
Abstract
Bradykinin (BK) was shown to stimulate the production of physiologically active metabolites, blood–brain barrier disruption, and brain edema. The aim of this prospective study was to measure BK concentrations in blood and cerebrospinal fluid (CSF) of patients with traumatic brain injury (TBI), subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), and ischemic stroke and to correlate BK levels with the extent of cerebral edema and intracranial pressure (ICP). Blood and CSF samples of 29 patients suffering from acute cerebral lesions (TBI, 7; SAH,: 10; ICH, 8; ischemic stroke, 4) were collected for up to 8 days after insult. Seven patients with lumbar drainage were used as controls. Edema (5-point scale), ICP, and the GCS (Glasgow Coma Score) at the time of sample withdrawal were correlated with BK concentrations. Though all plasma-BK samples were not significantly elevated, CSF-BK levels of all patients were significantly elevated in overall (n=73) and early (≤72 h) measurements (n=55; 4.3±6.9 and 5.6±8.9 fmol/mL), compared to 1.2±0.7 fmol/mL of controls (p=0.05 and 0.006). Within 72 h after ictus, patients suffering from TBI (p=0.01), ICH (p=0.001), and ischemic stroke (p=0.02) showed significant increases. CSF-BK concentrations correlated with extent of edema formation (r=0.53; p<0.001) and with ICP (r=0.49; p<0.001). Our results demonstrate that acute cerebral lesions are associated with increased CSF-BK levels. Especially after TBI, subarachnoid and intracerebral hemorrhage CSF-BK levels correlate with extent of edema evolution and ICP. BK-blocking agents may turn out to be effective remedies in brain injuries.
Introduction
A fatal sequel of various cerebral lesions, for example, traumatic brain injury (TBI), subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), or ischemic stroke, is barely controllable brain swelling that may cause mass effects and herniation, leading to life-threatening cerebral ischemia. 16 The small number of clinical studies measuring kinin levels after cerebral lesions showed elevated BK concentrations in the cerebrospinal fluid (CSF) after SAH, elevated plasma concentrations of kallidin after cerebral ischemia, and increased levels of the stable metabolite of BK (BK 1–5) in plasma and CSF after severe TBI (sTBI). 17 –19 Thus far, however, correlations of plasma-/CSF-BK levels with the formation of brain edema or clinical outcome are still missing. Therefore, the aim of the current study was to measure and evaluate time profiles of BK concentrations in blood and CSF of patients suffering from TBI, SAH, ICH, or ischemic stroke and to investigate whether these values correlate with edema formation, ICP, and clinical parameters.
Methods
Patients
From May 2008 to February 2010, 29 patients suffering from a sTBI, SAH, ICH, or ischemic stroke and provided with an external ventricular drainage (EVD) for ICP-monitoring and CSF drainage were prospectively examined. Patients selected for this study suffered from a severe cerebral lesion, in which formation of cerebral edema was anticipated at admission. Isolated infratentorial lesions were not included. For determination of BK time profiles, placement of the EVD and sample withdrawal should have been possible within a short period after cerebral ictus and patients should have been expected to have EVD in place for at least several days. Ventricular drainages placed secondary after decompressive craniectomy, hematoma removal, or even days after TBI because of ICP monitoring reasons of often intubated and sedated patients were not used for sample withdrawal. Because of the BK time profiles and the fact that various acute cerebral lesions should be represented, patient recruitment was done nonconsecutively. All patients were treated in the Department of Neurosurgery, Campus Grosshadern, Ludwig-Maximilians-University (Munich, Germany). For controls, blood and CSF samples were obtained from 7 patients with spinal degenerative diseases who underwent a lumbar puncture for spinal myelograms. Age and gender of these control patients were comparable to those of the 29 brain injury patients. Written informed consent was obtained from either the patient or an authorized relative in all cases. The study was performed with the approval of the local ethics committee.
BK measurements
Venous blood (5 mL) and CSF (5 mL) were collected into a precooled polypropylene syringe, containing 0.5 mL of cooled kinin-system inhibitor cocktail, consisting of protease and peptidase inhibitors, to obtain final concentrations of 21 μmol/L of aprotinin, 73 μg/mL of chicken-egg-albumin trypsin inhibitor, 305 μg/mL of hexadimethrine bromide, 4.5 mmol/L of 1,10-phenanthroline, and 4.5 mmol/L edetic acid. 20 After immediate centrifugation at high speed (1700g) at 4°C for 10 min, 0.5 mL of CSF/plasma and inhibitor solution were mixed with 2 mL of cold ethanol and stored at −80°C until analysis. For specific measurement of BK, a combination of liquid-phase extraction, high-performance liquid chromatography, and radioimmunoassay were applied, as described before. 21
Sample collection and evaluation of clinical and radiological factors
After initial computer tomography (CT) and assessment of Glasgow Coma Score (GCS), all patients were observed at an intensive care unit (ICU). Samples for determination of BK concentrations were obtained immediately after placement of the ventricular catheter and on days 3, 4, and 5 after insult. ICP, GCS, and blood pressure were assessed at the same times. The zero levels for blood pressure and ICP transducers were at heart and ear level, respectively, and were regularly calibrated. Additionally, Glasgow Outcome Score (GOS) at discharge was recorded. In case of neurological deterioration or an increase of ICP values of more than 20 mmHg exceeding 30 min, an additional CT scan and BK sampling were performed. CSF/plasma-BK concentrations were correlated with clinical and monitoring data, obtained at the time of sample withdrawal.
Quantitation of edema formation
Noncontrast CT was conducted either with a scanner in sequential mode (Toshiba Aquilion Multi; Toshiba Corporation, Tokyo, Japan), with slice thickness of 1 (skull base) and 4 mm (supraclinoidal), parallel to the skull base at 120 kV and 200 mA or a 64-slice CT scanner with 120 kV and 380mA (Siemens Sensation 64; Siemens AG, Erlangen, Germany) with a slice thickness of 0.6 mm.
Extent of edema formation (hypodense texture analysis, compared to unaffected brain regions by expert observation) was evaluated for each CT scan by two independent neuroradiologists who were blinded to the clinical results (H.B. and M.H.). Therefore, a 5-point scale was established. Inter-rater reliability of the two neuroradiologists was r=0.87. In case of discordant results, a reevaluation of the CT findings was performed and a consensus was found. CT scans were performed immediately at admission and at the following days in dependence on the underlying cerebral lesion and applied therapy on a regular base. BK levels, obtained immediately before or after CT examinations, were correlated with the radiologically determined extent (grade) of cerebral edema: 0, no edema; 1, focal unilobular edema; 2, plurilobular unilateral edema; 3, bilateral edema; 4, sulcal relief disappeared in global edema; and 5, basal cisterns disappeared in global edema. The grade of edema 1, 2, or 3 increased by one point, if an extensive lesion with surrounding edema caused a mid-line shift of the septum pellucidum ≥0.5 cm.
For differentiation of cerebral edema and perilesional ischemia, hypodense areas were classified as cerebral edema strictly in the case of hypodense, perilesional, or diffuse trauma/hemorrhage-related formations. Most patients underwent an additional magnetic resonance imaging (MRI) study, including diffusion-weighted sequences with apparent diffusion coefficient (ADC) mapping. If an MRI-based evidence of an additional ischemic damage, besides cerebral edema, after TBI, SAH, and ICH occurred, the lesion of ischemia has not been taken into account and only the extent of cerebral edema has been evaluated by the 5-point scale.
In patients with ischemic strokes, perilesional hypodense areas around the area of demarcated infarction were only then classified as edema when measurements of cerebral blood flow and ADC by MRI indicated lack of ischemic/penumbral conditions. We used this approach to differentiate between cerebral edema and perilesional ischemia to avoid an overestimation of cerebral edema in these patients. Potentially ischemic brain areas were not included into the analysis of brain edema.
Statistical analysis
Statistical analyses were performed with the use of SigmaStat statistical software (Version 3.0; SPSS Science, Inc., Chicago, IL). Student's t-test for unpaired values or Mann-Whitney's rank-sum test were used to assess the statistical significance of differences between controls and BK levels in patients with cerebral lesions. For evaluation of correlations, Spearman's rank-order test for not normally distributed values with constant variance and Pearson's product moment correlation for normally distributed data were performed. Time profile of BK concentrations was measured by one-way repeated-measures analysis of variance. Results are presented as means±standard deviation and in Figures 1 –3 as means±standard error of mean (SEM) for reasons of clarity. Median and range were given where appropriate. Significance level was set at p≤0.05.
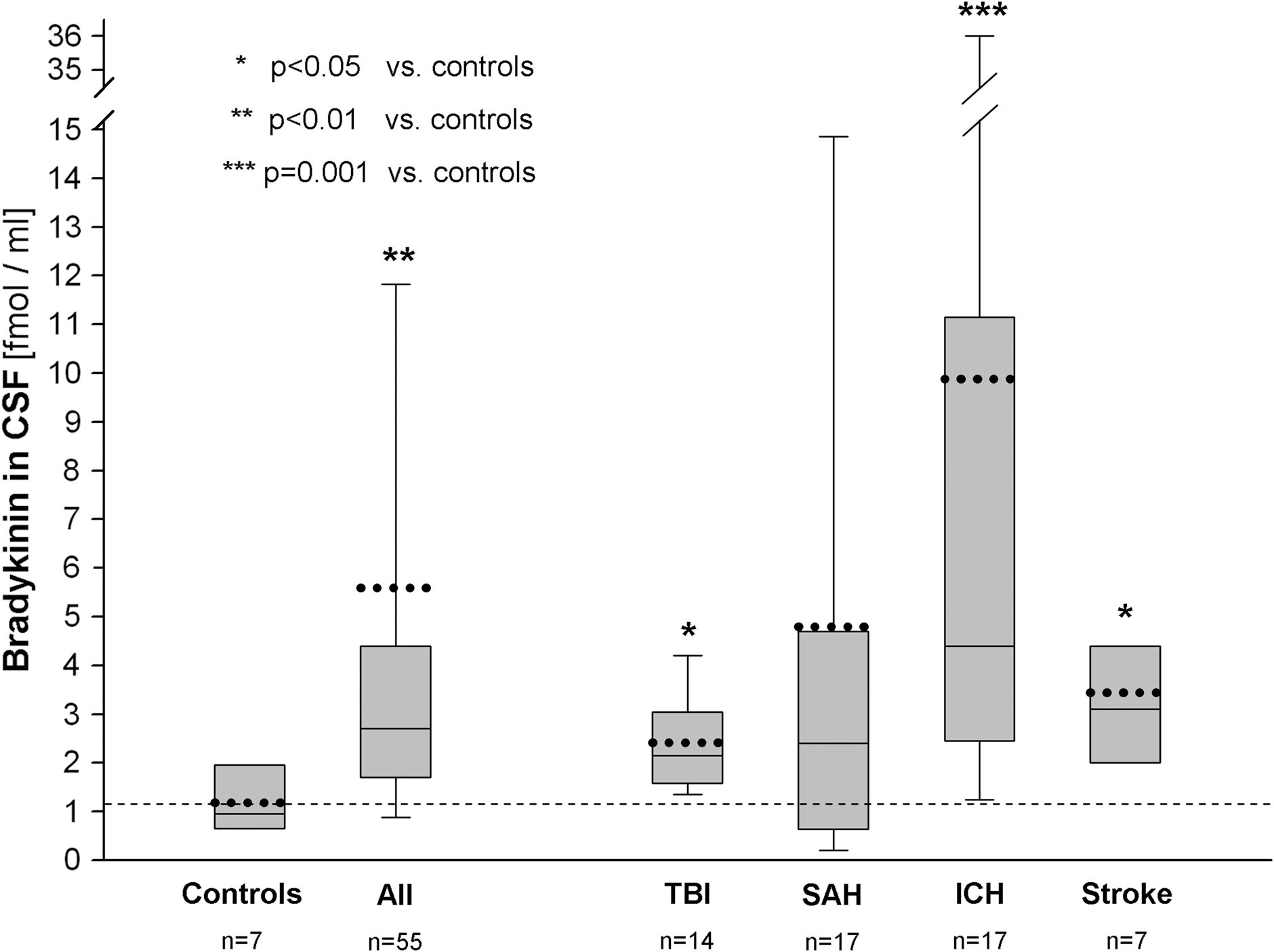
CSF-BK levels of all patients and of subgroups presented as early values (≤72 h) after ictus. Values given as box plot indicating median, lower, and upper quartiles and 10th and 90th percentile. Dotted lines represent mean values; dashed line represents the mean level of controls. Number of samples is given per group. CSF, cerebrospinal fluid; BK, bradykinin; TBI, traumatic brain injury; SAH, subarachnoid hemorrhage; ICH, intracerebral hemorrhage.
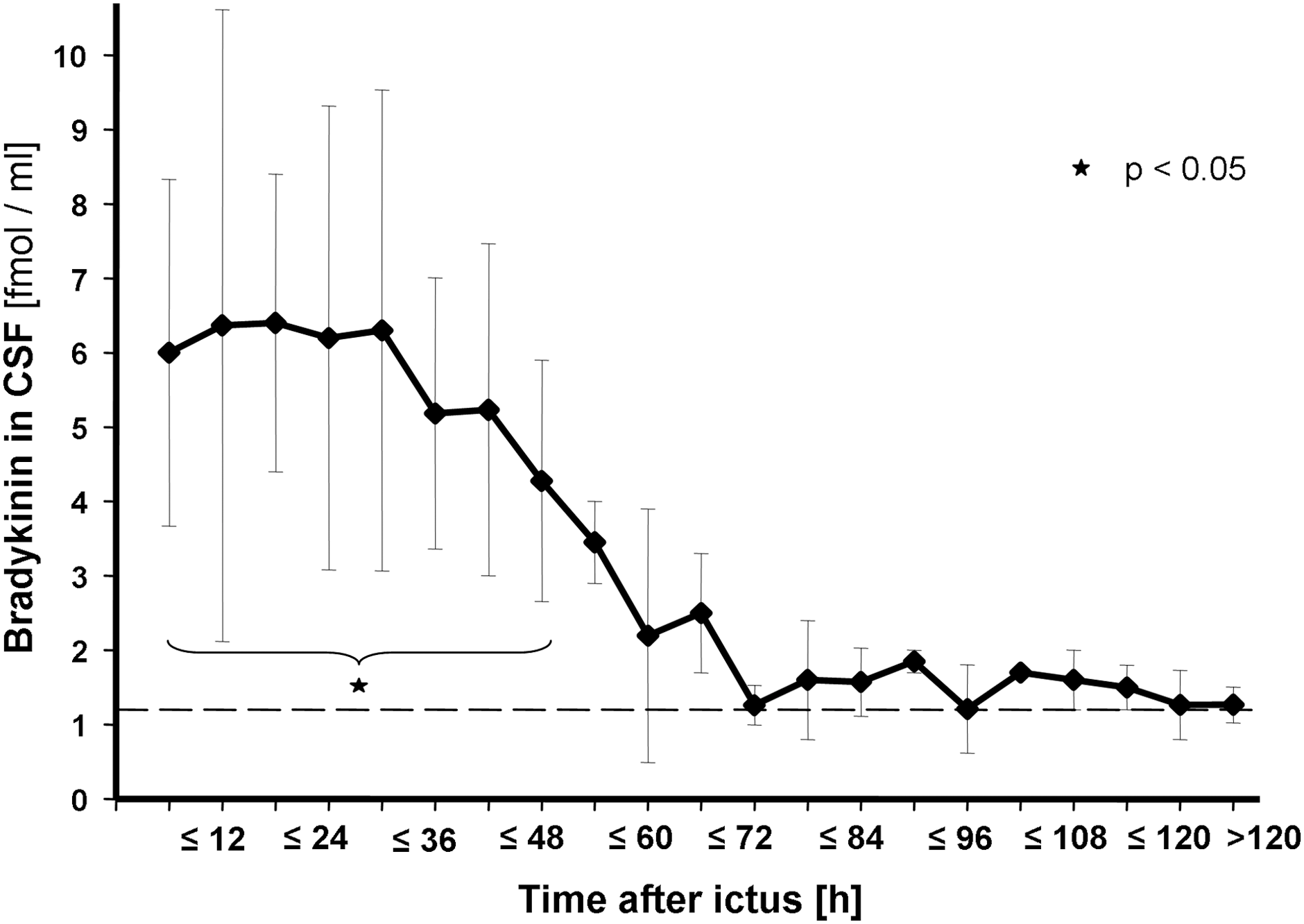
Time profile of CSF-BK concentrations within the observation period. Values are given as mean of number of samples obtained in each time interval (±SEM). Time intervals were 0 to ≤6 h (7 samples), >6 to ≤12 h (n=7), >12 to ≤18 h (n=4), >18 to ≤24 h (n=4), >24 to ≤30 h (n=5), >30 to ≤36 h (n=6), >36 to ≤42 h (n=3), >42 to ≤48 h (n=5), >48 to ≤54 h (n=4), >54 to ≤60 h (n=4), >60 to ≤66 h (n=3), >66 to ≤72 h (n=3), and so on. Within the first 48 h after onset of the acute cerebral lesion, CSF-BK levels were significantly elevated, compared to controls. Dashed line represents mean value of the control group. CSF, cerebrospinal fluid; BK, bradykinin; SEM, standard error of the mean.
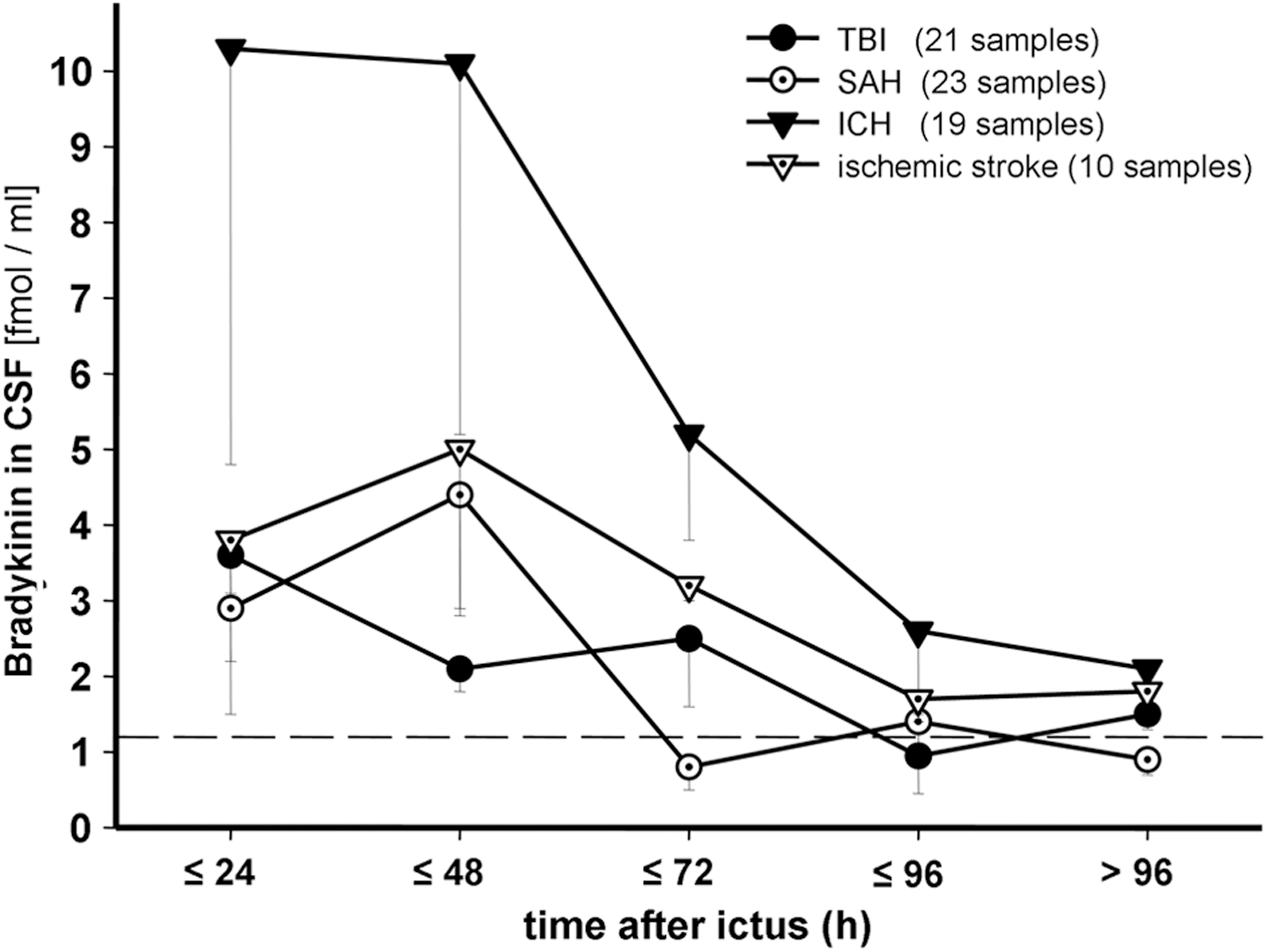
Subgroup analysis of the time profile of CSF-BK levels (±SEM) within the observation period. Number of samples of each time interval ranged from 2 to 7 within the subgroups. CSF, cerebrospinal fluid; BK, bradykinin; SEM, standard error of the mean; TBI, traumatic brain injury; SAH, subarachnoid hemorrhage; ICH, intracerebral hemorrhage.
Results
Patients and measurements
Twenty-nine patients (15 females, 14 males; mean age, 52.4±13.4 years) suffering from TBI (n=7), SAH (n=10), ICH (n=8), and ischemic stroke (n=4) were enrolled. Median GCS at admission was 4 (range, 3–14) in all patients. Duration of clinical observation was a mean of 17.5±8.9 days. A detailed analysis of the cerebral lesions is given in Table 1. Overall, BK levels were measured in 70 plasma and 73 CSF samples of brain-injured patients. Baseline BK levels were also determined in plasma and CSF of 7 controls. In the observed population, the first sample was collected after a median of 15 h (range, 4–48), the second at 54 h (range, 20–140), the third at 92 h (range, 52–150), and the fourth at 140 (range, 76–204) after insult.
hem., hematoma; WFNS, World Federation of Neurosurgery; CT, computed tomography; TBI, traumatic brain injury; SAH, subarachnoid hemorrhage; ICH, intracerebral hemorrhage; AVM, arteriovenous malformation; MCA, medial cerebral; ACA, anterior cerebral artery; CI, confidence interval.
The overall mortality rate was 13.8% (4 patients) as a result of brain herniation at a median of 63 h (range, 43–88) after admission; 2 patients died after ICH, 1 each after SAH and TBI. Sixteen patients (55.2%) had an unfavorable (GOS, 2 or 3) and 9 (31%) a favorable outcome (GOS, 4 or 5) at discharge.
Plasma BK levels
Mean plasma-BK concentrations of all samples of study patients and controls were 2.7±2.7 and 2.8±1.4 fmol/mL, respectively. Subgroup plasma-BK levels in patients with TBI (20 samples), SAH (22 samples), ICH (18 samples), and ischemic stroke (10 samples) were 2.2±1.6, 2.4±2.3, 3.6±4.3, and 2.9±1.8 fmol/mL, respectively. These values were not different from controls.
CSF-BK levels
The mean CSF-BK concentration of all samples obtained was higher in patients suffering from cerebral lesions at 4.3±6.9 fmol/mL (73 samples; median, 2.1; range, 0.18–36.0) than in controls (7 samples; 1.2±0.7 fmol/mL; median, 0.95; range, 0.53–2.1; p=0.05). CSF-BK levels were significantly elevated in patients suffering from ICH (19 samples; 7.8±10.1 fmol/mL; median, 3.4; range, 0.68–36.0; p=0.003) and ischemic stroke (10 samples; 3.1±1.8 fmol/mL; median, 2.9; range, 1.3–7.1; p=0.03), compared to controls. No significant increase from controls was observed in patients suffering from TBI (21 samples; 2.3±2.1 fmol/mL; median, 2.0; range, 0.28–8.4) and SAH (23 samples; 3.7±7.2 fmol/mL; median, 1.5; range, 0.18–35.5).
With respect to time profiles, early CSF-BK values (≤72 h after ictus) in the overall population (n=55) were 5.6±8.9 fmol/mL (median, 2.7; range, 0.18–36.0), compared to 1.2±0.7 fmol/mL in controls (median, 0.95; range, 0.5–2.1; p=0.006). Respective early mean values were 2.4±0.9 fmol/mL (median, 2.2; range, 1.3–4.4; p=0.01) in 14 TBI measurements, 4.8±8.4 fmol/mL (median, 2.4; range, 0.18–35.5; p=0.2) in 17 SAH measurements, 9.9±12.9 fmol/mL (median, 4.4; range, 1–36; p=0.001) in 17 ICH measurements, and 3.4±1.9 fmol/mL (median, 3.1; range, 1.3–7.1; p=0.02) in 7 ischemic stroke measurements (Fig. 1).
CSF-BK levels in all patients were a mean of 6.0±5.7 fmol/mL obtained within 6 h after injury, remained markedly elevated up to 48 h, and decreased thereafter, reaching levels comparable to those of the control group in less than 72 h (Fig. 2). CSF-BK levels were significantly elevated within the first 48 h after onset of the acute cerebral lesion, compared to controls (p=0.011).
CSF-BK concentrations peaked early in patients suffering from TBI and ICH (25±23 and 22±16 h after injury, respectively). In patients suffering from SAH and ischemic stroke, the highest levels were reached later, at a mean of 51±45 and 41±25 h, respectively (Fig. 3). However, peak time differences between these patient subgroups were not significant (p=0.11).
BK concentration and edema formation
Extents of edema were grades 0 (n=1), 1 (n=8), 2 (n=11), 3 (n=32), 4 (n=14), and 5 (n=7). The radiologically determined extent of edema significantly correlated with ICP, measured at time of CT examination, in all 73 examinations irrespective of the type of cerebral lesion (r=0.51; p<0.002), particularly in patients with TBI, ICH, and ischemic stroke (r=0.64, p=0.001; r=0.5, p<0.03; r=0.7, p=0.02). In 13 patients developing global cerebral edema (CT score, 4 or 5), a worse outcome was found, compared to patients with focal cerebral edema (CT score, 1–3; p=0.046); 2 patients had a favorable outcome (GOS, 4 or 5), 9 an unfavorable outcome (GOS, 2 or 3), and 2 died. Comparably, GOS of patients with focal edema revealed 7 patients with favorable outcome, 7 with unfavorable outcome, and 2 died.
CSF-BK concentrations of all patients (r=0.53; p<0.001; Fig. 4) and of those with TBI and SAH correlated with the extent of edema formation measured immediately before and after CT examination (r=0.46, p=0.04 and r=0.66, p<0.001, respectively), whereas in patients suffering from ICH, CSF-BK levels only tended to correlate with cerebral edema (r=0.4; p=0.09).

Correlation between the extent of cerebral edema and corresponding CSF-BK levels measured immediately before and after CT examination in patients after brain injury. The number of samples obtained in each edema category was n=1 in grade 0, n=8 in grade 1, n=11 in grade 2, n=32 in grade 3, n=14 in grade 4, and n=7 in grade 5. Markedly elevated CSF-BK concentrations were found when cerebral edema extended bilaterally (CT score, 3) or in global edema (CT score, 4 and 5). CSF, cerebrospinal fluid; BK, bradykinin; CT, computed tomography.
When comparing time profiles of CSF-BK levels with cerebral edema formation, a slightly earlier rise of CSF-BK concentration than edema evolution was found in the overall observation (mean CSF-BK peak of 35±32 h vs. maximum of edema formation of 42±40 h after injury) and in the subgroup of SAH patients (50.6±45 vs. 70.3±59 h); however, differences over time did not reach statistical significance. Maximum cerebral edema formation after SAH (mean, 70.3±59 h) tended to occur later than after TBI and ICH (25±23 h, p=0.08 and 25±15 h, p=0.054, respectively).
Overall, 13 patients exhibited global cerebral edema (CT score, 4 or 5), either at admission (n=10) or delayed (n=3). Corresponding mean CSF-BK levels of these patients at the time point of presence of global edema (10.6±12 fmol/mL) were increased, compared to controls (1.2±0.7 fmol/mL; p=0.002) and to patients (n=16) exhibiting focal cerebral edema (2.7±2.0 fmol/mL; p=0.015).
BK levels and ICP/clinical score
In 27 cases, elevated ICP values of more than 20 mmHg over 30 min were detected at the time point of sample acquisition. No correlations were detected between plasma-BK concentrations and ICP. In contrast, a significant correlation was found between all CSF-BK levels and the simultaneously measured ICP values (r=0.49; p<0.001; Fig. 5) and within the subgroups of TBI, SAH, and ICH, respectively (r=0.66, p=0.001; r=0.62, p=0.001; and r=0.47, p=0.04). In 62 of 73 sample collections, patients were sedated and ventilated. No correlation of plasma-/CSF-BK concentrations and clinical status (GCS) of patients at sample withdrawal was found.

Correlation between 73 CSF-BK levels and corresponding simultaneously measured intracranial pressure in patients with brain injury. CSF, cerebrospinal fluid; BK, bradykinin.
Discussion
The present prospective study is the first methodical analysis of BK plasma and CSF levels in humans after acute cerebral lesions. Our results demonstrate that TBI, SAH, ICH, and ischemic stroke are associated with increased BK levels in CSF, whereas the majority of plasma-BK concentrations remained within normal limits. Most important, from a clinical point of view, is the positive correlation of CSF-BK levels with the extent of cerebral edema formation and with ICP.
BK and BK receptors after experimental brain lesions
After formation from kininogen, BK is rapidly metabolized within the circulation with a half-time of less than 30 sec, 22 whereas a high rate of BK inactivation additionally occurs in the pulmonary circulation. 23 However, the kallikrein-kinin system, once activated, can stimulate itself for further production of BK to replace what was already rapidly degraded; in particular, kallikrein, once formed, is a potent activator of factor XIIa, which converts prekallikrein into the active proteolytic enzyme, kallikrein, therefore creating a positive feedback loop. 24 In TBI, kininogen levels within the traumatized hemisphere began to rise 1 h after, reaching their peak 15 h after insult and being still significantly elevated after 24 h. 25 A study of TBI in mice elucidated a maximal increase of tissue BK 2 h after trauma; B1 and B2 receptors were significantly up-regulated in the traumatic penumbra up to 24 h, whereas B2-receptor knockout (KO) mice had significantly less brain edema, smaller contusion volumes, and a better functional outcome. 26 After experimental ischemia, tissue BK was maximally increased after 12 h, 14 whereas B2-receptor messenger RNA up-regulation peaked after 24 h. After cerebral ischemia, BK levels in plasma and tissue corresponded to the cerebral water content (i.e., edema progression and regression). 27 Immunohistochemistry revealed up-regulation of B2 receptors 2 h after cells showed signs of ischemic damage, and up-regulation persisted in the penumbra up to 24 h after ischemia. Further, B2-KO mice had an improved outcome, smaller infarct volumes, and developed less brain edema, compared to wild type. 14 Experimental studies of traumatic and ischemic brain injury have shown that pharmacological blockade of BK generation or action by B2-receptor antagonists improves neurological recovery and inhibits brain edema formation as well as neuronal damage. 11 –13,27 –29
BK activation after cerebral lesions in clinical research
There are only very few clinical studies measuring kinin levels after acute cerebral lesions. Concerning plasma BK, Kasuya and colleagues 17 reported that BK levels after SAH were not significantly elevated, compared to the control group, and without significant changes over time. In 22 patients suffering from large territory infarction, plasma-BK and kallikrein levels did not differ from controls measured up to 8 days after stroke. 18 Accordingly, the present study revealed BK concentrations—regardless of time after ictus and the type of acute cerebral lesion—in all but 3 of 70 plasma samples, comparable to unaffected control patients and within the range of 22 healthy volunteers (mean, 2.2 fmol/mL; range, 0.2–7.1). 21
Explanations for plasma-BK levels remaining within normal limits while CSF-BK is significantly elevated may be the short half-life of BK in plasma, 22 which is rapidly degraded by the enzyme kininase located on the vascular endothelial cell surface, especially in the lungs or a higher activity of this BK-inactivating enzyme in plasma than in CSF. Further, overall damaged volume, and thus the amount of released BK, is comparably smaller in acute cerebral lesions than in generalized diseases, such as angioedema or anaphylaxis, 21,30,31 where elevated blood levels have been measured.
The time course of CSF-BK concentrations in humans has been examined very rarely, as yet. Kasuya and colleagues 17 found elevated levels on days 0 and 1 after SAH, but not after day 2. However, intracerebral and ventricular hemorrhage were not associated with elevated CSF-BK levels. In our study, CSF-BK levels peaked markedly later, within a mean of 51 h after the onset of SAH; further, intracerebal hemorrhage exhibited significantly higher CSF-BK levels, compared to controls. Differences of techniques of CSF sample collection and BK measurement may contribute to the difference: In the above-cited study, CSF was collected by lumbar puncture, ventricular drainage, or intraoperatively, 17 whereas our present study exclusively used CSF obtained from ventricular drainages alone. Irrespective of the type of cerebral lesion, we found that CSF-BK levels were markedly elevated less than 6 h after ictus, increased further up to 18 h, remained significantly elevated up to 48 h, and decreased thereafter, reaching levels of the control group within 72 h after ictus. CSF-BK levels peaked earlier in the group suffering from ICH and TBI, compared to SAH and ischemic stroke. However, this did not reach significance, possibly as a result of the low number of included patients.
Based on the promising experimental work, application of the selective B2-receptor antagonist, deltibant (Bradycor™), in patients suffering from TBI has been investigated. 32 A positive influence was observed with respect to ICP, clinical score, and outcome under study medication, next to a good tolerability and safety of the drug. In addition, Marmarou and colleagues 19 applied two different doses of the B2-receptor antagonist, LF16-0687Ms (Anatibant®), versus placebo in 25 patients with TBI. Levels of the stable circulating BK pentapeptide metabolite, BK1-5, were significantly elevated in plasma of patients, compared to controls. In fact, BK1-5 in plasma after TBI seems to be more stable and might reflect plasma concentrations over longer periods; however, inadequately high concentrations resulting from repetitive formations of BK cannot be excluded. In the current study, levels of BK itself in plasma remained within normal limits, probably as a result of the short half-life of BK in plasma. In Marmarou and colleagues' study, CSF measurements after TBI showed elevated BK1-5 levels, with an extreme variability ranging from 53 to 78,590 fmol/L, with a steep decrease within the first 80 h after TBI parallel to plasma BK1-5 levels. 19 The time profile in TBI is in accord with our data, displaying an early peak within the first 24 h and decreasing values thereafter to normal values in less than 96 h. However, no data have been reported by Marmarou and colleagues on correlations of the BK metabolite to cerebral edema, ICP, and clinical scores. Unfortunately, the following phase II placebo-controlled trial was abandoned after collecting data from just over half of the number of planned patients. 33 Accordingly, it has to be determined whether B2-receptor antagonists, such as deltibant, anatibant, or the angioedema remedy, icatibant, have beneficial effects in patients after acute and severe cerebral lesions.
BK and edema formation in clinical research
Thus far, it has never been elucidated whether elevated BK levels are associated with cerebral edema formation in humans. Additionally, a general classification to quantify cerebral edema after various acute lesions has not been defined, as yet; in the majority of studies, investigators focussed on particular imaging characteristics of defined acute lesions. Therefore, we established a feasible CT score for quantification of extent of brain edema after various acute and severe cerebral lesions. Grading of extent of cerebral edema in 73 cases significantly correlated with simultaneously measured ICP, independent of the type and extension of acute cerebral lesion. In accord with experimental results, we were able to demonstrate that CSF-BK concentrations significantly correlated with extent of cerebral edema formation, irrespective of the type of cerebral lesion. With regard to time profiles of edema development, CSF-BK peak levels slightly preceded maximum edema formation in the overall observation group and in patients suffering from SAH. However, these findings did not reach significance. Therefore, it remains speculative whether determination of CSF-BK levels might gain prognostic relevance for predicting the extent of cerebral edema formation (i.e., high CSF-BK concentrations as a warning of an upcoming high extent of cerebral edema). Overall, in patients exhibiting global cerebral edema, either at admission or delayed, a significantly worse outcome was found, compared to patients with focal cerebral edema. In patients with global edema, corresponding CSF-BK levels, measured at the time point of global edema occurrence, were significantly elevated, compared to patients suffering from focal cerebral edema (10.6 vs. 2.7 fmol/mL). Further, CSF-BK concentrations were significantly correlated to simultaneously measured ICP values.
One limitation of the current study was certainly the nonconsecutive nature of recruitment, which may have resulted in a biased patient selection. However, inclusion of patients to the study relied, in most cases, on the availability of personnel acquainted with the study protocol and trained to adhere to the rather complex protocol of sample collection and storage. Because this availability was based on ICU rotas designed by individuals unaware of the current study, selection bias is most likely only a minor issue of the current investigation. Another limitation of the current study is the relatively low number of patients included. This was a result of the complex, and therefore expensive, measurement of BK. For this reason, a correlative analysis of BK levels and outcome of each patient was not representative; at any rate, outcome after various cerebral lesions might be influenced by multiple factors. In view of the fact that the aim of the study was to investigate changes of BK development after various acute cerebral lesions over time, the limited budget allowed only measurement in a limited number of patients.
Conclusion
We show that acute cerebral lesions are associated with increased BK levels in the CSF. Particularly, the correlations of these BK levels with the extent of edema formation and ICP provide fundamental information and encourage subsequent studies in brain injuries evaluating the effects of BK-blocking agents.
Footnotes
Acknowledgments
This study was partly supported by a donation from Laboratoires Fournier S.A. (Dijon, France). The authors thank Mrs. Catherine Amstutz (Lausanne) for her excellent technical support in BK measurements.
Author Disclosure Statement
No competing financial interests exist.