
Abstract
Use of improvised explosive devices has significantly increased the incidence of traumatic brain injury (TBI) and associated neuropsychiatric deficits in the recent wars in Iraq and Afghanistan. Acute deleterious effects of single and repeated blast exposure can lead to long-term neurobiological effects and neuropsychiatric deficits. Using in vitro and in vivo shock tube models of blast-induced TBI, we studied changes in mitochondrial energy metabolism after blast exposure. Single and repeated blast exposures in vitro resulted in significant decreases in neuronal adenosine triphosphate (ATP) levels at 6 h post-blast that returned towards normal levels by 24 h. Similar changes in ATP also were observed in the cerebral cortices of mice subjected to single and repeated blast exposures. In neurons, mitochondrial glutamate oxaloacetate transaminase (GOT2) plays a critical role in metabolism and energy production. Proteomic analysis of brain cortices showed a significant decrease in GOT2 levels 6 h after repeated blast exposures, which was further confirmed by Western blotting. Western blot analysis of GOT2 and pyruvate dehydrogenase in the cortex showed direct correlation only between GOT2 and ATP levels. Activity of GOT2 in the isolated cortical mitochondria also showed significant decrease at 6 h supporting the results of proteomic and Western blot analyses. Knowing the significant role of GOT2 in the neuronal mitochondrial energy metabolism, it is quite likely that the down regulation of GOT2 after blast exposure is playing a significant role in mitochondrial dysfunction after blast exposure.
Introduction
Due to the complexity and unique physical forces responsible for blast-induced TBI, it is now widely believed that the TBI resulting from blast exposure is relatively distinct from other closed head or penetrating brain injuries. 4 The primary, secondary, tertiary, and quaternary injury phases of blast exposure are believed to contribute to the multifaceted mechanisms involved in blast TBI. Although several clinical and animal studies have explored the biochemical/histopathological changes and behavioral deficits resulting from blast exposure, 3,5 –11 the complex biochemical and molecular mechanisms of blast TBI and how it triggers subsequent secondary pathological processes and long-term neurobehavioral abnormalities are still not well understood. The lack of understanding of the precise mechanisms involved in blast TBI has hampered the development of personal protective gear and specific diagnostic tools for early detection and effective therapies for prevention/treatment.
Acute effects of blast exposure are not well studied because most of the significant pathological changes in the brain manifest during the secondary injury processes, which occur typically by 24 h post-blast exposures. Using an in vitro model of blast-induced TBI, we have shown that single and repeated blast exposures lead to transient changes in neuronal cell membrane integrity, which may be a potential mechanism contributing to the secondary injury processes. 12 Transient changes in cell membrane integrity also have been reported in the liver and muscle tissue after single and repeated blast exposures. 13 Acute deleterious effects of any direct or indirect insult on the brain can trigger secondary pathological changes, which lead to chronic neurobehavioral abnormalities. Thus, minimizing or preventing acute changes after brain injury is important to avert chronic neurobehavioral deficits.
In most of the cell types in the body, adenosine triphosphate (ATP) synthesis takes place through utilization of glucose, and mitochondrial pyruvate dehydrogenase (PDH) plays an important role. In the case of brain, amino acids—especially glutamate—also are utilized for the immediate and significant demands for ATP for energy. 14 –16 In one study, removal of glucose significantly increased the transamination of glutamate to aspartate in brain synaptosomes and suggested that brain cells utilize glutamate, the most abundant molecule in the brain, for energy production. 15 Mitochondrial glutamate oxaloacetate transaminase (GOT2) is the enzyme responsible for the transamination of glutamate to aspartate and α-ketoglutarate. Since α-ketoglutarate can directly enter the citric acid cycle to generate ATP, GOT2 is believed to play major role in ATP production in neurons. 14 –16
In the present study, using the in vitro and in vivo models of blast-induced TBI, we studied changes in mitochondrial energy metabolism after blast exposure. We first analyzed ATP levels in the neuronal cells in culture and brain cortex after blast exposure to demonstrate mitochondrial energy dysfunction. Decreases in ATP levels were further compared between single and multiple blast exposures to determine its dependency upon the severity of injury. Since GOT2 plays a central role in neuronal mitochondrial energy metabolism, we investigated its expression in the brain by proteomic analysis. Modulation of GOT2 was further analyzed by Western-blotting of the brain cortex and enzyme activity analysis in the isolated mitochondria from the cerebral cortex to demonstrate a potential role of GOT2 in the acute mitochondrial dysfunction after blast exposure.
Methods
Cell culture and blast exposure
SH-SY5Y human neuroblastoma cells, Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from American Type Culture Collection (Manassas, VA). One of the major advantageous of SH-SY5Y cells over primary cells and brain slices (derived from animals) is that the effect of blast exposure can be studied in cells of human origin. Cells were grown in DMEM with 10% heat inactivated FBS containing penicillin and streptomycin. The cells incubated at 37°C in a carbon dioxide (CO2) incubator kept at 5% CO2 and 95% air in a humidified atmosphere. Cells (4×104 cells/well) were grown on 96 well tissue culture plates 24 h before blast exposure. On the day of blast exposure, the medium was removed from the wells and fresh medium (360 μL) was added to fill the wells. Before blast exposure, the plates were sealed with gas permeable Mylar plate sealers as described previously. 12,17 The plates containing cells were subjected to single and triple blast exposures (21 psi) using shock tube as described earlier. 12,17 Intracellular adenosine triphosphate (ATP) content in the cells was determined at 6 and 24 h post-blast using ATPlite kits (Perkin Elmer, Waltham, MA) as previously described. 17 The ATPlite assay system is based on the production of luminescence caused by the reaction of ATP with added luciferase enzyme and D-luciferin substrate.
Animals and blast exposure
All animal experiments were conducted in accordance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to principles stated in the Guide for the Care and Use of Laboratory Animals (National Research Council Publication, 1996 edition). The animal protocol used was approved by Institutional Animal Care and Use Committee, Walter Reed Army Institute of Research. C57BL/6J male mice (8–10 weeks old) that weighed between 21–26 g (Jackson Laboratory, Bar Harbor, ME) were used in this study. Mice were exposed to single and triple blasts using a shock tube as described earlier. 3 Briefly, mice were anesthetized with 4% isofluorane gas (oxygen [O2] flow rate 1.5 L/min) for 8 min and restrained in the prone position with a net to minimize the movements during blast exposure. Animals were subjected to single or triple blast exposures (21 psi) and brain cortex was dissected after euthanasia at 1, 6, or 24 h post-blast.
ATP determination in the brain cortex
Homogenate (20% w/v) of brain cortex was made in tissue protein extraction buffer (Pierce Chemical Co, Rockford, IL) containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO). The homogenate was centrifuged at 5000 g for 5 min and the ATP content in the supernatant was measured using ATPlite kit as described earlier. 18
Proteomic analysis
Proteomic analysis of brain cortex was carried out as described by us earlier. 19 Briefly, proteins were isolated from the brain cortex of sham control and repeated blast exposed mice (3 animals/group) at 6 h post-blast using the ToPI-DIGE™ total protein isolation kit (ITSI-Biosciences, Johnstown, PA) and subjected to two-dimensional differential in-gel electrophoresis (2D-DIGE). After 2D-DIGE, the differentially-expressed (>2-fold change in abundance) protein spots were identified, selectively picked, and subjected to protein digestion followed by LC/MS/MS to identify the peptides. The obtained MS/MS spectra were searched against the National Center for Biotechnology Information non-redundant protein sequence database using the SEQUEST computer algorithm to establish the protein identity.
Western blotting analysis
Polyclonal rabbit antibodies against GOT2 and pyruvate dehydrogenase (PDH) were obtained from Sigma-Aldrich (St. Louis, MO) and Abcam (Cambridge, MA). Secondary antibody labeled with horse-radish peroxidase (HRP) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibody to β-actin conjugated with HRP (Sigma-Aldrich, St. Louis, MO) was used as a control. Polyacrylamide gel electrophoresis and Western blotting analysis of brain cortex was carried out as described by us previously. 19 Both GOT2 and PDH antibodies were used at a final dilution of 1:1000. After Western blotting analysis, the protein bands were detected using ECL-Plus Western blot detecting reagent (GE Healthcare, Piscataway, NJ) and the chemiluminescence was measured in an AlphaImage reader (Cell Biosciences, Santa Clara, CA).
Assay of GOT2 activity in the mitochondrial fraction of brain cortex
Mitochondrial fraction was isolated from the cerebral cortex using Mitochondria Isolation Kit obtained from Thermo Scientific (Rockford, IL) according to the manufacturer's instructions. The mitochondrial pellet was suspended in the enzyme assay buffer and disrupted using a sonifier for the enzyme assay. The activity of GOT2 was measured using the diagnostic kit for measuring GOT, obtained from Randox Laboratories (Kearneysville, WV) according to the manufacturer's instructions. The assay system utilizes the decrease in optical density at 340 nm due to the consumption of NADH during the formation of oxaloacetate and glutamate from aspartic acid and α-ketoglutaric acid catalyzed by GOT.
Statistical analysis
Statistical analysis was carried out by analysis of variance (ANOVA) using SAS software version 9.3 (SAS Institute, Inc., Cary, NC). For single variance, one-way ANOVA followed by t test was carried out. For multiple variance, two-way ANOVA followed by Tukey's post-hoc test using HSD multiple comparisons were used. A p value of less than 0.05 was considered significant.
Results
Blast exposure leads to acute decrease in neuronal ATP levels
Blast exposure causes a significant decrease in endogenous ATP levels in SH-SY5Y human neuroblastoma cells in a time-dependent manner with the highest decrease at 6 h post-blast, compared with 24 h post-blast (Fig. 1). Neuronal ATP levels were further decreased with multiple blast exposures. After single blast exposure, the ATP level decreased by 17.7% at 6 h, whereas no significant decrease was observed at 24 h post-blast. In the case of triple blast exposures, the ATP levels decreased by 44.6% and 27.7% by 6 h and 24 h, respectively.

Changes in adenosine triphosphate levels in SH-SY5Y cells at 6 h
ATP levels decreased in the cerebral cortex after blast exposures
Figure 2 shows the changes in ATP levels in the mouse brain cortex at different intervals after single and repeated blast exposures. Similar to the in vitro results, blast exposure of mice also resulted in a decrease in brain ATP levels, which varied relative to the time and number of blast exposures. Single blast exposure resulted in a 19.5% decrease in ATP levels at 6 h, whereas triple blasts resulted in a 23.4% decrease as early as 1 h. ATP levels were decreased by 39.7% at 6 h after repeated blast exposures, whereas the decrease was only 11.8% at 24 h post-blast. Thus, compared with by 6 h post-blast, the ATP levels were higher in the brain by 24 h post-blast but still lower than the sham controls which were not exposed to blast.
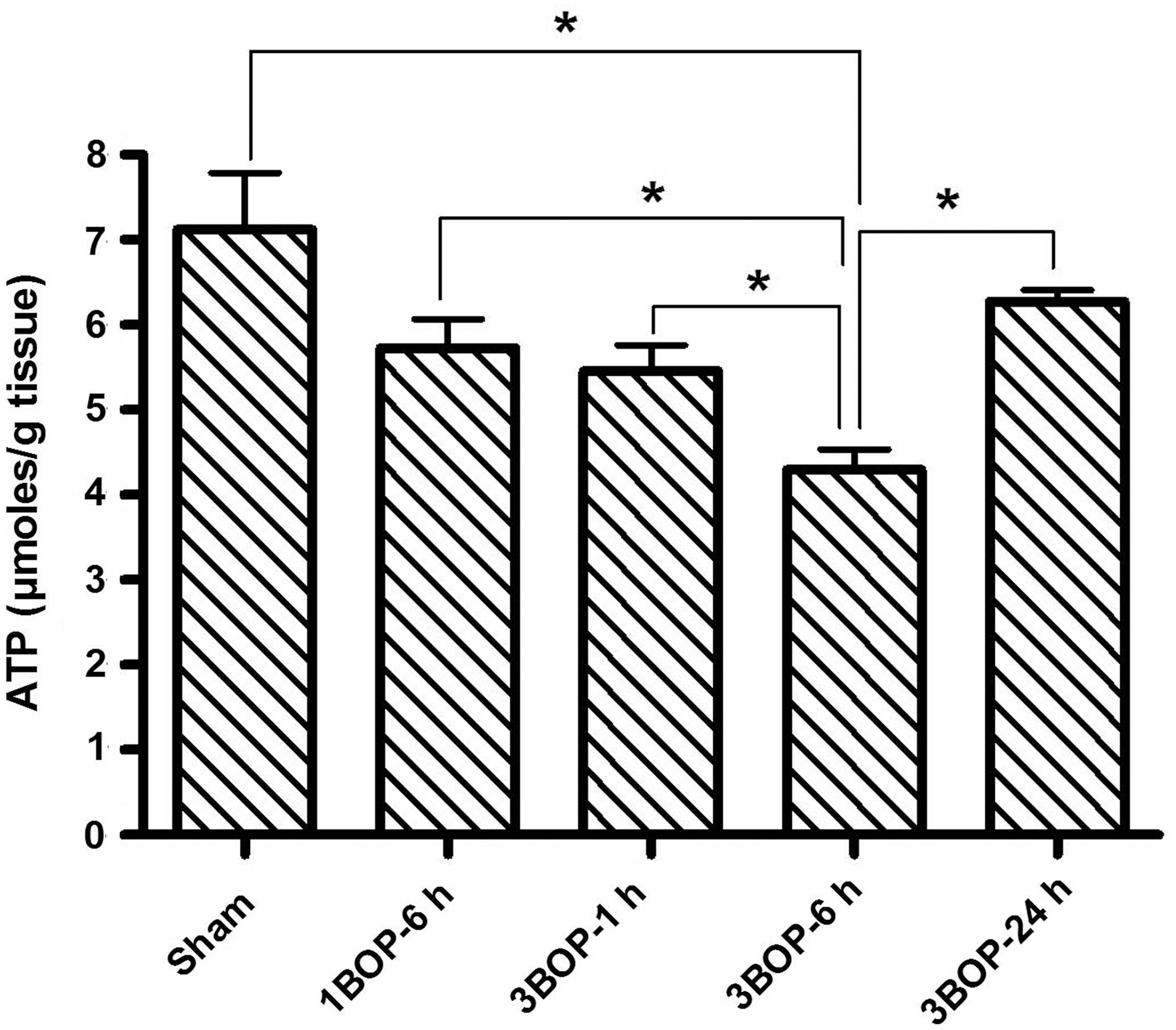
Changes in adenosine triphosphate levels in cerebral cortex at different intervals after single and repeated blast exposures. Values are expressed as mean±standard deviation. *p<0.05 (n=6). BOP, blast overpressure exposure.
Proteomic analysis of the cerebral cortex to identify modulation of GOT2 expression
Proteomic analysis data showed few proteins with >2 fold change in the cerebral cortex at 6 h after triple blast exposures. The proteins which showed >2 fold increase in expression after blast exposure include 14-3-3 protein gamma, calretinin, parvalbumin alpha, 14-3-3 protein zeta/delta, and calpastatin. One of the down-regulated proteins was identified by LC/MS/MS analysis as GOT2 by matching five peptides. Figure 3 shows the image of the 2D-gel portion showing the expression of GOT2 in the cortex after repeated blast exposures. GOT2 expression showed a statistically significant abundance ratio (3.13±0.76) between sham control and blast exposed mice suggesting that blast exposure alters the expression of GOT2.
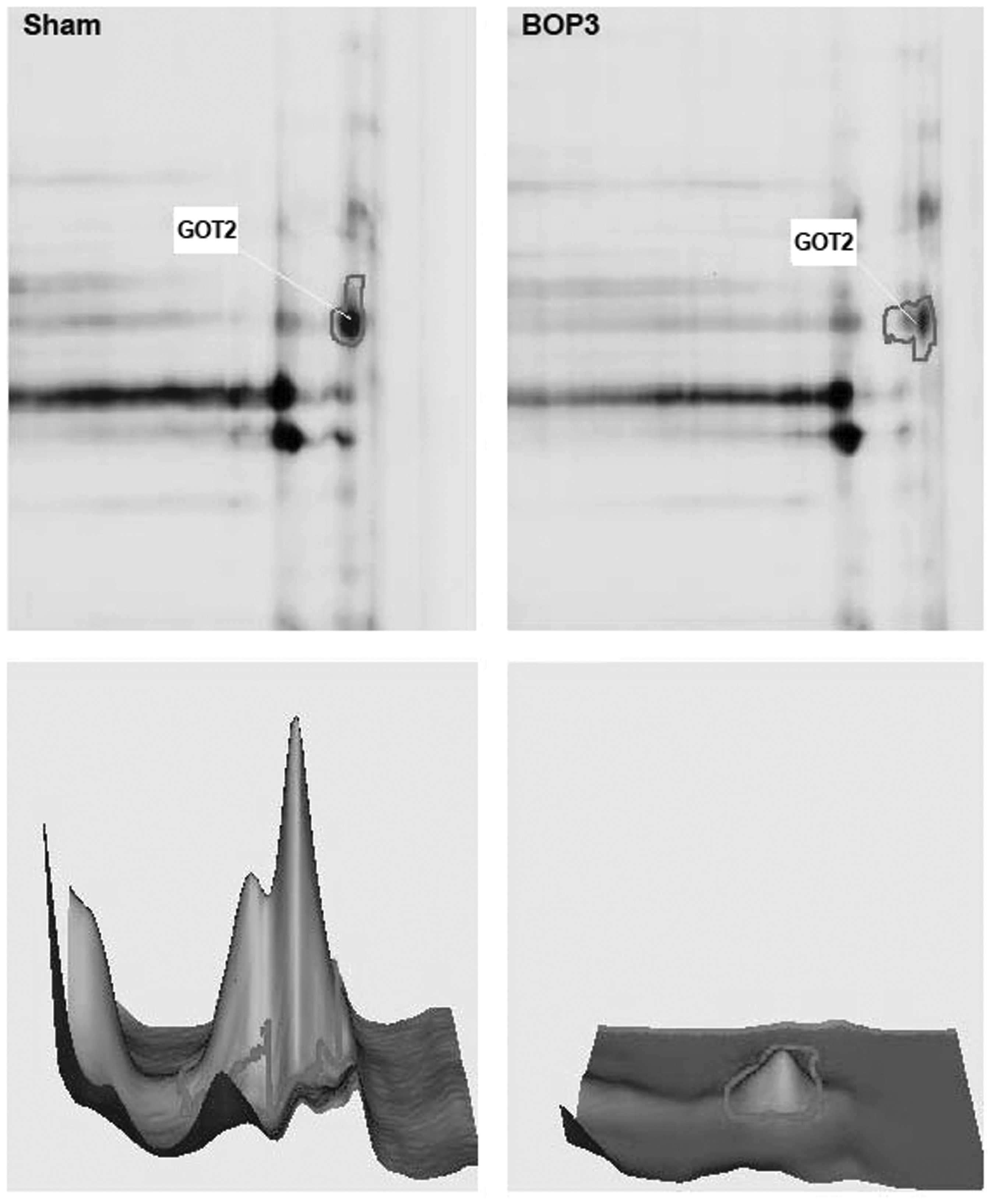
Representative two-dimensional differential in-gel electrophoresis figure from three different animals in the sham control and blast exposed groups focusing the spot of glutamate oxaloacetate transaminase. Procedures used are detailed in the methods section. The bottom two panels show the sum of pixel intensities within the boundary of each spot in the fluorescent image to demonstrate the level of the protein.
Western blotting of GOT2 and PDH in the cerebral cortex after blast exposure
Western blotting using antibodies specific to GOT2 showed a significant decrease (47.9%) in the expression of GOT2 in the cerebral cortex at 6 h after triple blast exposures, compared with sham controls (Fig. 4A). The ratio of GOT2 to actin was 0.161±0.025 for sham controls, whereas the ratios were 0.084±0.028 for 6 h (p=0.021) and 0.142±0.036 for 24 h (p=0.44; Fig. 4B). Compared with sham controls, single blast exposure at 6 h and triple blast exposure at 1 h, respectively, showed 20% (ratio of GOT2 to actin was 0.124±0.016, p=0.032) and 29.9% (ratio of GOT2 to actin was 0.113±0.032, p=0.036) decreases in the expression of GOT2 in the cortex. Thus, similar to the reduction in ATP levels, the expression of GOT2 decreased in the cerebral cortex after blast exposure. The decrease in GOT2 expression depended upon the number of blast exposures, as well as the time after blast exposure; lowest expression was recorded at 6 h and returned toward normal levels by 24 h post-blast.
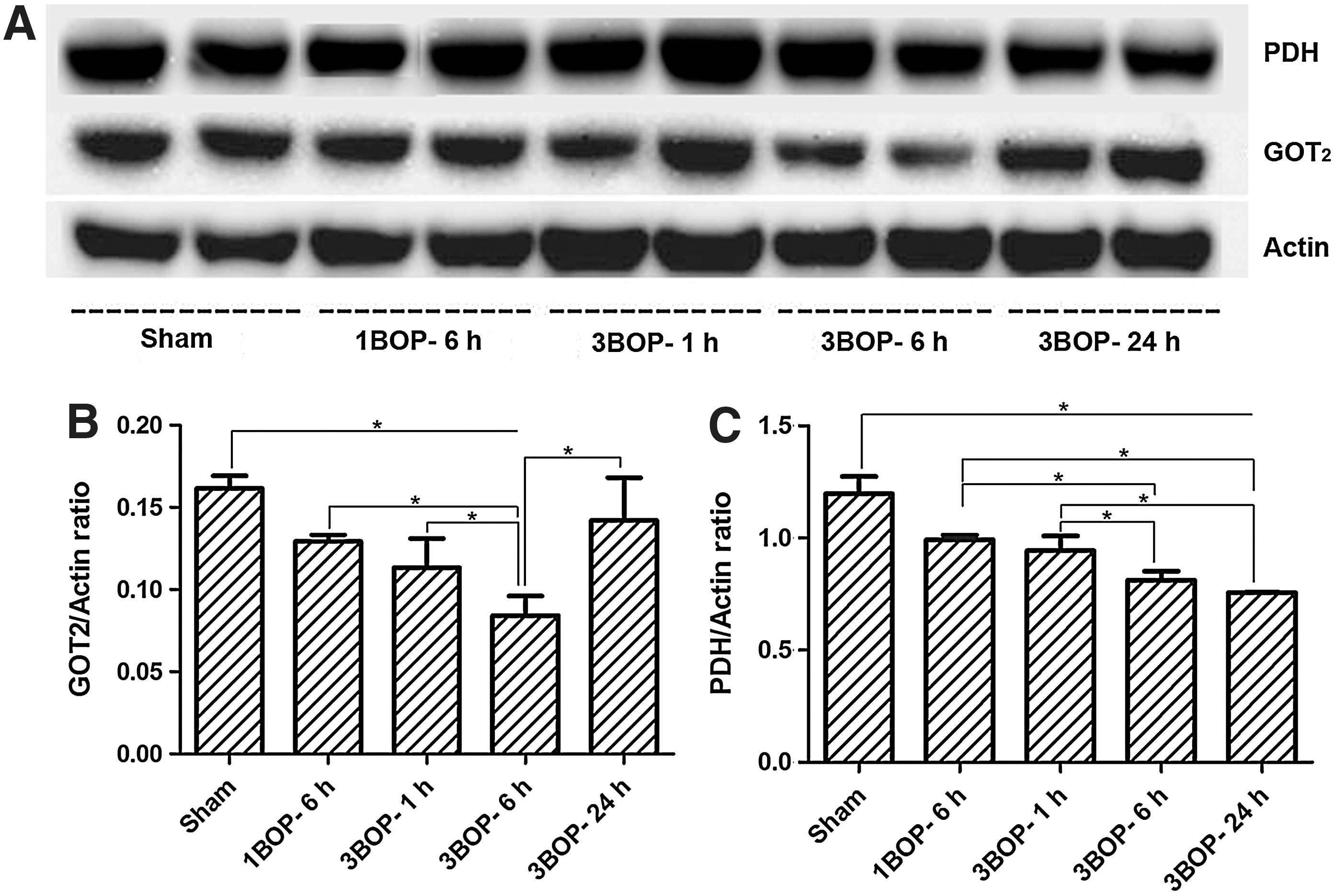
Western blotting analysis of mice cerebral cortex at different intervals after single and repeated blast exposures.
Western blotting of PDH with specific antibodies (Fig. 4A, C) showed significant decreases in the cerebral cortex at 6 h post-blast exposure. The decrease in the PDH level does not seem to correlate with ATP levels at 24 h after blast exposure.
Cerebral cortex mitochondrial GOT2 activity after blast exposure
To determine further that the cerebral cortex mitochondrial GOT2 activity was indeed decreased after blast exposure, the mitochondrial fraction was isolated from the cerebral cortex to avoid interference from the cytosolic isoform, GOT1. Disrupted mitochondrial sample was used for measuring GOT2 activity as described in the methods. The activity of GOT2 in the mitochondrial fraction showed a 28.8 % decrease at 1 h and a 41.4 % decrease at 6 h after triple blast exposures. No significant changes in mitochondrial GOT2 activity were observed at 24 h post-blast (Fig. 5).
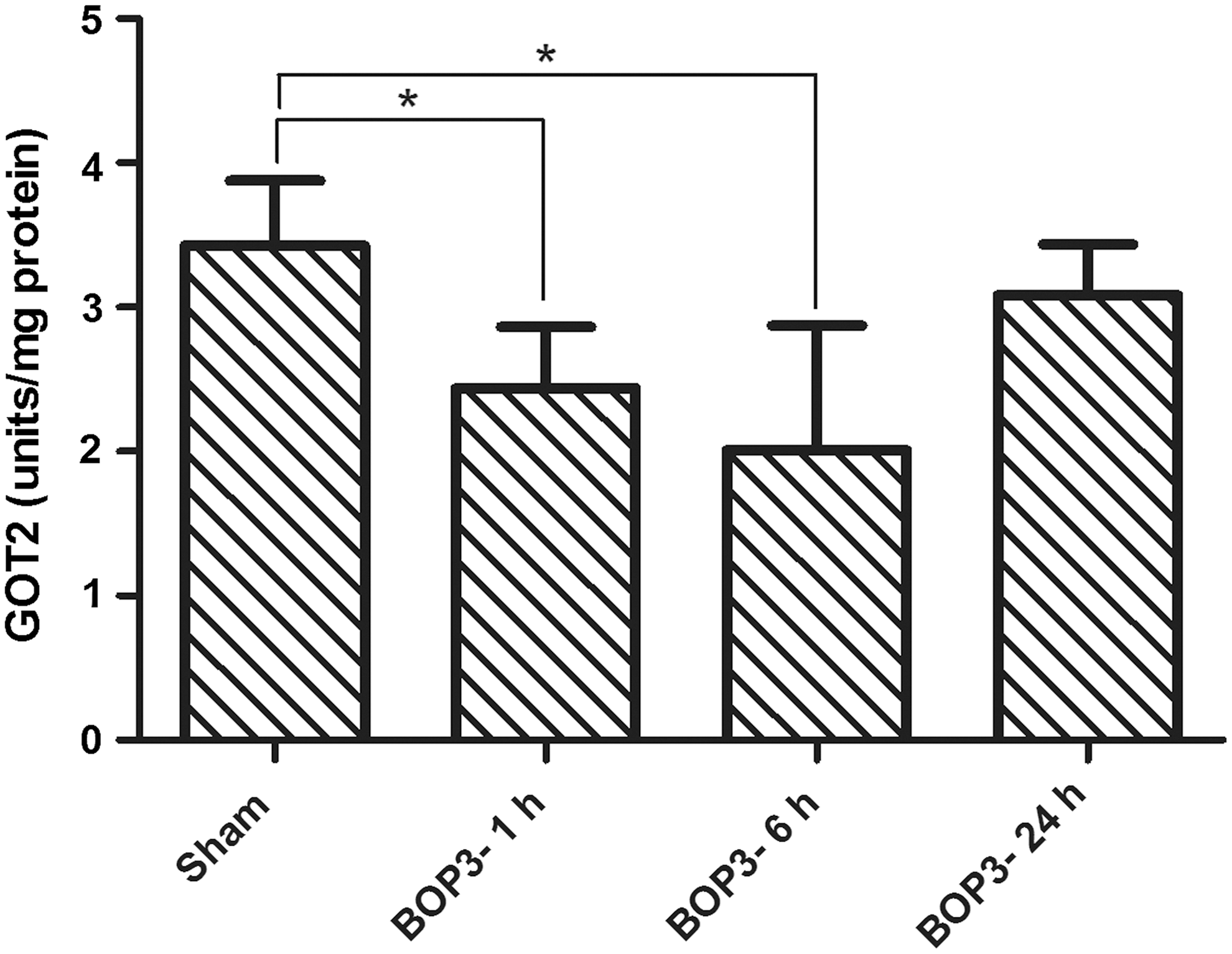
Activity of glutamate oxaloacetate transaminase in the mitochondrial fraction of mice cerebral cortex at different intervals after repeated blast exposures. Values are mean±standard deviation (*p<0.05, n=6).
Discussion
Our results show for the first time in an animal model that blast exposure leads to an acute mitochondrial dysfunction and an associated significant decrease in ATP levels in the brain. After a rapid decrease, the brain ATP levels return toward normal levels at 24 h post-blast exposures. Using the in vitro model of blast-induced TBI with NG108–15 cells (combination of neuroblastoma and glioblastoma cells), we have shown in a previous study that ATP levels decrease at 24 h after blast exposure. 17 Time points earlier than 24 h were not evaluated in that study. In the current study, we observed that a decrease in ATP levels after blast exposure occurs immediately and was most pronounced at 6 h, compared with at 24 h, indicating severe neuronal mitochondrial dysfunction acutely after blast exposure. The changes in ATP levels after blast exposure were similar in vitro and in vivo.
Acute mitochondrial dysfunction in the brain has been reported in different animal models of TBI. 18,20 –22 Unlike blast-induced TBI, the mitochondrial dysfunction and associated decrease in ATP levels persisted for longer durations in the other forms of TBI. In a controlled cortical impact model of TBI, the ATP levels ipsilateral to the injury were only about 22 % of sham control even after six days post-injury. 18 In that study, the ATP levels measured ipsilaterally may have been associated with significant cell/tissue loss and the cells adjacent to the site of injury might have been undergoing necrosis/apoptosis. In the present study, we have shown that mitochondrial dysfunction and impaired energy production in the brain after blast exposure takes place acutely and the energy levels return toward normal levels by 24 h post-blast. This could be due to the lack of significant tissue loss in the brain after blast exposure. 3 Since delayed neuronal degeneration occurs after blast exposure, 1 it is possible that there could be a secondary phase of mitochondrial dysfunction after blast exposure, which would become evident several days post-exposure.
In the present study, we found a correlation between the decrease in brain ATP levels and the reduced expression/activity of mitochondrial GOT2. The protein expression and activity of GOT2 were lowest in the brain at 6 h post-blast and were associated with the maximum decrease in ATP levels. The GOT2 levels returned to almost normal by 24 h and the ATP level also was significantly restored. These data suggest a potential role of GOT2 in the acute brain mitochondrial dysfunction associated with blast exposure. Detailed study is required to find out whether the synthesis or degradation of GOT2 in the brain is affected after blast exposure. Using the same model system, it has been reported that modest levels of neuronal cell death occur immediately after repeated blast exposures, which also could contribute to the decreased tissue levels of ATP and GOT2 measured after blast exposure. 3 The documented proliferation of astrocytes and microglia in the brain at 24 h after repeated blast exposure 11 can similarly contribute to the apparent recovery of ATP and GOT2 by 24 h after repeated blast exposures.
A significant role for PDH in mitochondrial dysfunction after TBI has been reported in different brain injury models other than blast TBI. 23,24 Following controlled cortical impact (CCI) in rats, Opii and colleagues showed that PDH activity decreases in the cortex due to oxidative modification of the enzyme. 24 In another CCI study, the level of PDH decreased in the ipsilateral and contralateral sides of the brain at 4 h and decreased further by 24 h post-injury, similar to our present results following blast exposure. 25 Phosphorylation of PDH (p-PDH) has been implicated in the decrease of its activity in the CCI model. 25 After CCI injury, the ratio of p-PDH/PDH decreased at 4 h, increased at 24 h and decreased again at seven days indicating that the activity of PDH will be less at 24 h, compared with at 4 h and at seven days. 25 An imbalance in the activities of PDH kinase and PDH phosphatase in the brain has been implicated in the decreased activity of PDH after CCI. 26 A significant decrease in PDH also was observed in the brain at 72 h following fluid percussion injury, which was the single time point studied. 27 Our results in the blast TBI model also showed decreased expression of PDH, but did not show a direct correlation with the changes in ATP levels.
The potential role of GOT2 in mitochondrial energy metabolism in the brain has been described earlier. 14,28 –30 Figure 6 shows the schematic representation of the role of GOT2 in brain energy metabolism involving the “mini citric acid cycle,” which was proposed by Yudkoff and colleagues in 1994. 16 Using radiolabeled aspartate and glutamine in brain synaptosomal preparations, Yudkoff and colleagues showed that amino acids could provide an alternate source of energy to help maintain ATP levels in the brain through “mini citric acid cycle”. 16 They also found that in the brain synaptosomes, the fastest reaction that provides metabolite input to the citric acid cycle is that of GOT2. 16 The “mini citric acid cycle” in the brain utilizes glutamine/glutamate for energy production instead of pyruvate, and bypasses a few initial steps in the regular citric acid cycle for faster energy production as required for neuronal activity. 14 –16 As shown in the Figure 6, GOT2 plays a major role in the truncated citric acid cycle. The excess aspartate formed through the “mini citric acid cycle” is used for N-acetyl aspartate (NAA) synthesis in the mitochondria, 14 which also explains the high concentrations of glutamate and NAA in the brain, where they are the most abundant molecules. By using GOT2 to convert glutamate to α-ketogutarate, neurons avoid the formation of toxic ammonia, which is critical in the absence of a urea cycle in the brain. 14,28 Thus, GOT2 plays a significant role in neuronal energy metabolism and its down regulation after blast exposure might contribute to the decreased ATP levels in the brain. Since low levels of ATP can significantly affect neuronal function, pharmaceutical interventions, which can rapidly restore or provide ATP rapidly to neurons, would potentially be beneficial after blast TBI. In this connection, supplementation to the brain of acetate, which can generate ATP, significantly increased brain ATP levels and improved motor performances in rats subjected TBI using CCI. 18
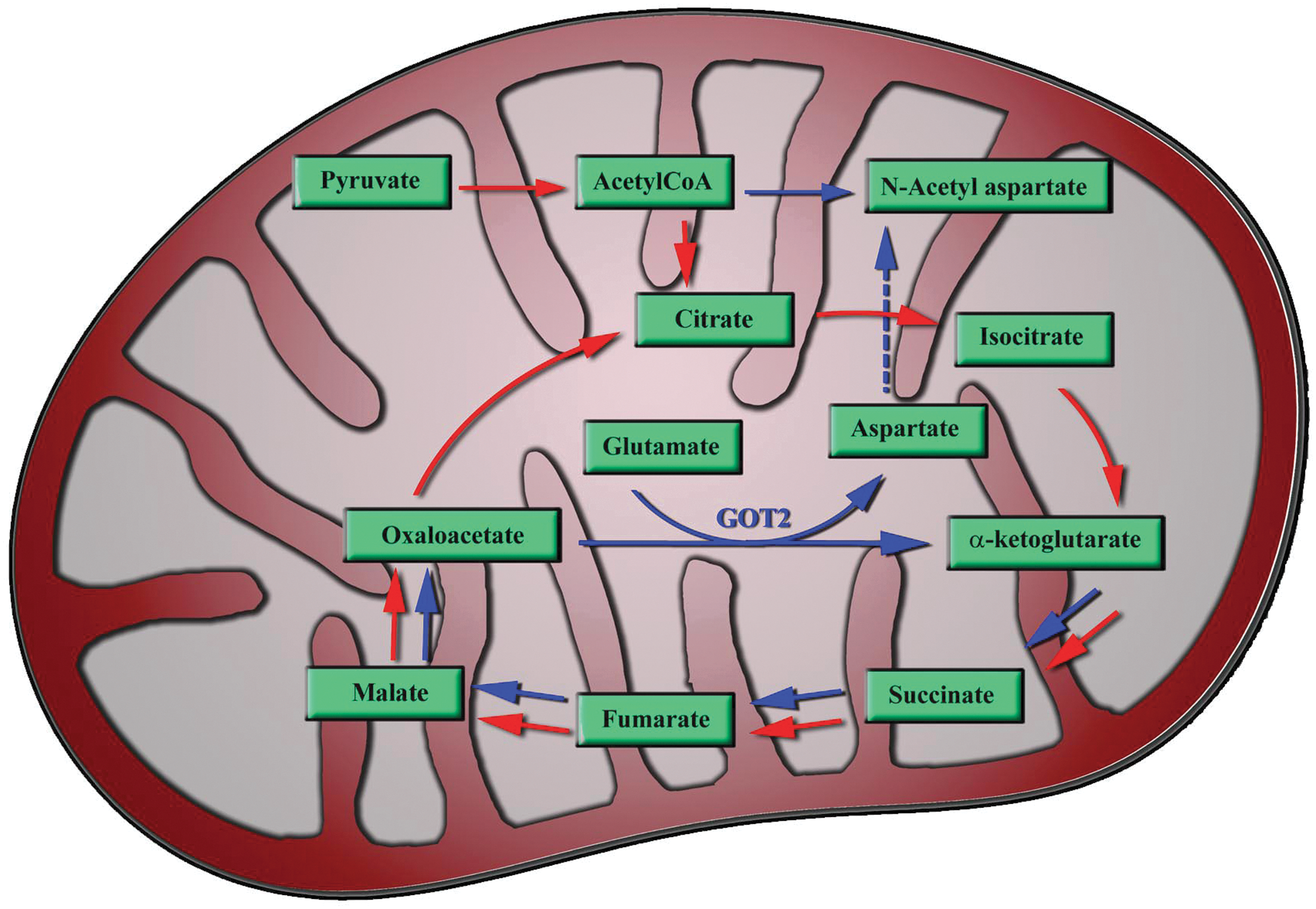
Schematic representation of the neuronal mitochondria showing the proposed “mini citric acid cycle” involving glutamate oxaloacetate transaminase. Red arrow shows the regular citric acid cycle pathway and the blue arrow shows the “mini citric acid cycle.” Most of the energy producing steps is in the “mini citric acid cycle.”
Disclaimer
The contents, opinions and assertions contained herein are private views of the authors and are not to be construed as official or reflecting the views of the Department of the Army or the Department of Defense.
Acknowledgments
Support from Mrs. Irene Gist, Blast-induced Neurotrauma Branch, COL, Paul Bliese, Director, Center for Military Psychiatry and Neurosciences at the Walter Reed Army Institute of Research, and Mrs. Patricia Stroy is gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist