
Abstract
Moderate to severe traumatic brain injury (TBI) in humans and rats induces measurable metabolic changes, including a sustained depression in cerebral glucose uptake. However, the effect of a mild TBI on brain glucose uptake is unclear, particularly in rodent models. This study aimed to determine the glucose uptake pattern in the brain after a mild lateral fluid percussion (LFP) TBI. Briefly, adult male rats were subjected to a mild LFP and positron emission tomography (PET) imaging with 18F-fluorodeoxyglucose (18FDG), which was performed prior to injury and at 3 and 24 h and 5, 9, and 16 days post-injury. Locomotor function was assessed prior to injury and at 1, 3, 7, 14, and 21 days after injury using modified beam walk tasks to confirm injury severity. Histology was performed at either 10 or 21 days post-injury. Analysis of function revealed a transient impairment in locomotor ability, which corresponds to a mild TBI. Using reference region normalization, PET imaging revealed that mild LFP-induced TBI depresses glucose uptake in both the ipsilateral and contralateral hemispheres in comparison with sham-injured and naïve controls from 3 h to 5 days post-injury. Further, areas of depressed glucose uptake were associated with regions of glial activation and axonal damage, but no measurable change in neuronal loss or gross tissue damage was observed. In conclusion, we show that mild TBI, which is characterized by transient impairments in function, axonal damage, and glial activation, results in an observable depression in overall brain glucose uptake using 18FDG-PET.
Introduction
I
Within the first week after a severe brain injury, both regional and cerebral hyperglycolysis (increase in glucose utilization) has been documented in several patients. 6,7 After peaking, cerebral glucose metabolism decreases to a broad minimum lasting from months to years, followed by a slow return to normal levels. 8 After an mTBI, patients have shown similar reductions in positron emission tomography (PET)-based measurements of glucose metabolism, 9 although post-injury hyperglycolysis has not been observed in any clinical studies of mTBI. In addition to temporal changes in glucose metabolism, acute and chronic changes in cerebral vascularity play a significant role in determining patient outcomes. Several studies have provided mixed results regarding the temporal change in cerebral blood flow (CBF) following mTBI, leading some to suggest a patient-specific, toxic cascade when blood flow, glucose, and oxygen utilization are uncoupled. 10 This uncoupling is transient, highly variable, and difficult to measure.
A similar temporal change in glucose utilization has been observed in several rodent models of moderate and severe TBI. For example, immediately after a moderate to severe lateral fluid percussion (LFP) or open or closed skull impact injury in rats, adenosine triphosphate (ATP) levels, N-acetylaspartate (NAA)/creatinine balance, and CBF decrease, and local cerebral metabolic rate of glucose (lCMRGlc) utilization increases. 11 –17 This apparent uncoupling between metabolism and CBF is short lived, lasting only ∼6 h in rats. After this time, a metabolic depression sets in with a minimum at 1–2 days post-TBI. CBF typically returns to normal levels within this time period, but glucose metabolism remains depressed for up to 10 days. 11 Very few studies have investigated the effect of mTBI on metabolism and glucose uptake in rodent models, however. Signoretti et al. 18 demonstrated a reduction in neuronal metabolites at 24 h after injury. In juvenile rats, depressed glucose metabolism was noted 24 h after an mTBI using 2-deoxy-D-glucose (2DG) autoradiography. 19
Glucose uptake and metabolism can be evaluated noninvasively in both humans and animal models using 18F-fluorodeoxyglucose (18FDG) with PET imaging. PET imaging provides exquisite sensitivity when compared with CT or magnetic resonance imaging (MRI), but with reduced resolution. Following a moderate LFP injury, two separate studies used 18FDG in rats to show: 1) a depression at 48 h that was mostly resolved within 10 days using PET, 11 and 2) an acute increase in glucose uptake at 42 min followed by depression at 12 h post-injury using autoradiography. 17 In more severe LFP, serial 18FDG-PET analysis showed a sustained depression in glucose metabolism in the affected cortex, hippocampus, and amygdala up to 6 months post-injury. 20 These changes in glucose uptake were associated with an increase in anxiety in rats, although other functional outcomes were not significantly correlated. (See Lin et al. 9 for a thorough review of PET-based TBI studies.)
To date, alterations in glucose uptake after mild brain injury have not been monitored in the controlled situation of adult animal models. Here, we demonstrate that an mTBI inflicted with a LFP injury device, similar to that characterized by Shultz et al., 21 resulted in sustained glucose uptake depression, transient motor deficits, axonal damage, and glial activation.
Methods
Mild traumatic brain injury
Adult male Sprague–Dawley rats (150–200 g) were given free access to food and water and a 12 h light/12 h dark cycle before mild TBI. All animal procedures were approved by the Uniformed Services University Institute for Animal Care and Control (IACUC). LFP was performed as previously described 22 with some modifications. Briefly, rats were anesthetized with isoflurane (4% induction, 2% maintenance), temperature was measured rectally and maintained at 36.5–37.5°C, and a 5 mm craniotomy was performed over the left parietal cortex midway between lambda and bregma and 2.5 mm from the vertex. A plastic female Luer Lock was cemented over the craniotomy and attached to tubing from the fluid percussion device (VCU Health System Custom Design and Fabrication Model 01-B). An impact with an estimated pressure of 1.2±0.1 atmospheres (atm) was achieved by dropping a pendulum onto the water tube from an angle of 22 degrees. Pilot studies determined that this angle and resultant pressure achieved a transient motor function impairment and histopathology consistent with previously published reports of mild brain injury. 21,23 –25 Because of the addition of a 30 cm length of tubing (0.071 cm ID, 0.142 cm OD) between the device and the female Luer Lock, the pressure was reduced from that reported by the manufacturer and was empirically determined to be 10-fold lower at the point of impact than at the point of measurement. Control rats were divided between craniotomy (sham; n=5) and non-craniotomy (naïve; n=4) groups. Sham animals underwent identical surgical procedures without impacts; naïve animals did not undergo any surgical procedure and were not exposed to anesthesia.
A total of 21 animals were used for this study. Of those, three were excluded from analysis because of surgical problems. Twelve rats (n=6/group; 3 sham/3 naïve) underwent PET imaging at 3 h, 24 h, 5 days and 9 days post-injury, in addition to functional testing at 24 h, 72 h, and 7 days post-injury, and histological assessment at 10 days post-injury. An additional group of 6 rats (n=3/group; 2 sham/1 naïve) underwent PET imaging at 48 h, 5 days, 9 days, and 16 days post-injury, in addition to functional testing at 24 h, 72 h, 7 days, 14 days, and 21 days post-injury and histological assessment at 21 days post-injury. As there was no significant difference between sham and naïve data, sham and naïve were combined for all analyses and are shown as sham/naïve. Where appropriate, data from all groups are combined (functional testing, PET imaging).
PET imaging
Prior to injury and at 3 and 24 h and 5, 9, and 16 days post-injury, glucose uptake was determined by PET imaging of the biodistributions of 18FDG. Studies have shown that animal handling during 18FDG uptake significantly influences biodistributions, and that isoflurane anesthesia administered during uptake reduces the overall variation in FDG distribution and results in a more homogenous brain uptake. 26,27 Therefore, for all PET-18FDG imaging, animals were anesthetized with isoflurane for tail vein injection of ∼1.5–2 mCi 18FDG and during the 45 min uptake period. After uptake, a static 30 min PET scan was acquired using a Siemens Inveon Multimodality scanner (Siemens Medical Solutions USA, Malvern, PA). Also, rats underwent a microCT scan for localization and attenuation correction immediately prior to the PET scan. The parameters for reconstruction were as follows: reconstruction algorithm - OSEM3D/MAP; projection filter – Ramp; scattered corrected; image size, 256×256; OSEM3D iterations, 2; MAP iterations, 18; resolution∼1.5 mm at center of the field of view.
After imaging, resultant images were analyzed with the Inveon Research Workplace software package (Siemens Medical Solutions USA, Malvern, PA). Three regions of interest (ROI) were assessed: cerebellum (ROI 50.6mm3), deep gray and periventricular brain (central ROI 256.4mm3), and ipsilateral/contralateral cortex (ROI 28.9mm3 each). 18FDG uptake in each ROI was calculated over the summed 30 min scan. Activity concentrations measured in the central and ipsi/contralateral ROIs were divided by the activity concentration measured in the cerebellum ROI from that same animal. This reference tissue ratio was then normalized to the animal's baseline reference tissue ratio. The final value presented represents a semiquantitative normalized reference tissue ratio from the central brain or ipsi/contralateral ROI.
Function testing
Animal motor function was assessed using the beam walk task. Briefly, rats were trained to cross 5 m beams of 4, 3, 2, or 1 cm thickness for 3 days prior to injury. Baseline scores including number of footfalls and time to cross each beam were obtained and compared with measurements at 1, 3, 7, 14, and 21 days post-injury.
In the second group of animals (n=3/group), rats also underwent a more vigorous motor task – the peg walk – at days 1, 7, 14, and 21 post-injury. To increase the sensitivity of the task, animals were trained to cross a 2 cm thick beam with 2 cm diameter 4 cm high round pegs at even intervals (10 cm) along the 5 m beam. Time to cross the beam and footfalls were measured in this task as well. All testing was performed by an investigator blinded to treatment group.
Animal cognitive function was assessed using the Barnes Maze task at 14 days post-injury. 28 Briefly, each rat was placed in the center of the maze, covered by a black box, for 60 sec. Animals were then given a maximum of 5 min to find an escape compartment, or were placed into the escape compartment, for 30 sec. Two training trials were run per day for 4 days, with a rest of at least 2 min between trials. The escape was kept constant for days 1–3, then rotated 135 degrees on day 4. ANY maze software (Stoelting Co., Wood Dale, IL) was used to assess latency to find the hidden platform and search strategy.
Histology
At 10 (n=6/group) or 21 (n=3/group) days post-injury, rats were euthanized and brain tissue perfused with 10% formalin before being dissected and sectioned at 20 μm. Tissue was then processed for standard hematoxylin and eosin (H&E) staining, Luxol fast blue/PAS, Bielschowsky's silver stain (Modified Bielschowsky's Stain Kit, American MasterTech Scientific, Inc, Lodi, CA), or immunolabeled for axons (MAP2, 1:50; Santa Cruz Biotechnology, Santa Cruz, CA), microglia (Iba-1, 1:100; Wako Chemicals USA, Richmond, VA), neurons (NeuN, 1:100; Millipore, Billerica, MA), and astrocytes (GFAP, 1:500; Abcam, Cambridge, MA). Fluorescent antibodies (1:1000; Alexa-Fluor Secondaries, Invitrogen, Grand Island, NY) were used to visualize primary antibodies. No-primary negative controls were used to confirm specificity of secondary antibodies. Specificity of primary antibodies was confirmed by visualization of appropriate and expected staining and demonstration of a single band in Western blotting (data not shown). Images were then captured on a NanoZoomer system (Hamamatsu, Bridgewater, NJ) or an Olympus BX43 microscope (Olympus America). Lesion volume from H&E images were then quantified using unbiased stereology and the Cavalieri method as previously described. 29 Fluorescent immunohistochemistry was quantified as previously described using pixel density measurement in Scion Image. 30
Statistical analysis
Quantitative data are presented as mean±standard error of the mean. PET values, functional data, pixel density, and lesion volume were obtained by an investigator blinded to treatment group. Data were analyzed using repeated measures ANOVA or one way ANOVA, as appropriate. Correlation analysis was performed using linear regression analysis, with Bonferroni correction for familywise error rate (for histology, a p value of 0.025 was considered significant). All statistical tests were performed using the GraphPad Prism Program, Version 5.0 for Windows (GraphPad Software, San Diego, CA). A p value<0.05 was considered statistically significant.
Results
PET imaging of TBI
As previously noted, regional ROI values were normalized to cerebellar uptake in order to account for differences in physiological parameters, depth of sedation, and activity injected. Baseline ROIs normalized to the cerebellum show a 4% standard deviation. Naïve and sham-injured rats showed variable changes in whole brain 18FDG uptake over time (Fig. 1), but this uptake did not drop below their baseline measurement. On the other hand, after a mild LFP injury, FDG uptake in the central ROI was significantly reduced compared with baseline levels and compared with sham/naïve animals at 24 h and 5 days post-injury (Fig. 1). This reduction was sustained through 9 days post-injury, with uptake returning to baseline or above baseline levels by 16 days post-injury.
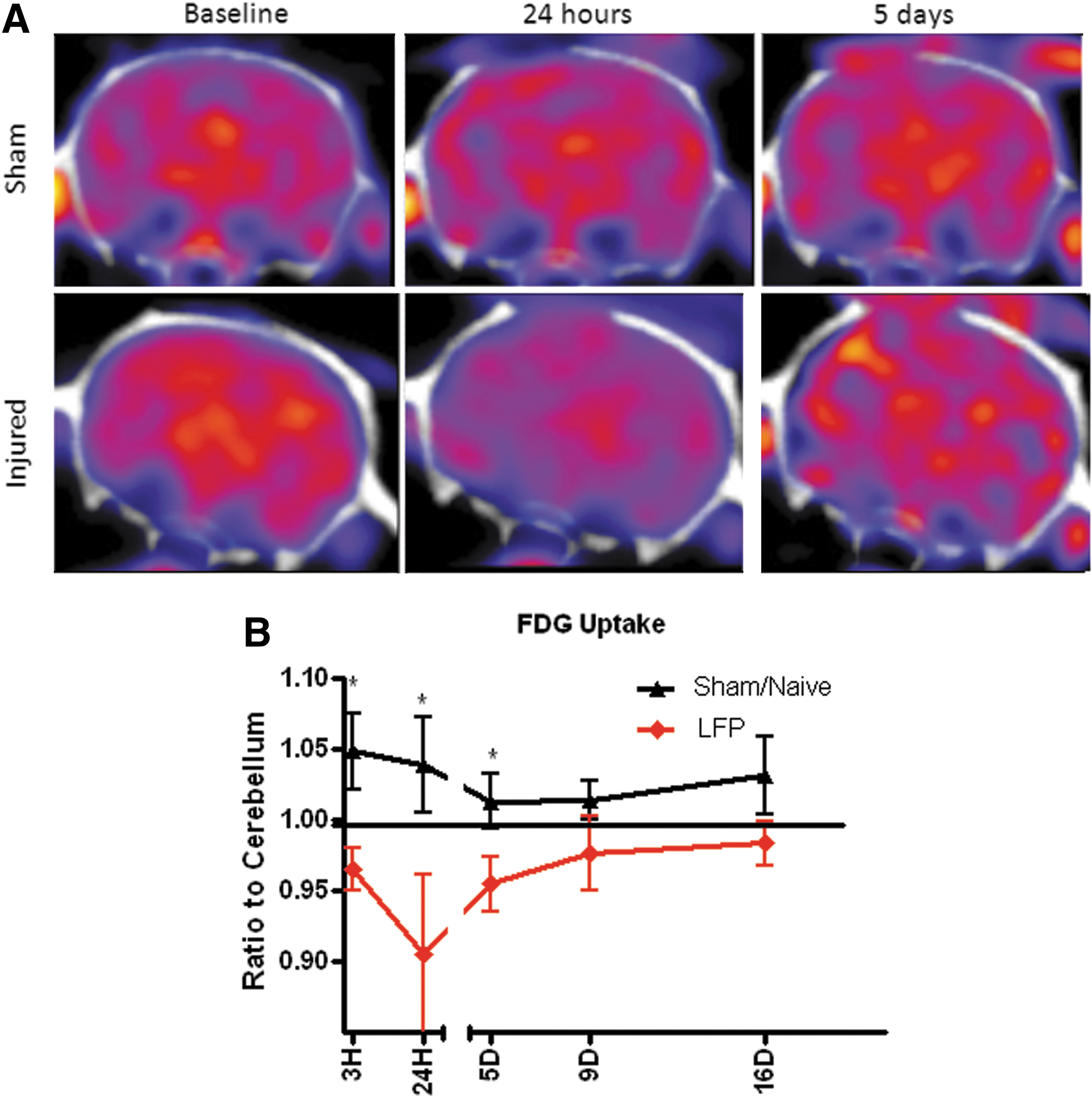
Mild lateral fluid percussion (LFP) injury results in a transient reduction in glucose uptake. 18F-fluorodeoxyglucose (18FDG) uptake was measured at 3 and 24 h and 5, 9 and 16 days after a mild LFP. 18FDG uptake in the whole brain was then normalized to the cerebellum and again to baseline to provide a view of glucose uptake at post-injury time points. Representative 18FDG-PET images from a sham/naïve and an injured animal at baseline and 24 h and 5 days after injury are shown
Smaller ROIs placed in the ipsi- and contralateral hemispheres were also assessed in sham/naïve rats and LFP-injured rats. Data represent ROI-specific glucose uptake, and are presented as a ratio of ROI activity concentration to the cerebellum and normalized to the baseline reference tissue ratio. Measurements demonstrate that both ipsilateral and contralateral hemispheres show alterations in glucose uptake after injury (Fig. 2). In both hemispheres, there is a depression in glucose uptake at 3 and 24 h post-injury, followed by a return to sham/naïve levels by 9 days post-injury. Beyond 9 days, glucose uptake shows a slight elevation above sham/naïve levels, although this did not reach statistical significance.

18F-fluorodeoxyglucose (18FDG) uptake is altered in both the ipsilateral and contralateral hemispheres after mild lateral fluid percussion (LFP) injury. 18FDG uptake was measured in the lesion and perilesional region using a sphere shaped region of interest (ROI) (shown in sagittal
Mild LFP induces a transient impairment in motor function
Animals were tested on two variations of the beam walk task to confirm the severity of the mild LFP injury. Rats that had been subjected to mild LFP demonstrated significant increases in crossing latency and foot faults at 24 h post-injury (Fig. 3A, B). Motor function then returned to normal levels (sham/naïve) for all subsequent time points.
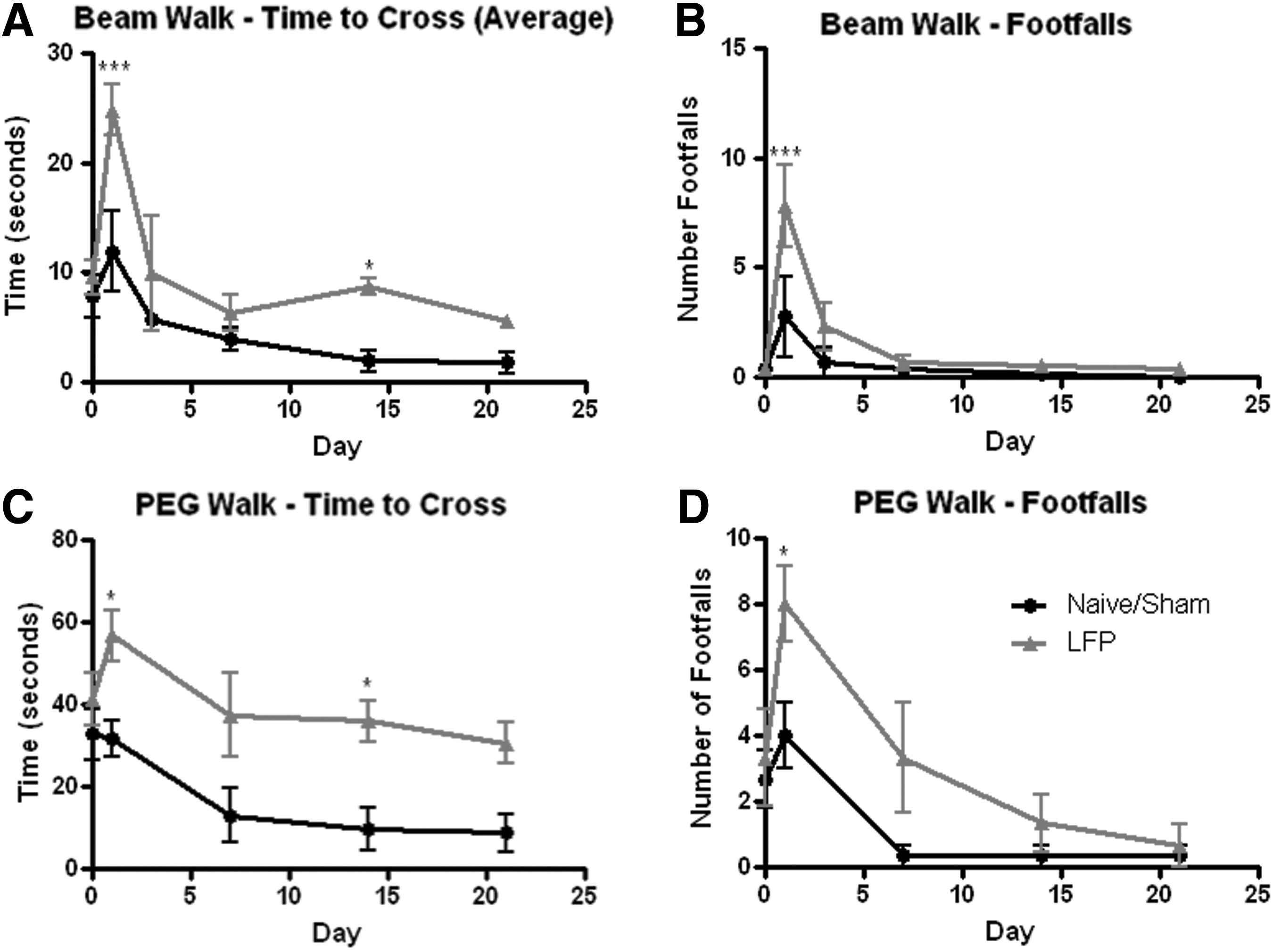
Mild lateral fluid percussion (LFP) injury results in a transient impairment in motor function. Motor function was tested in rats after a mild LFP injury using the declining width beam walk task
Increasing the difficulty of the beam walk task by attaching pegs to the 2 cm beam showed an even greater motor functional deficit (Fig. 3C, D). Crossing latency and foot faults were significantly increased in injured rats at all time points, or up to 14 days, respectively. The Barnes maze was used to assess subacute cognitive function. No significant deficit in cognitive function was found at 14–17 days after mild LFP (data not shown). A slight trend toward an impairment in short-term learning ability in the mild LFP group, as demonstrated by a reduction in latency to find the hidden exit between trials 1 and 2 on the first day of test, was observed. However this did not reach statistical significance.
Glucose uptake reduction is associated with diffuse axonal injury and evidence of inflammation
Gross histology, performed by analysis of H&E-stained tissue at 10 and 21 days post-injury, showed no significant alteration in tissue composition or lesion development after mild LFP (Fig. 4). However, on evaluation of the tissue when stained with Luxol fast blue, evidence of axonal loss and/or demyelination was found in the corpus callosum at 10 days post-injury, which corresponded with significant width reduction in both the ipsilateral and contralateral hemispheres (Fig. 4D). Similar results were obtained at 21 days post-injury (data not shown).
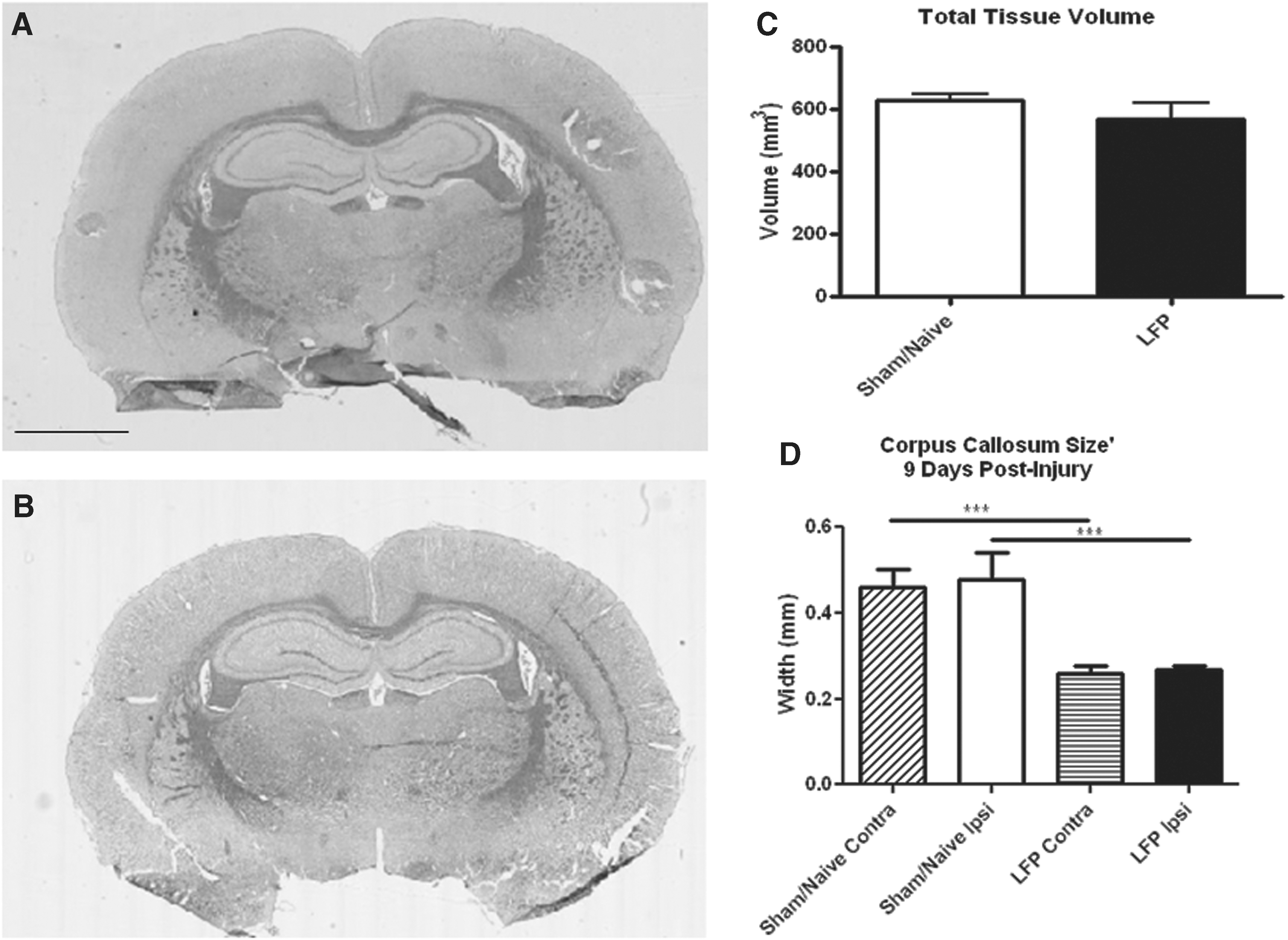
Mild lateral fluid percussion (LFP) injury does not result in a gross lesion, but does impact white matter tracts. After mild LFP injury, tissue was sectioned and stained with hematoxylin and eosin (H&E); representative sections from sham/naïve
Immunohistochemistry also demonstrated marked changes in cellular and axonal composition at the injury site, and in the contralateral hemisphere in regions associated with alterations in ipsilateral and contralateral ROI measurements (Fig. 2). Qualitative evaluation of MAP2 staining showed a reduction in MAP2 positive fibers by 10 days post-injury, which was further pronounced at 21 days post-injury (Fig. 5). Similarly, axonal damage, as detected by a qualitative evaluation of silver staining, was observed in the ipsilateral cortex at 10 days post-injury (Fig. 6). This staining could be visualized as black-labeled fibers within the ipsilateral cortex above the corpus callosum. No silver stained fibers were observed at 21 days post-injury (data not shown). Immunolabeling for neurons with the NeuN antibody, however, demonstrated no marked difference in cortical organization between injured and sham/naïve tissue at either 10 days or 21 days post-injury (data not shown).
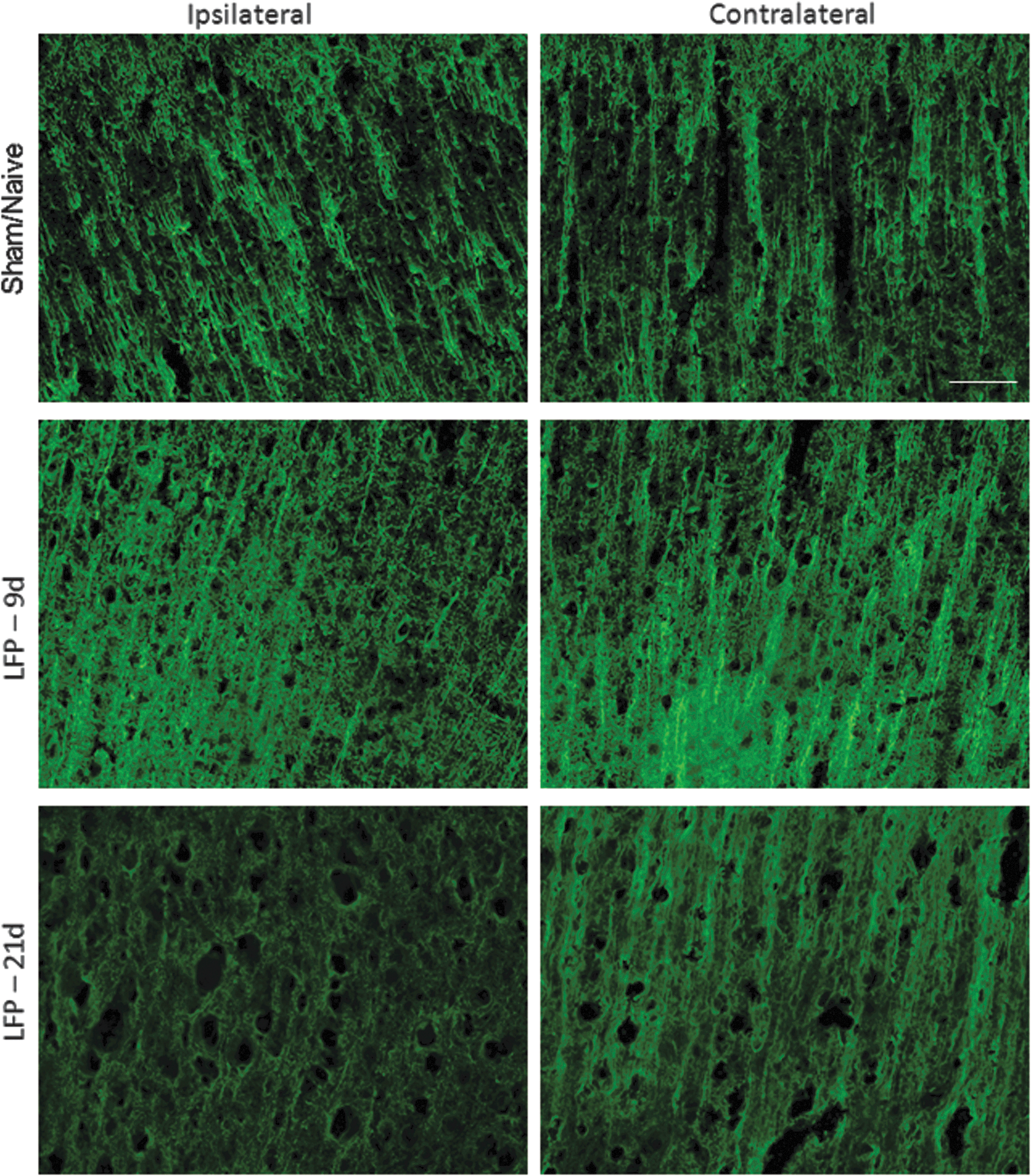
Mild lateral fluid percussion (LFP) injury reduces MAP2 staining in the ipsilateral cortex. MAP2 staining was performed at 10 and 21 days after sham/naïve or mild LFP injury. An apparent reduction in MAP2 staining was observed in the ipsilateral cortex at both 10 and 21 days post-injury. Bar=50 μm. Color image is available online at
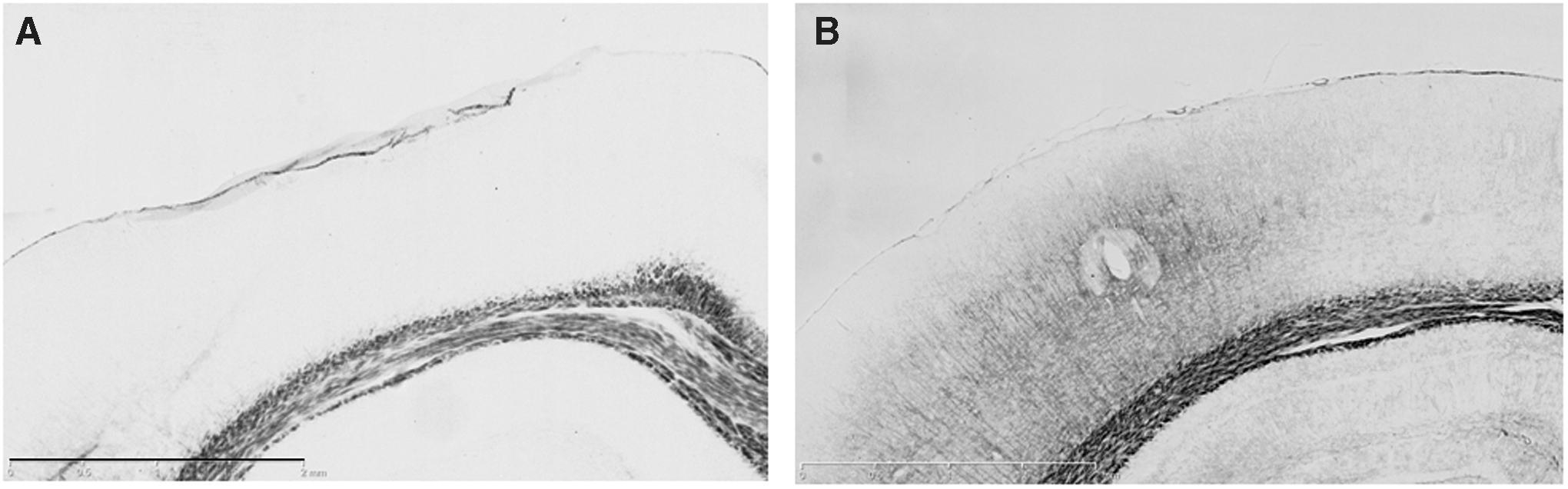
Mild lateral fluid percussion (LFP) injury increases silver stain in the ipsilateral cortex. Silver stain was performed at 10 days after sham/naïve
Glial changes were among the most marked after mTBI. GFAP immunolabeling for astrocytes demonstrated clear astrogliosis by 10 days post-injury, which was most pronounced in the ipsilateral hemisphere (Fig. 7). GFAP immunoreactivity was also more prominent than in sham/naïve animals in the contralateral hemisphere, but to a lesser degree than on the ipsilateral side. This astrocytic response remained at 21 days, but had declined in comparison with the 10 day post-injury group. At 10 days post-injury, this GFAP immunoreactivity was significantly elevated as measured by pixel density quantitation (Fig. 7B). Correlation of GFAP immunoreactivity at 10 days with FDG uptake at 24 h showed a significant correlation (r2 =0.9737, p<0.0001, which surpasses the Bonferroni correction threshold of 0.025).
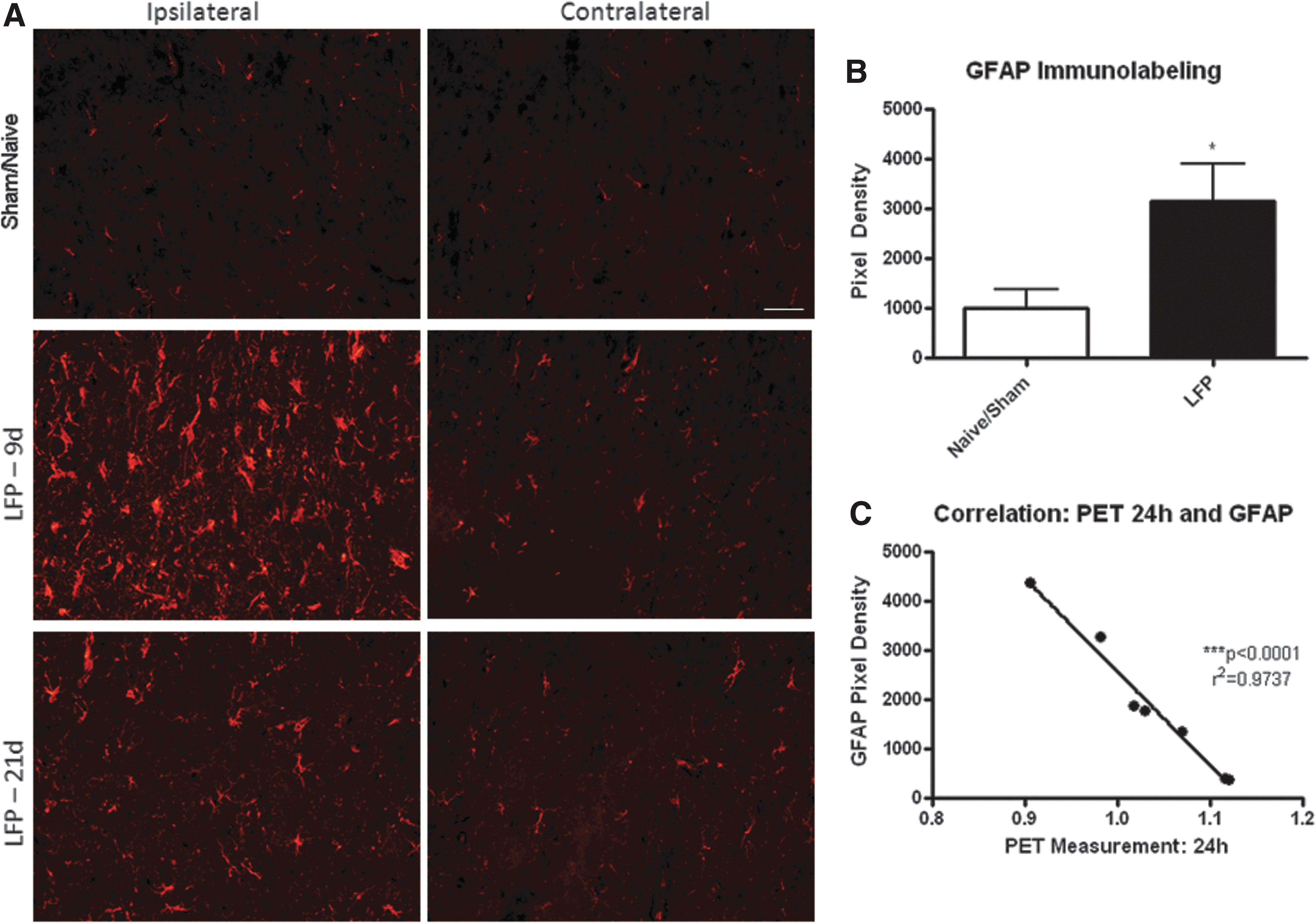
Mild lateral fluid percussion (LFP) induces an increase in GFAP labeling in the ipsilateral and contralateral cortex. Light GFAP staining was observed in sham (shown at 10 days post-surgery) and naïve tissue. However, by 10 days after LFP injury, there was a marked increase in GFAP labeling in the ipsilateral cortex and, to a lesser extent, in the contralateral cortex at 10 days post-injury. This increase was still observed, although lessened, by 21 days post-injury. Representative images shown in
Immunolabeling for microglia at 10 and 21 days post-injury showed a marked increase in cell number in the ipsilateral cortex and surrounding white matter (Fig. 8; 21 day data not shown). In addition to an overall increase in number, Iba-1 immunolabeling showed a marked increase in cellular size and a thickening of processes, indicating a hypertrophic activation state of the microglia. 31 Quantitation of the immunolabeling for Iba-1 at 10 days post-injury showed that injury resulted in a significant increase in this microglial marker in the ipsilateral cortex (Fig. 8E); however, this was not significantly correlated with acute 18FDG uptake. Interestingly, a slight increase in Iba-1 immunolabeling was also observed in the contralateral cortex after mild LFP. Furthermore, at 10 days post-injury, this increase in Iba-1 labeling was accompanied by a marked increase in ED1 immunoreactivity, indicating that regional microglia were phagocytic (Fig. 9).
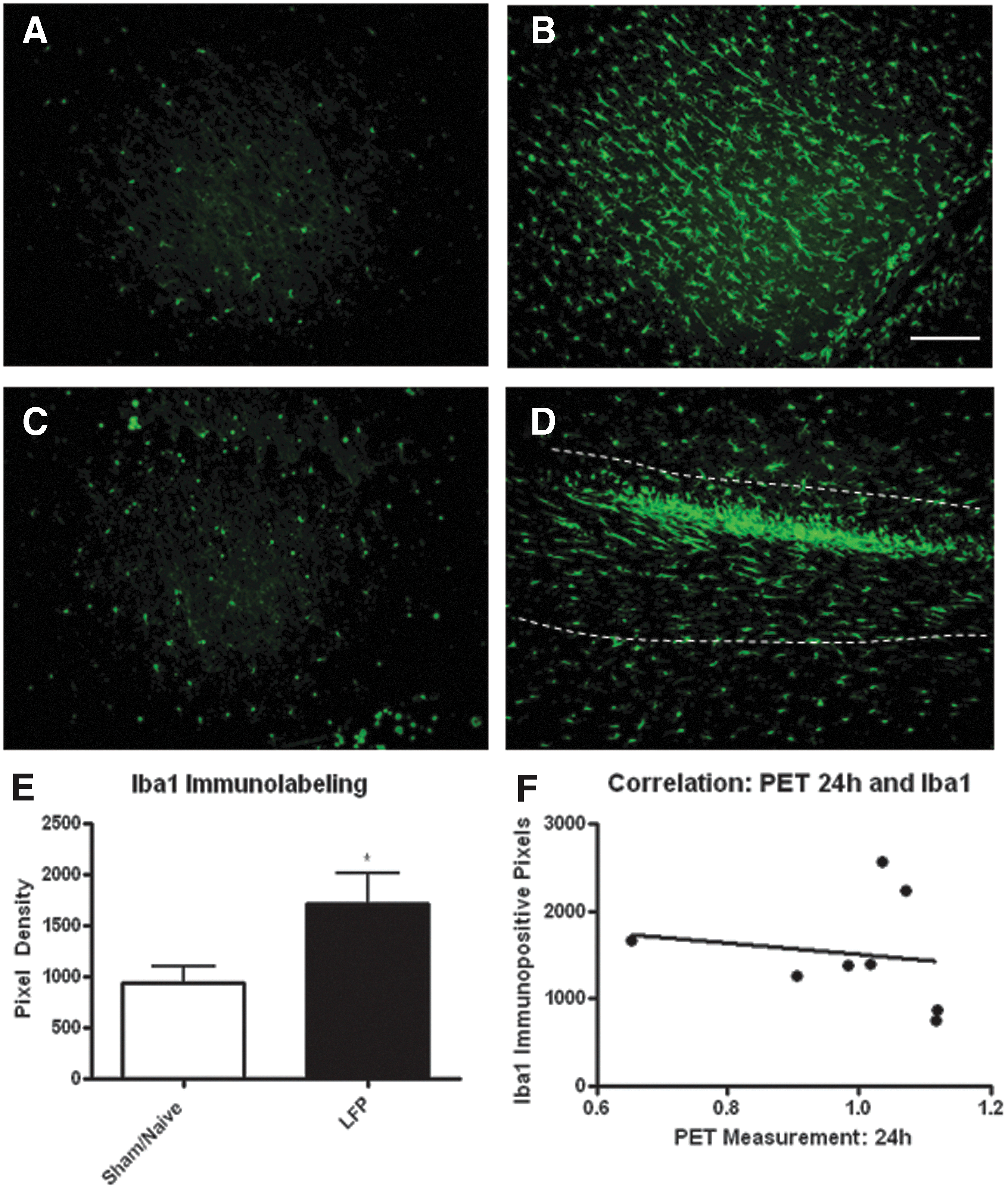
Mild lateral fluid percussion (LFP) injury induces an increase in microglia in the ipsilateral cortex. Microglia were immunostained with the Iba-1 antibody. Small cells with fine processes were observed in sham and naïve rats
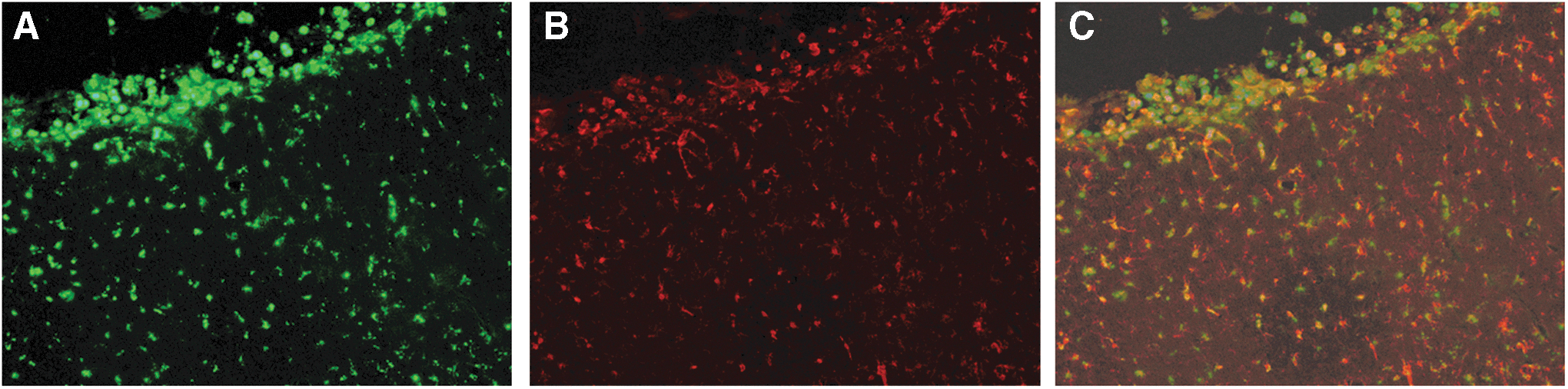
Mild lateral fluid percussion (LFP) injury induced an increase in microglial activation. Microglial activity, as indicated by an upregulation in ED1 immunolabeling
Discussion
This study demonstrates that mild LFP is an adequate model of mTBI, resulting in transient functional impairment and glial activation without neuronal loss or gross tissue damage. In this model of mTBI, a transient reduction in glucose uptake in the ipsilateral and contralateral cortices was observed. We also demonstrate that this depression in glucose uptake is apparent bilaterally and is, acutely, greater in the ipsilateral cortex than in the contralateral cortex. Although these measurements are of glucose uptake, not metabolism, these findings are comparable to the glucose hypometabolism that has been reported in human mTBI studies, 9,32 in rodent models of moderate and severe TBI, 11,16 and in a juvenile model of mTBI. 19 In general, 18FDG uptake is proportional to glucose metabolism when controlling for uptake and scan time and assuming an unchanging lumped constant (LC). However, several studies show that TBI induces a metabolic cascade that could change the LC and complicate lCMRglc measurements. 33 Therefore, analysis of this and other 18FDG-PET data should be considered carefully.
Like our work, previous animal studies showing hypometabolism or reduced glucose uptake used diffuse injury models, utilizing either the fluid percussion or a weight drop model. In all studies, an acute decline in glucose uptake or metabolism was found to peak at∼24 h post-injury. In one model of mild TBI, depressed glucose metabolism reached ∼19%, 19 which is comparable to our detected depression in glucose uptake of ∼15% in comparison with sham/naïve animals at 24 h. After a mild to moderate injury, the glucose uptake/metabolism was found to return to baseline levels by ∼10 days, 19,17,11 whereas after a more severe injury, the glucose uptake remained depressed for months. 20 For example, after a severe LFP, glucose uptake remained 16% lower than baseline at 3 months post-injury. 20
Unlike studies from moderate and severe TBI models, we found no detectable early increase in glucose uptake in our mTBI model, which is similar to human studies of mTBI. Despite some suggestion that the method of calculating glucose uptake, or, more specifically, local cerebral metabolic rate of glucose, 33 may influence the detection of this early increase, several studies have suggested that acute hyperglycolysis is likely related to an attempt by neurons to stabilize ion balance following membrane disruption. 34 As our mTBI model does not produce neuronal loss, it may not induce the activation of energetic pathways needed for acute repair that may play a role in this acute increase in glucose uptake. More likely, however, the acute induction of increased glucose uptake may take place earlier than 3 h post-injury, and was not observed at the time points imaged during our study. Future research will explore earlier post-injury time points with PET to confirm this outcome.
In rodent models and in human studies, after TBI there is a marked reduction in several markers of metabolism, such as ATP, NAA, and other metabolites. 15,18,35 –37 After mTBI, this reduction in NAA and ATP is short lived, with a maximal impairment at 24 h post-injury, 11,18 which associates well with our finding of maximal glucose uptake depression at 24 h post-injury. Previous research after LFP injury has suggested that both neurons and astrocytes contribute to these metabolic changes at 3 and 24 h post-injury, 38 and suggest that by 24 h post-injury, astrocytes are the primary cellular source of depressed glucose metabolism. However, as this was a longitudinal 18FDG-PET study, the cellular and connectivity correlates of the glucose uptake depression were not investigated in this study. Future studies will aim to clarify the cellular contribution to this outcome.
Studies have also shown that brain perfusion is reduced after mild, moderate, and severe TBI in both humans and rodent models. 12,39,40 The contribution of CBF to glucose uptake after mild LFP in rats was not considered in the current study, but will be investigated in future studies. The review article by Len and Neary 10 provides a summary of cerebrovascular pathophysiology after mild TBI in humans and animal studies. Overall, early dysfunction in CBF, oxygenation, glucose utilization, and cerebrovascular reactivity may contribute to short-lived and long-term outcomes associated with mTBI. 10 A small uncoupling between any of these essential systems may result in a chemical cascade that alters the local microenvironment leading to chronic neuronal dysfunction. Future studies should focus on understanding this critical cascade.
Motor abnormalities after mTBI have been reported in a subset of the human population, and include abnormalities in gait, 41 balance, 42 and walking speed. 42 Our mTBI model did show a significant transient impairment in motor function, particularly at 24 h post-injury, utilizing two variations on the classical beam walk task. Our study, however, did not demonstrate subacute (2 weeks) cognitive impairment following a cognitive learning task, the Barnes Maze. Likewise, studies using similar injury parameters 21 have not shown subacute to chronic (4 weeks) cognitive deficits following mTBI based on learning tested with the Morris Water Maze. In human TBI, glucose metabolism deficits have been correlated with chronic cognitive impairments in some patients. 32
To investigate the cause of the perturbations in glucose uptake, immunohistochemistry was performed to evaluate neuronal, astrocytic, and microglial responses to injury. Despite the lack of neuronal loss, evidence of glial activation, including astrocytic and microglial proliferation and morphological alterations, and of axonal disruption was observed at both 10 and 21 days after injury in regions observed to demonstrate glucose uptake alterations. This is in agreement with previous studies. 21,43 Interestingly, Shultz et al. found that the microglial changes were significantly correlated with cognitive impairments observed in the Morris Water Maze test. 21,23 It is well known that a primary response to injury in the brain is inflammation, including activation of resident microglia and invasion of blood-borne macrophages. 44,45 After mTBI, an increase in microglial/macrophage immunolabeling has been reported by 4–5 days post-injury, 21,46 and increased expression of microglial markers has been observed for weeks to months after mild, moderate, and severe TBI. 47 Further, models of neuroinflammation without direct injury to the central nervous system have shown that microglial activation can directly result in neuronal damage and glucose metabolism alterations. 48 Alternatively, microglial and macrophage activation have been shown to directly influence 18FDG-PET results, showing high 18FDG uptake in areas of inflammatory lesions. 49 Our data now show that there is a significant increase in microglial activation in this model of mTBI, but a lack of significant correlation between 18FDG uptake at 24 h and quantitation of microglial activation.
On the contrary, astrocyte activation has been shown to be correlated with 18FDG changes after TBI and other models of neuronal damage, 50,51 and our data show a significant correlation between GFAP quantitation and 18FDG uptake at 24 h post-injury. Several TBI studies have shown a pronounced upregulation of astrocyte markers of activation after injury, including GFAP and vimentin, with a time course that strongly parallels the recovery in glucose uptake observed in our current study. 44,52,53 It is important to note that immunohistochemistry was performed at 10 and 21 days post-injury, suggesting that these glial changes were sustained for weeks after the initial injury, and after the glucose uptake returned to baseline levels. It is unknown whether the return to normal uptake levels is the result of neuronal recovery or of an increase in energy demand from glial cells, and is a question for future research.
Diffuse axonal injury after mTBI is reported to occur at hours to days after the injury in both humans and rodent models (for review, see Smith et al. 54 ). After mTBI by LFP, we found an increase in silver stain and a decrease in corpus callosum width, as well as a decrease in MAP2 staining, indicating diffuse axonal injury. Axonal damage, particularly in the corpus callosum, may be a cause for the bilateral spread of the observed glucose uptake depression.
Conclusion
In conclusion, we now show that a mild LFP based mTBI model can induce transient functional impairment, axonal damage, glial activation, and acute glucose uptake depression. This depression appears to be quantitatively related to the injury severity, both in terms of degree of decreased uptake and in length of time that uptake is impaired. 11,20 Further, acute 18FDG uptake is depressed and significantly correlated with future astrocyte activation. This study provides the initial proof of principle for 18FDG-PET as a method to noninvasively detect persistent metabolic dysfunction as an indicator of chronic effect of mTBI, and correlates with a behavioral effect that may be more difficult to quantify in humans.
Footnotes
Acknowledgments
This work was funded by the Department of Defense in the Center for Neuroscience and Regenerative Medicine. The authors thank Yujia Zhao, Guzal Khayrullina, and Alexander Gordon for technical assistance, and the Uniformed Services University Biomedical Instrumentation Center/Histopathology department for assistance with the Fast blue and silver staining procedures.
The opinions or assertions contained herein are the private ones of the author(s) and are not to be construed as official or reflecting the views of the DoD or the USUHS.
Author Disclosure Statement
No competing financial interests exist.