
Abstract
This study evaluated the effects of clinically relevant concentrations of amantadine (AMT) on cognitive outcome and hippocampal cell survival in adult rats after lateral fluid percussion traumatic brain injury (TBI). AMT is an antagonist of the N-methyl-D-aspartate-type glutamate receptor, increases dopamine release, blocks dopamine reuptake, and has an inhibitory effect on microglial activation and neuroinflammation. Currently, AMT is clinically used as an antiparkinsonian drug. Amantadine or saline control was administered intraperitoneally, starting at 1 h after TBI followed by dosing three times daily for 16 consecutive days at 15, 45, and 135 mg/kg/day. Terminal blood draws were obtained from TBI rats at the time of euthanasia at varying time points after the last amantadine dose. Pharmacokinetics analysis confirmed that the doses of AMT achieved serum concentrations similar to those observed in humans receiving therapeutic doses (100–400 mg/day). Acquisition of spatial learning and memory retention was assessed using the Morris water maze (MWM) on days 12–16 after TBI. Brain tissues were collected and stained with Cresyl-violet for long-term cell survival analysis. Treatment with 135mg/kg/day of AMT improved acquisition of learning and terminal cognitive performance on MWM. The 135-mg/kg/day dosing of AMT increased the numbers of surviving CA2–CA3 pyramidal neurons at day 16 post-TBI. Overall, the data showed that clinically relevant dosing schedules of AMT affords neuroprotection and significantly improves cognitive outcome after experimental TBI, suggesting that it has the potential to be developed as a novel treatment of human TBI.
Introduction
T
Currently, there are no approved pharmacological interventions to treat the acute or chronic phases of TBI. In the acute phase, nonsurgical treatment of severe TBI (sTBI) is mainly focused on relieving intracranial pressure (ICP), because elevated ICP pressure is an independent predictor of poor outcome. These treatments include intravenous mannitol, cerebral spinal fluid drainage, hypertonic saline, and hypothermia. Though some of these therapies have been demonstrated to relieve ICP, their effects on improving outcome have yet to be demonstrated in large, well-designed randomized trials. 6,7 Treatment of the long-term, chronic effects of TBI has been primarily symptomatic in focus. Various studies have evaluated the use of methylphenidate to address motor and attention deficits, 8 donepezil to improve visual and verbal memory, 9 and sertraline to relieve depression and pseudobulbar palsy. 10,11 However, solid data on the symptomatic treatment of TBI in patients from well-designed clinical trials were lacking until recently.
Of the pharmacological interventions evaluated for the treatment of TBI, amantadine (AMT) is the only drug that has demonstrated class I evidence of clinical benefit in both the acute and chronic phases of TBI. In a landmark study by Giacino and colleagues, 12 184 patients with sTBI in the acute phase were randomized to treatment with AMT or placebo. Recovery during the acute phase was significantly faster in the AMT group versus the placebo group, as measured by an improvement on the Disability Rating Scale (difference in slope, 0.24 points per week; p=0.007), indicating a benefit for treatment with AMT. Further, Hammond 13 evaluated the effects of AMT on irritability and aggression among individuals with chronic TBI in a controlled, randomized trial of 76 patients. Irritability was measured before and after treatment using the Neuropsychiatric Inventory (NPI) Irritability and Aggression Domains, which measure the frequency and severity of these symptoms. Mean change in NPI-I was −4.3 in the AMT group and −2.6 in the placebo group (p=0.0085). In the AMT group, 81% improved at least 3 points on the NPI-I, compared to 44% who improved at least 3 points in the placebo group.
AMT, an antiviral and antiparkinsonian drug, has pleiotropic activity, including (1) activation of dopaminergic neurons by increasing presynaptic levels of dopamine and increasing the number of postsynaptic dopamine receptors, 14 –16 (2) uncompetitive inhibition of N-methyl-D-aspartate (NMDA) receptor to modulate glutamate-induced excitotoxicity, 17 (3) increasing serotonergic function, 18 and (4) inhibition of the release of proinflammatory factors and stimulation of the release of glial cell-line–derived neurotropic factor (GDNF). 19 These activities are consistent with the demonstrated benefits of AMT in clinical trials and suggest that AMT may provide both symptomatic benefit and neuroprotection after TBI.
Although a number of investigational drugs have demonstrated good efficacy in experimental models of TBI, the fact that many of these drugs have failed in the clinic and that none have been licensed for TBI create an uncertain picture of the quality and utility of animal models. Further, despite convincing clinical evidence for AMT's benefit in treating acute and chronic TBI, there is little evidence in the literature for a corresponding benefit in animals. A previous experimental TBI study found only modest cognitive improvement and no benefit in neuroprotection with a once per day administration with a low dose (10 mg/kg intraperitoneally [i.p.]) of AMT. 20 Based on our understanding of the pharmacokinetics (PK) half-life of AMT in rats and humans (1.2 vs. 18 h, respectively), we evaluated the efficacy of AMT in experimental TBI using doses and dosing schedules that simulate human exposures in rats. In this study, we report that AMT, at clinically relevant doses, provides dose-dependent improvement in cognition and neuroprotection after experimental TBI in rats. These results are consistent with the recent clinical data for AMT in TBI and provide pharmacological validation of experimental TBI in rats.
Methods
Animals
A total of 42 adult male Sprague-Dawley rats (300–350 g; Harlan Laboratories, Hayward, CA) were used for this study. Animals were housed in individual cages in a temperature-(22°C) and humidity-controlled (50% relative) animal facility with a 12-h light/dark cycle. Food and water were continually available. Animals remained in the animal facility for at least 7 days before surgery. All animal protocols and procedures were approved by the University of California Davis Institutional Animal Care and Use Committee.
Drug preparation and experimental design
AMT hydrochloride (Sigma A1260) was purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in sterile saline.
For efficacy analysis, rats were randomly assigned to one of four groups: AMT (15, 45, or 135 mg/kg/day administered three times daily in equal doses) or saline control. Specific doses and dosing schedules were based upon analysis and simulation of the single-dose PK data (data not shown), and confirmed to bracket human exposures from 100 mg BID dosing (see below). Rats were administered three daily i.p. injections of AMT (5, 15, and 45 mg/kg in a volume of 1 mL/kg) or equal volume of saline at intervals of every 8 h for a total 16 days. The first injection was administered at 60 min after TBI. Body weight was recorded daily for 16 days postinjury. Rats were tested on the Morris water maze (MWM) over days 12–16 postinjury. Animals were euthanized and brains collected for histology analysis after the final MWM trial. Terminal blood samples were obtained just before transcardial perfusion euthanasia and the time since last drug injection was recorded. Serum concentrations of amantadine were determined by liquid chromatography plus tandem mass spectrometry (LC/MS-MS), and the concentration-time profiles were used to calculate the steady-state area under the curve (AUCss(0–8)) for amantadine at each dose level. Investigators were blind to the treatment group assignment throughout the experiment and data analysis.
Surgical procedure
Rats were initially anesthetized with 4% isoflurane in a carrier gas mixture of nitrous oxide/oxygen (2:1 ratio), intubated, and mechanically normoventilated with a rodent volume ventilator (model 683; Harvard Apparatus, Holliston, MA) with isoflurane reduced to 2%. Depth of anesthesia was routinely monitored every 10 min by testing for suppression of hind-paw withdrawal in response to a toe pinch. Sterile techniques were used during surgery. Rats were mounted in a stereotaxic frame, a scalp incision was made along the mid-line, and a 4.8-mm-diameter craniectomy was performed with a trephine on the right parietal bone (centered at 4.5 mm posterior from bregma and 3.0 mm right lateral to the sagittal suture). A rigid plastic injury tube (modified Luer-loc needle hub, 2.6 mm inside diameter) was secured over the exposed, intact dura with cyanoacrylate adhesive. Two skull screws (2.1 mm diameter, 6.0 mm length) were placed into burr holes, 1 mm rostral to bregma and 1 mm caudal to lambda. The assembly was secured to the skull with cranioplastic cement (Plastics One, Roanoke, VA). Rectal temperature was continuously monitored and maintained within normal ranges (37±0.5°C) during surgical preparation by a feedback temperature controller pad (model TC-1000; CWE Inc., Ardmore, PA). Brain temperature was measured (model TH-5, Physitemp Instruments Inc., Clifton, NJ) by insertion of a needle temperature probe (model MT-29/2; Physitemp Instruments) between the skull and temporalis muscle and recorded just before and after injury.
Induction of experimental brain injury
Lateral fluid percussion TBI was produced using a fluid percussion device (VCU Biomedical Engineering, Richmond, VA) 21 using the lateral orientation. 22 The device consists of a Plexiglas cylindrical reservoir filled with isotonic saline. One end of the reservoir has a Plexiglas piston mounted on O-rings, and the opposite end consists of a transducer housing with a 2.6-mm-inside-diameter male Luer-loc opening. Injury was induced by the descent of a pendulum striking the piston, which injects a small volume of saline epidurally into the closed cranial cavity, producing a brief displacement and deformation of brain tissue. The resulting pressure pulse was measured in atmospheres (atm) by an extracranial transducer (model SPTmV0100PG5W02; Sensym ICT, Milpitas, CA) and recorded on a digital storage oscilloscope (model TDS 1002; Tektronix Inc., Beaverton, OR). The rat was disconnected from the ventilator, the injury tube was connected to the fluid percussion device, and a moderate fluid percussion pulse was delivered within 10 sec. Immediately after TBI, the rat was again ventilated with a 2:1 nitrous oxide/oxygen mixture in the absence of isoflurane. The plastic injury tube and skull screws were removed, and the scalp incision was closed with 4.0 braided silk sutures. As soon as spontaneous breathing was observed, the animal was disconnected from the ventilator and transferred to a heating pad. Assessment of the righting reflex was begun by placing the rat in a supine position at regular intervals (∼20 sec) to test the rat's ability to spontaneously recover to a prone position. The duration of suppression of the righting reflex was used as an indicator of traumatic unconsciousness and an additional indicator of injury severity. After TBI, animals were individually housed and weighed daily.
Behavioral analysis
Acquisition of spatial learning and memory retention was assessed with a MWM on days 12–16 after TBI. The maze apparatus consisted of a large circular pool (183 cm in diameter by 60 cm high) filled with water to a depth of 22 cm. Water temperature was maintained at 24–28°C. A transparent Plexiglas escape platform (12.8 cm in diameter by 20 cm high) was placed 2 cm below the water surface. Consistent visual cues were located in the test room outside of the maze. Rats were released from one of four starting points (selected randomly on each day for each rat) and allowed 120 sec to find and mount the escape platform. If the rat did not find the platform within 120 sec, the experimenter guided the rat to the platform. Subjects remained on the platform for 30 sec before being removed from the maze. Subjects received a 4-min intertrial interval in a warmed holding cage before being returned to the maze for subsequent trials. A total of four trials per day were performed across 5 consecutive days. After the final MWM trial, a probe test was performed for 60 sec without an escape platform to assess spatial memory retention. After completion of the probe trial, a visual acuity test was performed using a black-circular platform 1 cm above the water surface located in a different quadrant from the escape platform. Data from all trials were recorded using a video tracking system (Poly-Track Video Tracking System v 2.1; San Diego Instruments Inc., San Diego, CA).
Tissue collection and sectioning
Rats were euthanized after completion of MWM testing on day 16 after TBI by deep sodium pentobarbital anesthesia (100 mg/kg, i.p), followed by transcardial perfusion with 100 mL of 0.1 M of sodium phosphate buffer (PB; pH, 7.4) and then 350 mL of 4% paraformaldehyde (PFA; pH 7.4). Brains were removed and stored overnight in 4% PFA at 4°C. Brains were cryoprotected in 10% sucrose for 1 day followed by 2 days in a 30% sucrose solution, frozen on powdered dry ice, and coronal sections were cut at 45-μm thickness with a sliding microtome (model 860; American Optical Company, Southbridge, MA). Every serial section, starting at −2.12 mm bregma and ending at −4.80 mm bregma, was saved in 24-well cell-culture plates for subsequent mounting.
Histology
Long-term neuronal survival in CA2–CA3 hippocampal fields was assessed using cresyl violet-stained tissue from animals euthanized on postinjury day 16. Tissue sections were mounted on gelatin-coated slides and dried overnight before staining. Sections were dehydrated at room temperature by a series of ethanol immersions: 70% (2 min×1); 95% (2 min×2); and 100% (2 min×2), followed by immersion in xylene (16 min). Sections were then rehydrated in a series of ethanol immersions: 100% (2 min×2); 95% (2 min×2); and 75% (2 min×1), then rinsed with distilled water (30 sec×2). Sections were next stained with cresyl violet acetate (0.1%) for 6 min, followed by rinsing in distilled water (15 sec×2), differentiated by immersion in 95% ethanol with 0.15% acetic acid (8 min), and dehydrated in a series of ethanol immersions: 95% (30 sec×2); 100% (30 sec×2); and cleared by immersion in xylene (5 min×2). Sections were coverslipped with Permount (Fisher Scientific, Hampton, NH).
Anatomical regions of interest and stereological cell counts
The region of interest (ROI) for measurement of surviving neuronal cells encompassed the stratum pyramidale of the hippocampus CA2 and CA3, bounded on one end by its entry into the dentate gyrus at the lateral tips of the dorsal and ventral blades of the granule cells and at the other end by the narrowing of the stratum pyramidale at the boundary of the CA2 to CA1. Systematic random sampling techniques were used for selecting tissue sections for staining and stereological analysis. Every fifth section was sampled starting at a section randomly determined from the first through fifth rostralmost sections. The tissue sections were then mounted onto gelatin-coated slides using a 1:1 distilled water/0.1-M PB mixture and allowed to dry.
Pyramidal neuron counts of the CA2–CA3 hippocampal fields were performed by investigators uninformed of the group assignment. Sections were examined on a microscope (Nikon E600; Nikon, Tokyo, Japan) with a motorized stage (MAC5000 System; Ludl Electronic Products, Ltd., Hawthorne, NY) using computer software (Stereo Investigator™ 8.0; Microbrightfield, Inc., Williston, VT). Criterion for counting pyramidal neurons required visualization of the nucleus of morphologically distinct cell bodies. Neuronal cell counting was performed with a 100×oil objective (Plan Apo, NA 1.40; Nikon). The total number of neurons in the ROI was quantified using optical fractionator stereological methods. 23 The spacing of the optical dissectors produced an average area sampling fraction of 0.030. The guard height was set at 0.40 μM, producing a tissue sampling fraction of 0.70. Target cells in every fifth section were counted, producing a section sampling fraction of 0.20.
Statistical analysis
Differences between body weights, injury magnitudes, righting times, and temperature measurements were analyzed using one-way analysis of variance (ANOVA) models with treatment group as the factor.
Two primary outcome measures were analyzed: overall performance on the MWM (days 12–16) and the numbers of surviving CA2–CA3 cells. Data from the overall MWM performance were analyzed using one-way analysis of covariance (ANCOVA) models with treatment group as a factor and the baseline (day 12) value as a covariate. Differences in the numbers of cresyl violet-stained surviving neurons in the CA2–CA3 of the ipsilateral hippocampus between groups were analyzed using one-way ANOVA. Comparisons between each AMT group and the saline control group were made using Fisher's least significant difference (LSD). Because the LSD does not adjust the p value for multiple comparisons, adjustments to the p values were made by multiplying the nominal p values by the number of comparisons for each test, and a significance level of p<0.05 was used for each test. As sensitivity analyses, Wilcoxon-Mann-Whitney's test was also used to compare each pair of treatment groups. In addition, three secondary outcome measures were evaluated: terminal performance in the MWM (days 15–16); rate of learning the MWM (slope of days 12–14); and MWM probe trial. To reduce the possibility of a type I error from multiple comparisons, the secondary outcome measures were not subjected to statistical analysis.
Results
Pharmacokinetics analysis
To confirm that target levels of AMT were reached during the TBI study, terminal blood draws were obtained from TBI rats at the time of euthanasia at varying time points after the last dose. Based on the concentration-time profiles from the terminal blood draws (Fig. 1A), the area under the curve at steady-state (AUCss(0–8)) was calculated for each dose level (Fig. 1B). The AUCss for the 45 mg/kg/day dose level was 5,359 ng*h/mL, which closely approximates the AUC for 100 mg BID amantadine in humans of 6,414 ng*h/mL. 24 At the 135 mg/kg/day dose level, the AUCss(0–8) was 32,339 ng*h/mL, which is approximately 5 fold the human exposure. These data confirm that clinically relevant exposures were obtained at the two higher dose levels in rats during the TBI study.

Pharmacokinetic analysis of amantadine serum concentration from terminal blood draws from TBI rats.
TBI descriptive characteristics
There were no significant differences between groups in TBI magnitude, righting time, and temperature (Table 1). The number of rats per group was as follows: saline control, n=14; 15 mg/kg/day AMT, n=9; 45 mg/kg/day AMT, n=12; and 135 mg/kg/day AMT, n=7.
Mean±standard deviation.
atm, atmospheres; TBI, traumatic brain injury; AMT, amantadine.
Body weight
Mean group body weights on the day of surgery and TBI ranged from 308±9 to 319±11 g and were not significantly different between groups. All groups lost body weight over the first 3 days post-TBI and subsequently began gaining weight by day 6 post-TBI (Fig. 2). There was no difference in body weight between groups after TBI (F(3,37)=1.791; p=0.166).

Comparisons of total body weight over the course of 16 days postinjury. All groups had typical body-weight loss for the first 3 days post-TBI and subsequently began gaining weight by day 6. There was no difference in body weight between groups after TBI. Values are means±standard error of the mean. TBI, traumatic brain injury; AMT, amantadine.
Morris water maze assessment
For the primary outcome measure, the overall performance in the MWM was assessed by comparing mean latency to find the hidden platform over days 12–16 post-TBI (F(3,37)=4.414; p=0.009; Fig. 3A). Treatment with 135 mg/kg/day of AMT resulted in significantly shorter latency to find the hidden platform, compared to the saline control group (46.2±3.36 and 59.5±4.25 sec, respectively; p=0.030), whereas there was a trend toward decreasing latency time with the 45-mg/kg/day group (45.5±2.77 sec; p=0.123). The 15-mg/kg/day AMT group was not significantly different from the saline group (66.6±7.27 sec; p=1.000).
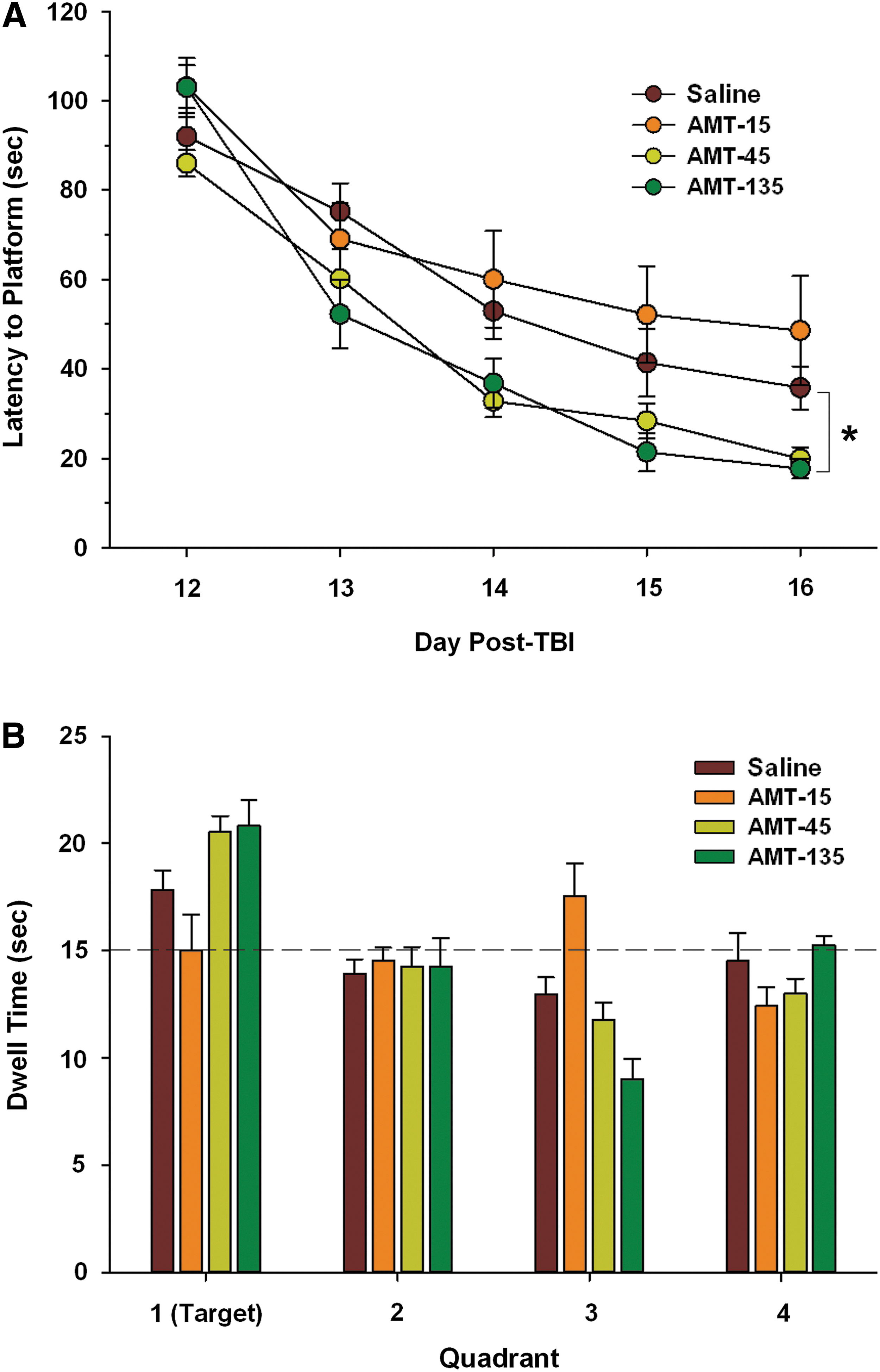
Acquisition of spatial learning and memory performance in the Morris water maze (MWM) over days 12–16 after TBI. (
For the secondary outcome measures, the terminal performance in the MWM was assessed by comparing mean latency to find the hidden platform on days 15–16 post-TBI, which represent the last 2 days of MWM testing. The 45- and 135-mg/kg/day AMT groups had shorter latencies to find the hidden platform (24.1±2.91 and 19.6±2.53 sec, respectively), compared to the saline control group (38.6±5.67 sec), whereas the 15-mg/kg/day AMT group did not (50.4±10.1 sec). Similarly, when the rate of learning was assessed by comparing the slopes of the learning curves calculated from the latency to platform over days 12–14 post-TBI, which represents the linear portion of the curve, treatment with AMT at the two higher doses resulted in steeper learning curves (−26.6±1.53 and −33.1±4.60 for 45 and 135 mg/kg/day, respectively), compared to placebo (−19.5±3.66) and the low-dose AMT group (−21.5±5.07). Memory retention was assessed on day 16 post-TBI with a 60-sec probe trial in which the submerged escape platform was removed (Fig. 3B). Again, there was longer retention times in the target quadrant with the higher doses of AMT, compared to placebo or low-dose AMT. There were no significant differences between groups in performance on the visible platform trial, indicating that animals in all groups had normal visual processing and thus the differences in MWM performance were not confounded by visual impairment (data not shown).
Histology measures
For the second primary outcome measure, stereological counts of surviving neurons in the CA2–CA3 fields of the dorsal hippocampus were restricted to the saline control group and the two AMT dose groups that affected MWM performance (45 and 135 mg/kg/day). There was a dose-dependent increase in the number of surviving CA2–CA3 pyramidal neurons (F(2, 30)=4.705; p=0.017; Fig. 4). Treatment with 135 mg/kg/day of AMT significantly increased the numbers of surviving CA2–CA3 pyramidal neurons, compared to the saline control group (p=0.01). There was no significant difference between the 45-mg/kg /day AMT group and the saline group (p=0.680).
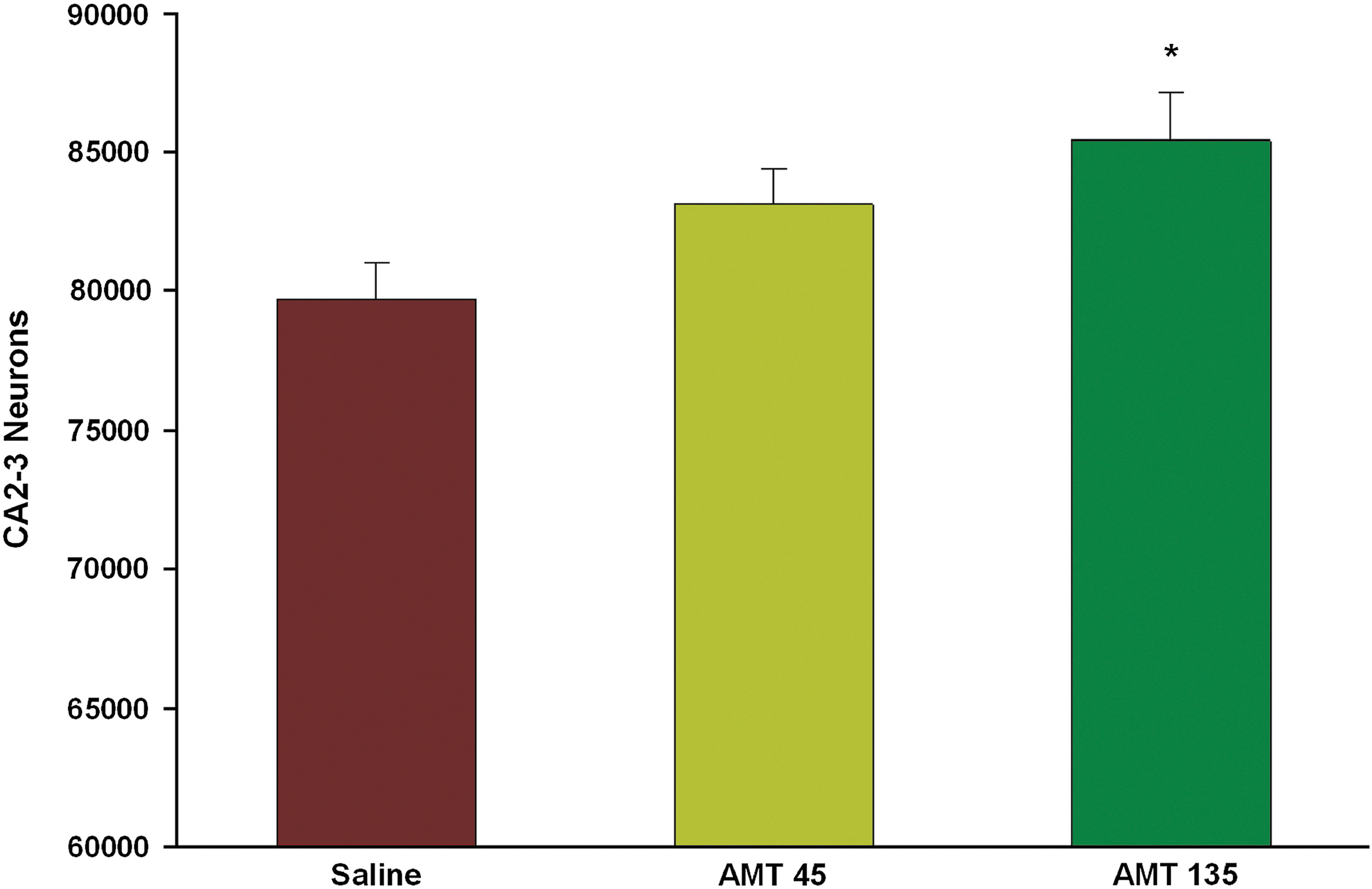
Quantification of cresyl violet-stained cells in the ipsilateral CA2–CA3 region of hippocampus between groups at 16 days after traumatic brain injury. More pyramidal neurons survived in the AMT-135 group, compared to the saline group. Values are mean±standard error of the mean. *p<0.05, compared to saline. AMT, amantadine.
Discussion
This study investigated the effects of post-TBI administration of AMT on cognitive deficits and long-term neuronal cell loss in the hippocampus after moderate lateral fluid percussion TBI in the rat. AMT was administered i.p. every 8 h for 16 days starting at 1 h after TBI. Treatment with 135 mg/kg/day of AMT improved cognitive performance in the MWM and reduced hippocampal CA2–CA3 neuronal cell loss after experimental TBI, with a trend toward improvement by both measures with the intermediate 45-mg/kg/day dose. Amantadine serum levels determined from terminal blood draws demonstrated that clinically meaningful doses were utilized in this study. These data suggest that postinjury treatment with clinically relevant doses of AMT is a potential neuroprotective treatment for TBI that could attenuate associated cognitive dysfunction.
The MWM is a widely used test of spatial learning and memory function in studies of rodent experimental TBI. 25 Four important aspects of MWM acquisition performance were evaluated in this study, including mean latency to find the platform over all 5 testing days, terminal performance on the final 2 testing days, and slope of the learning curves over the first 3 days. The results of the present study were not confounded by differences in visual acuity between groups. All treatment groups swam at similar speeds and performed similarly on the visible platform trial, indicating that motor swim deficits and visual acuity were not influential factors in performance between groups. On each of these measures, only the higher dose AMT regiments (45 or 135 mg/kg/day) provided improvements in cognitive performance, compared to the saline control group. Statistical analysis of the primary measure—latency to find the platform over all five testing days—demonstrated that the difference between 135 mg/kg/day and saline was significant. The PK data from the present study indicated that only the higher AMT dosing regimens (45 and 135 mg/kg/day) achieved serum concentrations approaching those of clinically effective studies. 12,26 Taken together, our present study data indicated that clinically relevant doses of AMT treatment reduced cognitive deficits, compared to the low-dose (15 mg/kg) AMT and saline-treated groups.
Progressive neuronal death in the hippocampal CA2–CA3 region as well as shrinkage of the hippocampal pyramidal cell layer after brain injury has been documented as a hallmark of head injury in humans 27 as well as in experimental TBI animal models. 28 –30 Long-term neuronal loss was evaluated in the ipsilateral CA2–CA3 region of the dorsal hippocampus at 16 days postinjury using cresyl violet-stained sections and stereological counting methods. Treatment with the 135-mg/kg/day dosing regimen of AMT significantly increased the numbers of surviving CA2–CA3 pyramidal neurons at day 16 post-TBI, compared to the saline group. The result of reducing neuronal cell loss combined with the ameliorative effects on cognitive performance suggested treatment with clinically relevant doses of AMT may reduce certain pathological features associated with TBI as well as improve CNS function after TBI.
The present study did not specifically examine the mechanisms by which AMT provided improvement in cognitive performance and neuroprotection. However, several of the actions of AMT are likely contributors to the improved outcome measures observed in this study. These include inhibition of the NMDA receptor, 17 which may prevent excessive excitatory activation of glutaminergic pathways and calcium influx, ultimately resulting in apoptosis. 31 In addition, AMT has been demonstrated to be involved in the inhibition of the release of proinflammatory factors and stimulation of the release GDNF, 19 which may result in neuroprotection. These activities are consistent with the demonstrated benefits of AMT in this study.
In summary, this study reports improvements in cognitive outcome along with neuroprotection after administration of a clinically relevant dosing paradigm of AMT after experimental TBI. Further, given that AMT is the only drug with class I evidence of clinical benefit in TBI, these results provide pharmacological validation of the fluid percussion injury model of TBI, enabling the evaluation of other investigation therapies for the treatment of TBI. Altogether, the data presented here provide a rationale for the further evaluation of AMT for the treatment of TBI. Future studies should investigate the timing of administration to determine the therapeutic window in which AMT provides neuroprotective and cognitive benefit.
Footnotes
Acknowledgment
The research was supported by funding from Adamas Pharmaceuticals, Inc.
Author Disclosure Statement
No competing financial in interests exist.