
Abstract
The formation of brain edema and subsequent intracranial hypertension are major predictors of unfavorable outcome following traumatic brain injury (TBI). Previously, we reported that arginine vasopressin (AVP) receptor antagonists reduce post-traumatic and post-ischemic brain edema in mice. The aim of the current study was to investigate further the contribution of arginine vasopressin V1a receptors to TBI-induced secondary brain damage in V1a receptor knock-out mice. V1a receptor knock-out (V1a -/-) and wild-type mice were subjected to controlled cortical impact (CCI), and edema (brain water content measured before and 24 h after CCI), primary and secondary contusion volume (15 min and 24 h after CCI), neurological function (one day before and seven days after CCI), body weight (before and seven days after CCI) and mortality were measured. Twenty-four h after CCI, V1a receptor knock-out mice had significantly less brain water content than wild-type mice (mean±standard error of the mean: 79.8%±0.3 vs. 80.6%±0.2, respectively), and secondary contusion volume was significantly smaller (38.2±1.7 mm3 vs. 45.1±1.5 mm3 in wild-type mice). Furthermore, the V1a receptor knock-out mice had less neurological dysfunction (3.2±0.8 vs. 7.0±1.4 in wild-type mice) and weight loss (1.0±1.0% vs. 4.9±1.8% in wild-type mice) seven days after CCI. Our data show that mice lacking V1a receptors have less secondary brain damage following experimental traumatic brain injury. We therefore conclude that V1a receptors may represent a novel drug target for preventing post-traumatic brain edema.
Introduction
Arginine vasopressin (AVP), also known as antidiuretic hormone, is a cyclic nonapeptide that is produced primarily in the magnocellular neurons of the hypothalamic paraventricular and supraoptic nuclei and is stored in the posterior pituitary gland. Systemically, AVP regulates water homeostasis via renal V2 receptors and maintains arterial blood pressure via vascular V1a receptors. 11 –14
In addition to these systemic functions, AVP also exerts various effects on the central nervous system. AVP was identified previously in extrahypothalamic neurons by Bargmann and Scharrer in 1951 15 and it is believed to function as a neurotransmitter or neuromodulator. 16,17 AVP increases the water permeability of brain capillaries, 18 –20 thus modulating central ion homeostasis. 21,22 In the choroid plexus, AVP may reduce blood flow and subsequently decrease the production of cerebral spinal fluid (CSF) production. 23 In in vitro studies, AVP can increase the volume of astrocytes. 24,25 Based on all of these findings, AVP and its V1 receptors have been suggested to play an important role in several brain pathologies that are associated with the formation of brain edema, including focal cerebral ischemia, intracerebral hemorrhage, and TBI. 26 –32
The majority of these studies administered peptide-based or small-molecule AVP receptor antagonists by intra-ventricular delivery. Thus, it is not possible to preclude non-specific activities of these compounds such as binding to other G-protein-coupled receptors or non-specific binding to V1a versus V2 receptors. Therefore, the aim of the current study was to investigate the role of V1a receptors in TBI-induced secondary brain damage in specific V1a receptor knock-out mice (V1a -/-). We monitored the physiological parameters and measured brain water content, primary and secondary contusion volume, neurological function and mortality in knock-out and wild-type mice following controlled cortical impact, a well-established rodent model of cerebral contusion.
Methods
Animals
Homozygous male V1a receptor knock-out (V1a −/−) 33 and wild-type C57/BL6 mice (Charles River, Sulzfeld, Germany) with a body weight of 18–25 g were used in this study. The animals were housed at 22°C with 60% relative humidity and had free access to food and water. All animal experiments were performed in accordance with our institutional guidelines and were approved by the government of Upper Bavaria (protocol number 55.2-1-54-2531-117-05).
Measurement of physiological parameters
Male C57/BL6 (n=7) and V1a receptor knock-out mice (n=4) were anesthetized by an intraperitoneal injection of medetomidine (0.5 mg/kg body weight), midazolam (5 mg/kg body weight) and fentanyl (0.05 mg/kg body weight). After oro-tracheal intubation, the animals were mechanically ventilated (Minivent 845, Hugo Sachs Elektronik, March - Hungstetten, Germany) with 30% oxygen in room air. Ventilation was monitored using microcapnometry (Micro Capnograph CI240, Columbus Instruments, Columbus, Ohio) and adjusted accordingly throughout the experiment. Systemic blood pressure and blood gases were monitored via a catheter placed in the femoral artery. Cerebral blood flow was measured over the contralateral hemisphere using laser Doppler flowmetry (Periflux 4001 Master, Perimed, Stockholm, Sweden) with a glass fiber probe (Probe 418/1, Perimed) affixed perpendicular to the middle cerebral artery territory with cyanoacrylate glue after partially removing the insertion of the temporal muscle. Intracranial pressure was measured as described previously using a custom-made intraparenchymal probe (Mammendorfer Institute for Physics and Medicine, Mammendorf, Germany). 34,35
Anesthesia and trauma induction
Anesthesia was induced with 4% isoflurane in a chamber and maintained with 2% isoflurane in 30%:68% oxygen:nitrous oxide delivered via a face mask. Core body temperature was maintained at 37°C using a feedback-controlled heating pad connected to a rectal probe. To induce trauma, a craniotomy was prepared over the right parietal cortex as described previously. 35 Controlled cortical impact (CCI) was delivered perpendicular to the surface of the brain with a custom-made CCI applicator for mice (MouseKatjuscha 2000, L. Kopacz, University of Mainz, Germany) using the following parameters: 8 m/s velocity, 3 mm diameter, 1 mm brain displacement and 150 ms duration. Following CCI, the skull was closed by affixing the removed bone flap using veterinary-grade tissue glue (Vetbond, 3M, St. Paul, MN). The animals recovered from anesthesia in an incubator heated to 33°C.
Measurement of brain water content
Brain water content was measured in C57/BL6 (n=17) and V1a receptor knock-out mice (n=14) by experimenters who were blinded with respect to the genotype. The brain was removed from mice that did not undergo CCI (n=7 WT and n=6 V1a −/− mice) or 24 h post-CCI mice (n=10 WT and n=8 V1a −/− mice) and placed on a cooled brain matrix. The olfactory bulb and cerebellum were removed, and the cerebral hemispheres were separated. The wet weight (ww) of each hemisphere was measured, after which the hemispheres were dried at 120°C for 24 h and dry weight (dw) was then measured. Brain water content was calculated as a percentage using the following formula: ((ww-dw)/ww))*100.
Quantification of contusion volume
The brain was removed 15 min or 24 h after CCI and immediately frozen on crushed dry ice and stored at −80°C. Coronal sections (10-μm thick) were cut from rostral to caudal using a cryostat (CryoStar HM 560; Microm, Walldorf, Germany), and one in every 50 sections was prepared for further analysis. The sections were stained with cresyl violet for quantifying the area of contused brain, and contusion volume was calculated as described previously. 31
Measurement of body weight
Each mouse was weighed before surgery and then daily for seven days following CCI. Changes in body weight are expressed as a percentage of the pre-surgery weight to correct for variations in pre-traumatic weight.
Evaluation of neurological outcome
Neurological function was measured in a randomized and blinded manner using the Neurological Severity Score (NSS) one day before surgery and daily for seven days following CCI as described previously. 34 The worst NSS score possible is 19 points, and the best score possible is 0 points. Behavior and movement were assessed using the following tests: exiting an open field within 2 min exhibiting spontaneous motor activity, walking a beam, and balancing on a beam.
Experimental groups
Brain water content was measured in four groups as follows: wild-type and V1a a−/− mice pre-surgery and 24 h after CCI. The group sizes were: wild-type pre-surgery (n=7), 24 h post-CCI (n=10); V1a −/− pre-surgery (n=6), 24 h post-CCI (n=8).
Contusion volume was measured 15 min after CCI in wild-type mice and 24 h post-CCI in wild-type and V1a −/− mice (n=8 mice per group). Body weight, NSS, and seven-day mortality rate were measured after seven days (n=7 each).
Each animal was randomly assigned to one of the treatment groups. The investigator who performed the surgery and the follow-up measurements was blinded with respect to the genotype of the animals and to the treatment group.
Statistical analysis
All summary data are presented as mean±standard error of the mean. Statistical analyses were performed using SigmaStat 2.0 (Jandel Scientific, Erkrath Germany). Groups were compared by analysis of variance (ANOVA) on ranks followed by the Student Newman-Keuls (SNK) test as a post-hoc analysis. Changes over time were analyzed by two-way ANOVA for repeated measures followed by SNK test. Differences were considered significant if p<0.05.
Results
Physiological parameters
Mean arterial pressure (MAP), intracranial pressure (ICP) and contralateral cerebral blood flow (CBF) 3 min prior to and both 9 and 30 min after controlled cortical impact (CCI) were not significantly different between wild-type and V1a receptor knock-out mice (Table 1).
MAP, mean arterial pressure; ICP, intracranial pressure; CBF, cerebral blood flow.
Baseline brain water content in V1a receptor–deficient and wild-type mice
Brain water content was not significantly different between V1a receptor knock-out mice and wild-type mice (78.2±0.1% versus 78.0±0.1%, respectively; Fig. 1, left bars).
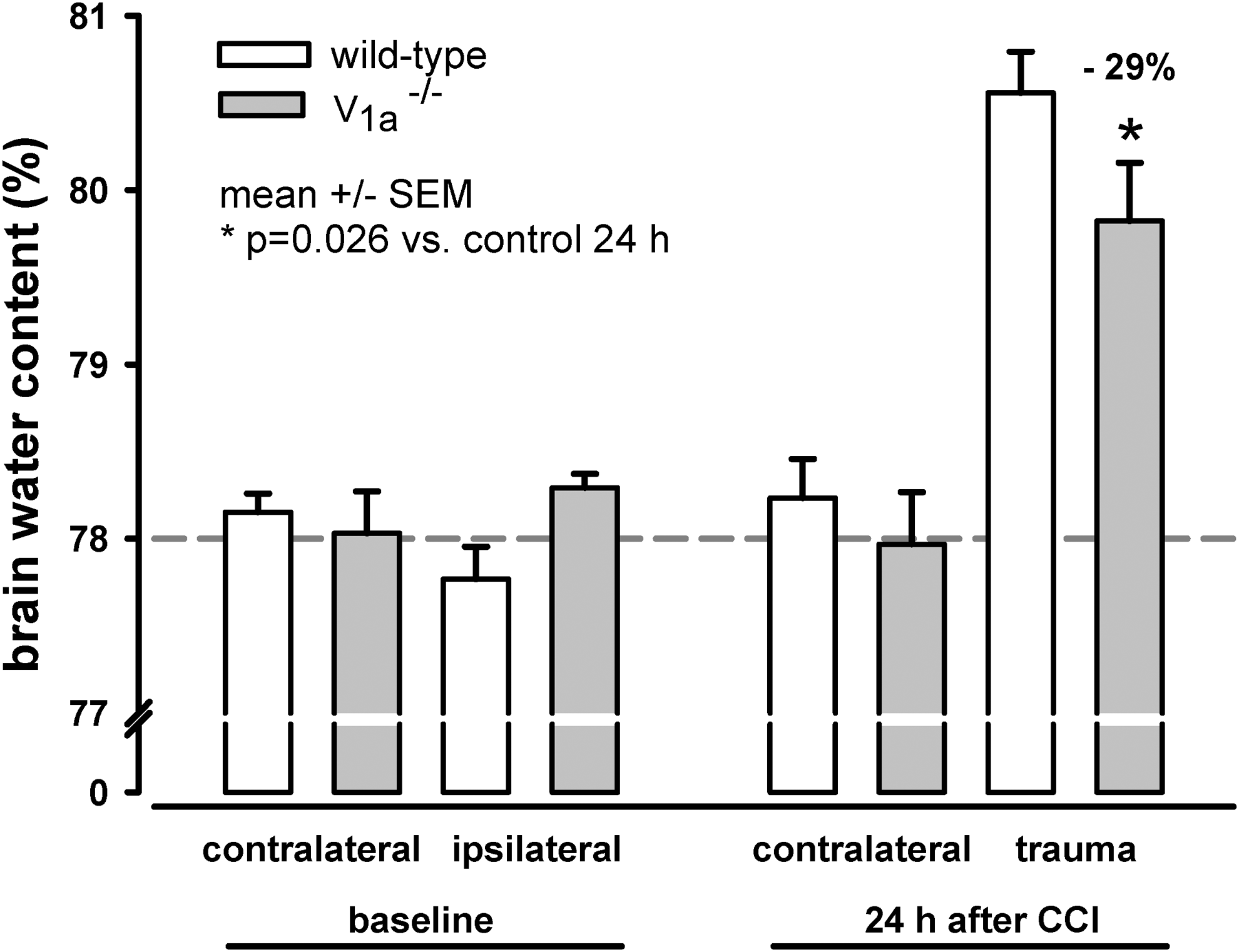
Brain water content. Baseline brain water content is similar between wild-type and V1a receptor knock-out (V1a −/−) mice. Wild-type mice had a baseline brain water content of 77.96±0.13%, and V1a receptor knock-out mice had a baseline brain water content of 78.16±0.13%. Brain water content increased significantly in the traumatized (ipsilateral) hemisphere in both groups 24 h after controlled cortical impact. However, the increase in brain water content was significantly lower in the V1a receptor knock-out mice (p=0.026). Referring to mean brain water content of ipsilateral hemispheres (both groups, baseline), there is a reduction of brain water content 24 h after controlled cortical impact by approximately 29% in V1a −/− mice.
Decreased brain edema in V1a receptor knock-out mice following controlled cortical impact
Both V1a −/− and wild-type mice developed significant brain edema in the traumatized (i.e., ipsilateral) hemisphere 24 h after CCI, whereas brain water content in the contralateral hemisphere was similar to baseline 24 h after CCI in both groups (Fig. 1). Importantly, however, 24 h after CCI, brain water content was significantly lower in the ipsilateral hemisphere of the V1a −/− mice, compared with wild-type mice (i.e., post-traumatic brain edema formation was reduced by 29% in V1a-deficient mice; Fig. 1, right bars).
Decreased contusion volume in V1a receptor knock-out mice 24 h after controlled cortical impact
In the wild-type mice (n=8), primary contusion volume measured immediately (15 min) after TBI was 21.1±0.6 mm3 and increased secondarily to 45.1±1.5 mm3 (p<0.001) during the next 24 h. In contrast, contusion volume 24 h after CCI was only 38.2±1.7 mm3 (n=8) in V1a −/− mice, thus representing a reduction of secondary contusion volume referred to primary lesion by 29% (Fig. 2).
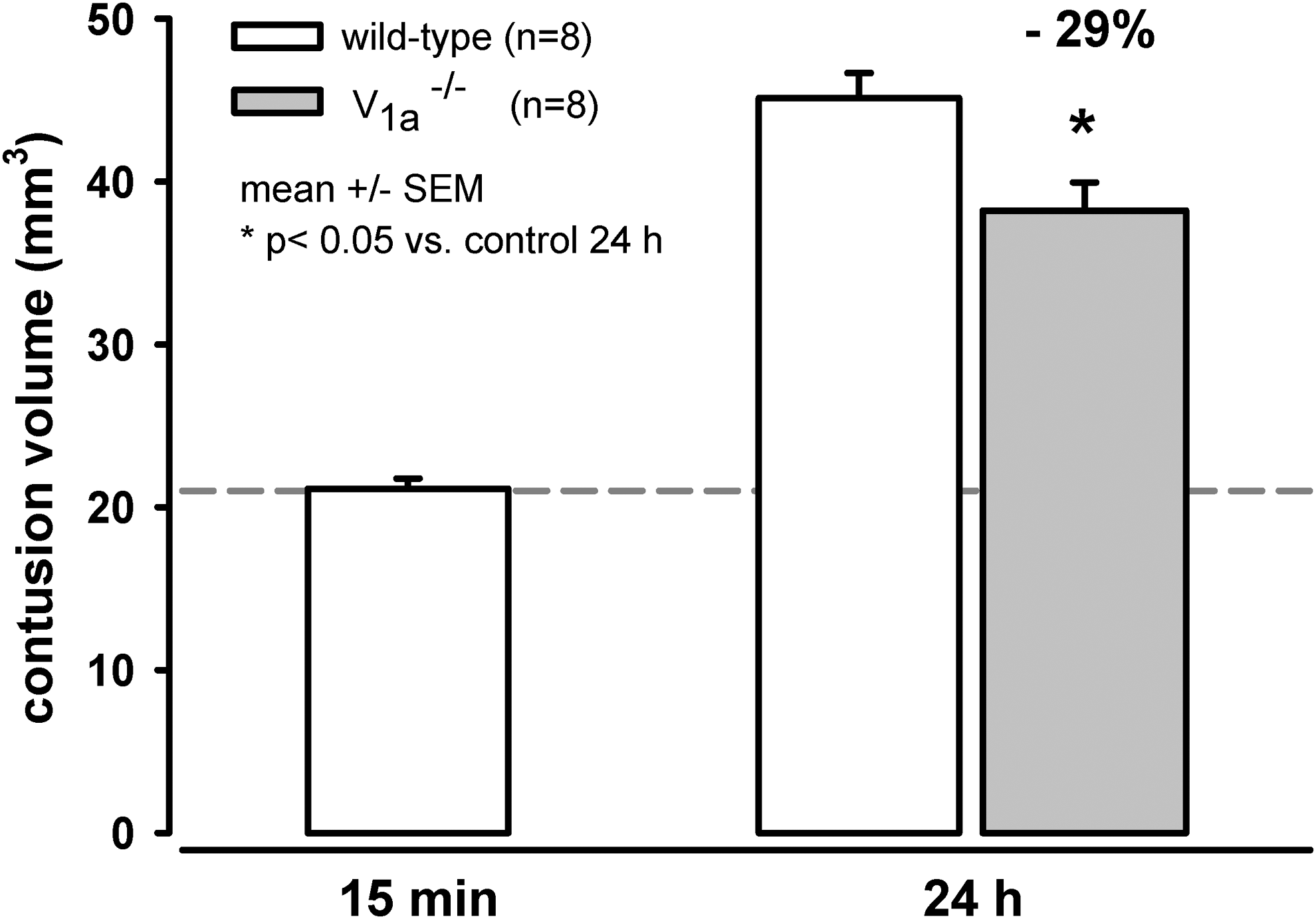
Primary and secondary contusion volume. The volume of the primary lesion was measured 15 min after controlled cortical impact (CCI) in wild-type mice, and the volume of the secondary contusion was measured 24 h after CCI in the wild-type and V1a receptor knock-out mice. The V1a receptor knock-out mice had significantly smaller secondary contusion volume 24 h after CCI (p<0.05), which is a reduction of secondary contusion volume referred to primary lesion by approximately 29%.
V1a receptor knock-out mice have reduced weight loss following controlled cortical impact
Both the V1a −/− and wild-type mice experienced major weight loss within one day of CCI. However, the V1a −/− mice recovered their body weight significantly faster than wild-type mice (p=0.02); seven days post-CCI, the body weight of the wild-type mice was still significantly lower than before CCI; in contrast, the body weight of the V1a −/− mice was not significantly different than their pre-CCI weight (Fig. 3).
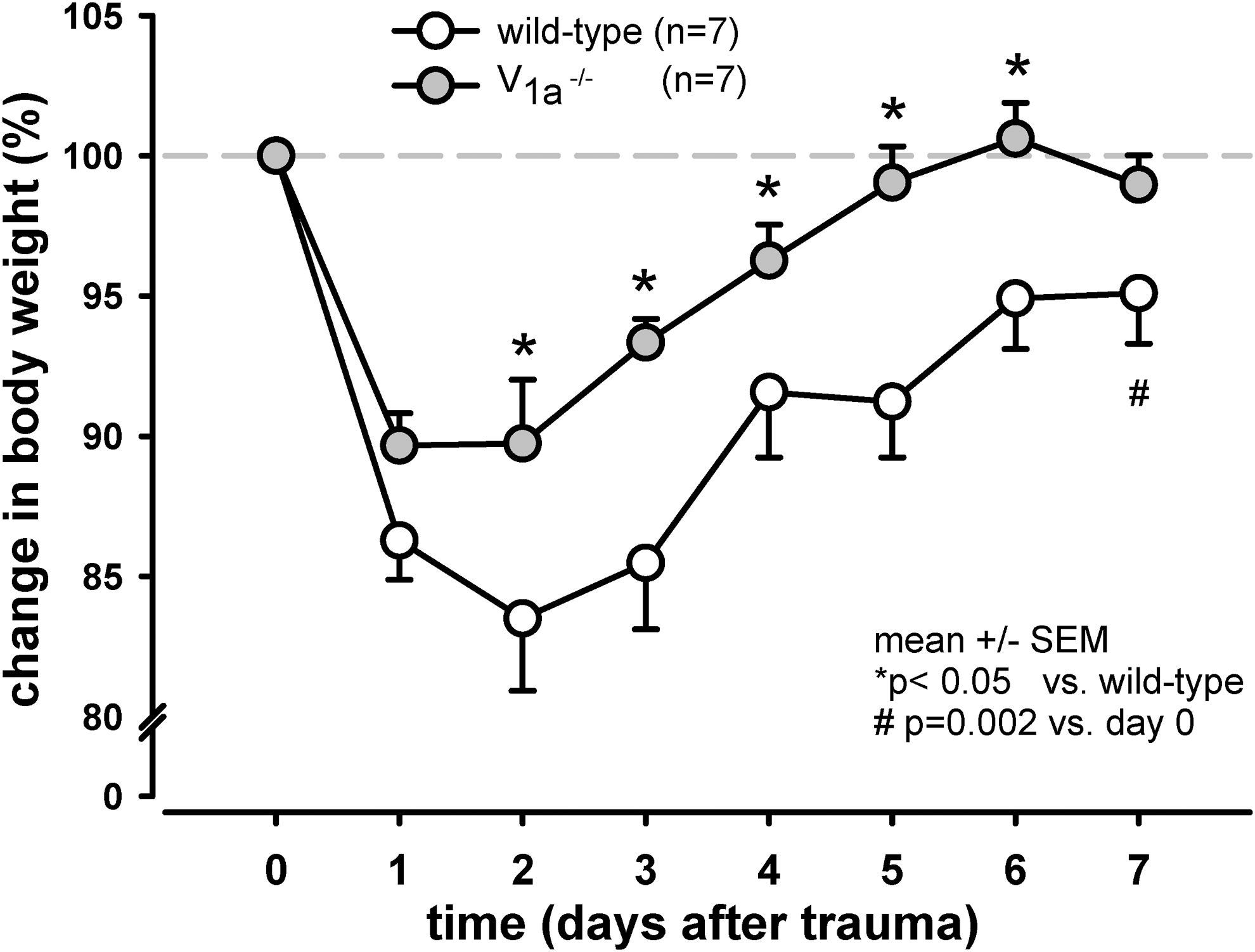
Changes in body weight following controlled cortical impact (CCI). Body weight was measured before and following CCI as a measure of the animals' general state of health. Both the wild-type and V1a receptor knock-out mice had significant weight loss within 1 day of CCI; however, at each time point thereafter, the wild-type mice recovered their body weight significantly less than the V1a receptor knock-out mice (p<0.05). By day 4, the V1a receptor knock-out mice had recovered their pre-trauma body weight, whereas the body weight of the wild-type mice remained significantly decreased even after 7 days (p=0.002).
Improved functional performance in V1a receptor knock-out mice following controlled cortical impact
Although the two groups did not differ in their initial neuroscores after CCI the V1a −/− mice had significant better NSS scores over the subsequent seven days (p<0.05). Even after one day, the V1a −/− had significantly better neurological function than wild-type mice (p=0.017). At seven days post-CCI, the wild-type mice still had significantly impaired neurological function relative to both the V1a −/− mice (p<0.05) and their own baseline performance (p=0.002); in contrast, the neurological function of the V1a −/− recovered within several days, and by seven days post-CCI, NSS was not significantly different than baseline (Fig. 4).
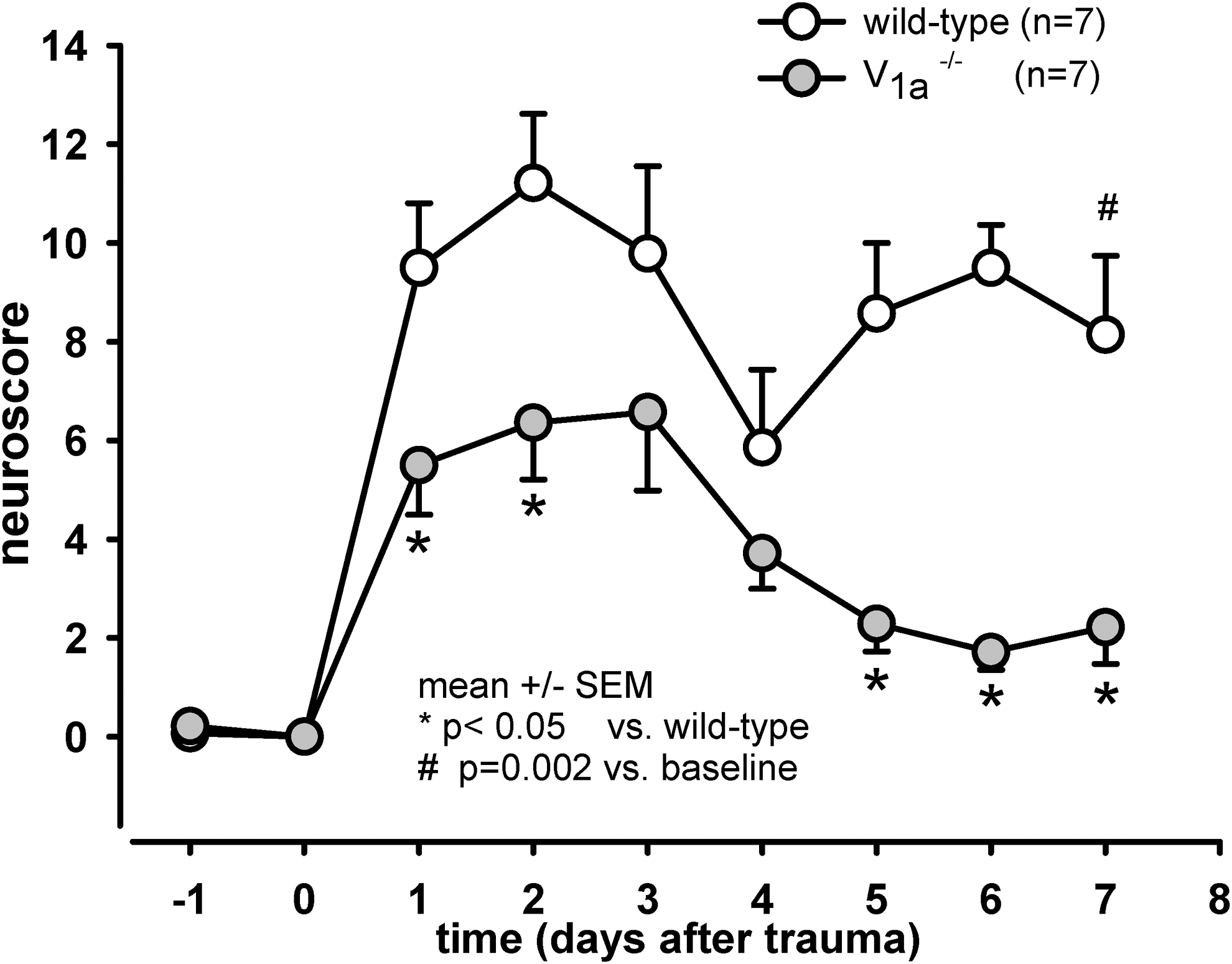
Neurological outcome following controlled cortical impact (CCI). Neurological function was measured using the Neurological Severity Score, in which a score of 19 reflects the worst performance and a score of 0 reflects no neurological dysfunction. Relative to the wild-type mice, the V1a receptor knock-out mice had significantly improved neurological function 1, 2, 5, 6, and 7 days after CCI (p<0.05). After 7 days, the wild-type mice still exhibited significantly impaired neurological function (p=0.002), whereas the knock-out mice recovered by day 4.
Mortality
After seven days (the experimental endpoint), no mice in either group died as a result of CCI (n=7 mice per group).
Discussion
In this study we investigated whether AVP V1a receptors play a role in the development of brain edema and subsequent secondary brain damage following TBI. Our findings demonstrate that mice lacking V1a receptors (V1a −/−) have significantly reduced brain edema and smaller contusion volume compared with wild-type mice, thereby implicating AVP V1a receptors in the development of secondary brain damage following TBI.
Animal model
CCI was developed originally by Lighthall and colleagues and was later adapted for use in mice in order to utilize the power of transgenic mouse lines. 36,37 CCI is now well-characterized and is widely used as a model for TBI. CCI is specifically suited to the investigation of post-traumatic brain edema, as it causes a combination of vasogenic and cytotoxic edema that is highly similar to the outcome observed in patients following TBI. 8
In our knock-out mouse line, the V1a receptor gene was inactivated by removing portions of the first exon and intron. These knock-out mice are reported to have somewhat lower blood pressure without notable changes in heart rate, reduced circulating blood volume, a decreased response to adrenocorticotropic hormone, and possibly reduced social interactions; in contrast, these mice have normal plasma AVP levels and normal AVP responses compared with wild-type mice. 33
Our data suggest that the observed reduction in brain edema in the V1a receptor knockout mice is not due to either systemic or vascular effects of the genetic deletion, as these mice did not differ from wild-type mice with respect to their baseline or post-CCI physiological parameters, including mean arterial blood pressure, cerebral blood flow, and intracranial pressure. These findings, however, need to be interpreted cautiously, since we measured MAP in anesthetized mice and after waking up from anesthesia V1a knock-out mice may well have a slightly lower blood pressure than wild type mice. 33 Whether the previously observed difference of 7% is pathophysiologically relevant remains, however, questionable.
The role of vasopressin in the pathophysiology of traumatic brain injury
Arginine vasopressin (AVP) likely plays a role in several neurological disorders, as serum AVP levels were elevated in humans following traumatic brain injury, and AVP levels were positively correlated with severity of brain damage. 38 –40 Barreca and colleagues reported elevated serum AVP levels that were independent of osmo- and baro-mechanisms in post-ischemic human conditions. 41 In addition, elevated AVP levels in the cerebral spinal fluid (CSF) were found specifically in neurological disorders that have increased intracranial pressure. 42 These reports raise the following questions: What are the mechanisms that lead to increased serum AVP levels, and what mechanisms cause increased AVP concentration in the CSF? Previous studies using cats found increased activity of the hypothalamus-hypophysial-neurosecretory system that was likely due to ischemic anoxia during a sudden rise in intracranial pressure. Strikingly, correlations between intracranial hypertension, systemic arterial pressure and plasma AVP level were observed in experimental cerebral compression that was unrelated to blood osmolality and sodium concentration, but not in experimental subarachnoid hemorrhage (SAH). 43 The latter observation suggests that a different mechanism is involved in TBI—which primarily involves brain edema—than in SAH, which in addition to an acute increase in ICP followed by the central release of AVP likely also requires the Cushing reflex. 44
Recent experimental studies using CCI in rats revealed increased AVP production in the hypothalamus and a dramatic increase in AVP in the cortex adjacent to the post-traumatic lesion, the hippocampus and the corpus callosum. Specifically, AVP production was found predominantly in the microglia and macrophages that are associated with cortical microvessels and—to a lesser degree—in the cerebrovascular endothelium. 45 In addition to these findings, both AVP-containing axonal processes leading from the hypothalamus directly to the lateral ventricles 16 and AVP synthesis in the choroid plexus endothelium have been described, suggesting that AVP can be released into the CSF and modulate autocrine and/or paracrine functions. 46 Taken together, these results suggest that AVP released from the posterior pituitary gland as well as centrally released AVP from microglia, macrophages and cerebrovascular endothelium may play a role in post-traumatic and post-ischemic pathophysiology.
Distribution of cerebral arginine vasopressin receptors
Arginine vasopressin is a ligand for three distinct receptors that belong to a large family of G-protein-coupled heptahelical membrane receptors. 47 The primary focus of the present study was V1a receptors, which are expressed on smooth muscle cells in blood vessels and drive vasoconstriction; in addition, these receptors are expressed widely throughout the central nervous system 48 and are likely involved in the formation of brain edema. 31,32,49 A recent animal study by Szmydynger-Chodobska and colleagues comparing the fronto-parietal cortex in uninjured and brain-injured rats found V1a receptors expressed in neuronal nuclei and astrocytes in uninjured brains. In non-parenchymal cells, the V1a receptor was highly expressed apically in the choroid plexus epithelium. In the injured rat brain, brief transient up-regulation of the V1a receptor was observed in the endothelium, including the microvasculature and large-diameter blood vessels. Furthermore, increased expression of V1a receptors lasting up to 14 days was found in perilesional cortical astrocytes, and the receptors redistributed from the cell bodies to the processes. 50 This is a highly relevant finding, as the time course is on par with the post-traumatic duration of brain edema. Moreover, astrocytic processes form close contacts with the endothelium in the blood-brain-barrier (BBB), and perivascular astrocytic end feet are the principal site where AQP4, the most abundant water channel in the brain, is expressed. 51 –54 In addition, increased V1a receptor expression was found in neurons with enlarged varicosities in their axonal processes, perhaps involving AQP4 regulation. Remarkably, the time courses of AVP and V1a receptor up-regulation are quite similar, suggesting AVP-mediated disruption of the BBB and providing a possible cause for the exacerbated post-traumatic brain edema. 55
Molecular mechanisms
AVP plays an important role in the pathophysiology of brain injury by activating a variety of intracellular signaling cascades and contributing to the downstream consequences of the injury. AVP acts via two major pathways, the V1a receptor-mediated protein kinase C (PKC) and the V2 receptor–mediated protein kinase A pathways. 47 The PKC pathway triggers several downstream events. First, this pathways triggers increased intracellular calcium and vasoconstriction. 11,13 Second, Fos/Jun family proteins, the transcription factor activator protein 1 and the cAMP-responsive element in the AVP promoter region are all activated, inducing transcription of the AVP gene. 56 Third, the transcription factor NF-κB becomes activated. 57 Lastly, as shown by in vitro studies using Xenopus oocytes, a shirt-lived V1a receptor-dependent internalization of AQP4 occurs via a phosphorylation event. 58
In contrast to the study in Xenopus oocytes, studies using mice found that intraventricular V1a receptor antagonists led to up-regulated AQP4 and decreased brain water content 24 h after inducing middle cerebral artery occlusion. 27 Furthermore, following TBI, AVP acts synergistically with tumor necrosis factor α to up-regulate pro-inflammatory mediators such as chemokines and matrix metalloproteinase 9 via the c-Jun N-terminal kinase pathway, leading to increase influx of inflammatory cells and a disruption in the BBB, among other effects. 59
In addition to the aforementioned mechanisms, AVP stimulates the V1 receptor–mediated and calcium-dependent activity of the cerebral microvascular endothelial Na+/H+ exchanger as well as the Na+-K+-2Cl– co-transporter, both of which play a role in brain edema. 60,61 These results reveal several molecular mechanisms that need to be investigated. For example, further investigation of the intracellular V1a receptor pathway is needed. In particular, studies of the short- and long-term regulation of aquaporins could reveal their contribution to the development of posttraumatic brain edema.
Conclusions
Our experiments using V1a receptor knock-out mice show that inhibiting the V1a receptor may serve to reduce posttraumatic brain edema and secondary brain damage following TBI. We therefore suggest that the V1a receptor may have potential as a novel drug target for preventing acute post-traumatic brain edema. Moreover, our findings suggest that vasopressin should not be administered to patients following TBI. Future studies will analyze the time course and further characterize the molecular mechanisms that regulate brain aquaporins.
Footnotes
Acknowledgments
We thank Gozoh Tsujimoto, Kyoto University, Japan, for providing the V1a receptor knock-out mice, Eckart Thein and staff members for careful breeding of our knock-out animals and Uta Mamrak for excellent technical assistance. The presented results are part of KR's doctoral thesis.
Author Disclosure Statement
No competing financial interests exist.