1 MPa with durations on the order of microseconds, 59,61,62 which are not applicable to real-world blast events.
Following blast exposure, significantly increased permeability of 10 kDa dextrans in our exposed BBB cultures suggested impaired ability of the monolayers to restrict the passage of solutes equal to or below this molecular weight. However, because our endothelial cultures do not form permeability barriers as restrictive as those observed in vivo or in primary cell culture systems, 30,39,63 the baseline permeability of the 3 kDa dextrans was high in shams and not significantly elevated in blast-injured cultures. Noteworthy is that the permeabilities of other in vitro BBB models (incorporating bEnd.3 cells) to similar dextran tracers ranging from 3–70 kDa were in close agreement with permeability values observed in our sham cultures. 10,28,31 Clinical studies have used dynamic magnetic resonance imaging of gadolinium diethylenetriaminepentaacetate (Gd-DTPA; 550 Da) 64 to identify areas of BBB breakdown in neurological diseases such as multiple sclerosis. 65,66 Permeability values for multiple sclerosis lesions in patients ranged from 4–17×10−6 cm/sec, 65 which is comparable to our measured permeabilities for 3 and 10 kDa solutes in blast-exposed cultures. Increased solute permeability post-injury holds implications for neurotoxic serum constituents that may infiltrate the brain, potentially leading to neuronal dysfunction or death.
A potential limitation of the current study is that, at one of the highest exposures tested, endothelial cells may have detached from the Transwell membrane. The counting of nuclei (representing individual cells) in immunostaining images of sham and injured cultures resulted in a non-significant difference in cultures exposed to the 571 kPa peak overpressure blast, but a significant difference (p<0.05) in cultures exposed to the 396 kPa overpressure blast. Detachment was minimal, however, as the average number of cells between sham and injured images differed by no more than 11 cells (12% of total cells in shams) and is likely only applicable to the highest impulse exposure level tested in this study (396 kPa overpressure, 2.40 ms duration, 276 kPa*ms impulse, nitrogen driver gas). Others utilizing in vitro cell monoculture systems have reported some degree of cell detachment after shock wave exposure, 61,62 which is thought to resemble in vivo lesions associated with pathophysiological processes of bTBI. 62 Denuded regions of cell loss have been observed after exposure to shock wave pulse trains, sometimes in conjunction with cavitation, in extracorporeal lithotripsy models. 62 We did not observe denuded patches in our cultures using our blast injury model.
BBB dysfunction and the disruption of tight junctions is a common phenomenon following TBI. 6,67 –69 The ZO-1 accessory protein is extensively studied because its interactions with the cytoskeleton are thought to dictate the localization and functional expression of tight junctions. 70 –73 Activation of pro-inflammatory factors and MMPs are implicated in elevated BBB permeability following TBI, causing degradation of tight junction proteins including ZO-1, an MMP-9 substrate. 38,74 –76 ZO-1 levels have been shown to decrease by up to 50% of baseline at 24 h post-injury using a controlled cortical impact model. 38 This effect is comparable to our approximately 55% decrease in ZO-1 staining after exposure to a 276 kPa*ms impulse blast, suggesting similar BBB disruption under dramatically different physical loading conditions.
The bEnd.3 mouse brain microvascular cell line was used in our in vitro BBB model for its ability to retain phenotypic stability, preserve physiologic cell architecture, and form a functional paracellular barrier. 10,30,35,39 While our model consisted of only one cell type, the BBB is commonly thought of as a three-cell archetype that includes the brain capillary endothelial cell, astrocyte, and supporting pericyte, to help mediate full expression of the BBB phenotype. 30 Brain endothelial tight junctions in rats establish a rate-limiting diffusion barrier resulting in sucrose (342 Da) 77 permeability ranging from 0.03–0.10×10−6 cm/s. 6,30,78 By comparison, the measured permeability of 3 kDa dextrans through our sham control cultures was at least an order of magnitude higher, at 3.3×10−6 cm/s. TEER values reported in vivo exceed 1000 Ω*cm 2 , 30,79 whereas typical values achieved by our in vitro culture system are approximately 20–30 Ω*cm 2 . 10,28,31 Primary cerebromicrovascular endothelial cells more closely resemble the BBB phenotype in vivo due to their ability to form tight monolayers with higher TEER and lower permeability; 10,28,30,39 however, their vulnerability to contamination by other neurovascular unit cells, lack of phenotypic stability over multiple passages in vitro, and inter- and intra-batch variability between cultures may hamper sensitivity for identification of injury thresholds. 30,39
The endothelial monolayer exhibited an intrinsic capacity for spontaneous recovery as demonstrated by the full recovery of TEER at 3 days post-injury. In vivo bTBI studies reporting BBB damage marked by increased IgG levels in the brain have shown return to control levels 3 days post-exposure, which is in close agreement with our TEER recovery period. 13,15 Self-repair mechanisms leading to restoration of BBB integrity after the biomechanical insult have been described in other TBI models like fluid percussion injury, where initial increases in BBB permeability (detected by IgG brain localization) was followed by barrier recovery preventing further influx of the circulating proteins. 80 –82 These results help provide insight on standing controversy as to whether damaged endothelial cells can undergo functional recovery 83 and suggest value in developing acute therapeutic strategies to repair the blast-injured BBB.
In summary, this study is the first to demonstrate disruption of an in vitro model of the BBB after exposure solely to primary blast injury. The acute dose-dependent TEER response following exposure to a range of blast loading conditions revealed compromised monolayer integrity that was most strongly correlated with impulse. BBB opening after exposure was confirmed by significantly increased hydraulic conductivity and solute permeability. The physical breakdown of tight junctions was identified with compromised ZO-1 immunostaining. The temporal recovery of TEER post-blast highlighted that acute treatments to repair the blast-injured BBB may provide clinical utility. Taken together, these results indicate that BBB damage could be a major mechanism contributing to vascular and neuronal pathology of bTBI at blast levels above a critical threshold, and hold implications for novel helmet designs to mitigate the effects of blast in humans.
Footnotes
Acknowledgments
We would like to thank Aaron Huang and Charles Levin for their assistance with experimental setup and operation of the blast injury device. This work was supported by a Multidisciplinary University Research Initiative from the Army Research Office (W911MF–10–1–0526) and a National Science Foundation Graduate Research Fellowship (to C.D.H.; DGE–07–07425). Portions of this work were previously presented at the 2012 International Research Council on the Biomechanics of Injury (IRCOBI), the 2012 National Neurotrauma Symposium, and the 2012 Annual Biomedical Research Conference for Minority Students (ABRCMS). The current manuscript has been significantly improved and expanded upon with new data compared to prior conference presentations of this work.
Author Disclosure Statement
No competing financial interests exist. None of the authors listed have received personal financial support resulting from any information generated in this submission.