
Abstract
Traumatic brain injury (TBI) is associated with loss of cerebrovascular autoregulation, which leads to cerebral hypoperfusion. Mitogen activated protein kinase (MAPK) isoforms ERK, p38, and JNK and endothelin-1 (ET-1) are mediators of impaired cerebral hemodynamics after TBI. Excessive tissue plasminogen activator (tPA) released after TBI may cause loss of cerebrovascular autoregulation either by over-activating N-methyl-D-aspartate receptors (NMDA-Rs) or by predisposing to intracranial hemorrhage. Our recent work shows that a catalytically inactive tPA variant (tPA-S481A) that competes with endogenous wild type (wt) tPA for binding to NMDA-R through its receptor docking site but that cannot activate it, prevents activation of ERK by wt tPA and impairment of autoregulation when administered 30 min after fluid percussion injury (FPI). We investigated the ability of variants that lack proteolytic activity but bind/block activation of NMDA-Rs by wt tPA (tPA-S481A), do not bind/block activation of NMDA-Rs but are proteolytic (tPA-A296–299), or neither bind/block NMDA-Rs nor are proteolytic (tPA-A296–299S481A) to prevent impairment of autoregulation after TBI and the role of MAPK and ET-1 in such effects. Results show that tPA-S481A given 3 h post-TBI, but not tPA-A296–299 or tPA-A296–299S481A prevents impaired autoregulation by upregulating p38 and inhibiting ET-1, suggesting that tPA-S481A has a realistic therapeutic window and focuses intervention on NMDA-Rs to improve outcome.
Introduction
T
Among the possible mediators, glutamate is known to bind to each of three ionotropic receptor subtypes named after synthetic analogues: N-methyl-D-aspartate (NMDA), kainate, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA). Activation of NMDA receptors (Rs) contributes to excitotoxicity, 7 but also elicits cerebrovasodilation and represents a mechanism by which local metabolism is coupled to CBF. 8 Glutamatergic hyperactivity has been implicated in neurotoxicity after TBI, whereas NMDA antagonists are protective in some models. 9,10 Although CBF is thought to contribute to neurologic outcome, little attention has been given to the role of NMDA-Rs in the regulation or dysregulation of this process. In our studies, NMDA-R–mediated vasodilation is reversed to vasconstriction after FPI in the piglet. 11
Similarly, upregulation of endogenous wild type (wt) tissue plasminogen activator (tPA) after TBI not only enhances excitotoxic neuronal cell death through excessive activation of NMDA-Rs, 12,13 but also contributes to impaired NMDA-R–mediated vasodilation, loss of autoregulation during hypotension, and histopathology 14 –17 via mitogen activated protein kinase (MAPK), 18 a family of three kinases (ERK, p38, and JNK) critically important in hemodynamics after TBI. 14 We found that activation of ERK and JNK contributed to impaired NMDA-R cerebrovasodilation after FPI, while p38 was protective. 18 Endothelin-1 (ET-1) is also upregulated and antagonizes NMDA-R–mediated vasodilation after FPI. 19 The link between activation of NMDA-Rs and autoregulation is supported by several interventions at the receptor level. The NMDA antagonist MK 801 protects against cerebral dysregulation after FPI, 20 but its toxicity limits use in humans. Alternatively, glucagon minimizes the surge in glutamate after TBI in mice and pigs that is partially responsible for over-activation of NMDA-Rs and preserves autoregulation during hypotension by blunting upregulation of tPA, which prevents tissue damage. 15
Excessive tPA released after TBI may impair cerebral hemodynamics by over-activating NMDA-Rs as well. Post-TBI (30 min) administration of a catalytically inactive tPA variant (tPA-S481A) that competes with wt tPA for binding to NMDA-R through its receptor docking site but cannot activate it, prevents activation of ERK MAPK and thereby prevents impairment of autoregulation after FPI. 21 The potential deleterious role of tPA-mediated fibrinolysis and intracerebral hemorrhage (ICH) after TBI, however, may also contribute to adverse outcome. Also, a therapeutic window of greater than 30 min is needed for any agent to have clinical utility for this indication.
We hypothesize that over-activation of NMDA-Rs is a major contributor to adverse outcome by the rise in endogenous tPA after TBI. To test this hypothesis, we used a model in which ICH does not predominate 14,17,21 and examined the effect of tPA variants that are either: (1) not proteolytic (do not cleave/activate NMDA-Rs or fibrin) but bind/block activation of NMDA-Rs (tPA-S481A); (2) do not bind NMDA-Rs but are proteolytic (tPA-A296–299); or (3) neither bind/activate NMDA-Rs nor are proteolytic (tPA-A296–299S481A), to prevent impairment of autoregulation after TBI, as well as the roles of p38 MAPK and ET-1 as mediators of these effects.
Methods
tPA variants
To generate tPA-S481A, tPA-A296–299 and tPA-A296–299S481A, mutations were introduced in wt tPA by polymerase chain reaction using the QuickChange Mutagenesis kit (Stratagene, La Jolla, CA), and the complete sequences were verified. 22 –24 Each protein contains two extra amino acids, RS-, at the NH2 terminus, resulting from the introduction of the Bgl II cloning site. Proteins were expressed in S2 Drosophila Expression System (Invitrogen) according to the manufacturer's protocol and were purified by affinity chromatography using anti-tPA coupled to CN-Br activated Sepharose.
We constructed three tPA variants with mutations in either the NMDA-R binding site, the catalytic triad, or both that were designed to: (1) selectively block proteolytic activation of NMDA-Rs by endogenous wt tPA; (2) selectively block proteolysis; or (3) both (Table 1A). The first variant, tPA-S481A, has a single mutation in the catalytic site that enables it to bind to but not to cleave and activate NMDA-Rs or fibrin (Table 1A). At therapeutic concentrations, tPA-S481A will compete with high levels of endogenous wt tPA for binding to NMDA-Rs and fibrin (i.e., tPA binds to fibrin through the finger and K2 domains, outside both the NMDA-R docking site and the catalytic site) and protect both from proteolysis, and it will neither activate NMDA-Rs nor cause fibrinolysis (Table 1). 25 The second tPA variant has a mutated docking site, tPA-K296A/H297A/R298A/R299A (tPA-A296–299), that prevents it from binding to NMDA-Rs, while maintaining binding to fibrin, and it will augment the proteolytic activity of endogenous wt tPA (Table 1A). The third is a double-variant tPA-A296–299S481A that is designed not to bind to NMDA-R and lacks proteolytic activity. The double variant is used to compete with endogenous tPA for binding to fibrin and thereby prevent ICH (Table 1A), but not bind to NMDA-R and therefore not compete with receptor activation by endogenous wt tPA (Table 1B). We hypothesized that tPA-S481A would protect against the toxicity of endogenous wt tPA in this model by inhibiting the binding and activation of NMDA-Rs without causing ICH (Table 1B). The difference between the protective effect of tPA-A296–299 and tPA-A296–299S481A compared with tPA-S481A would help to elucidate the relative contribution of the two pathways (Table 1B).
tPA, tissue plasminogen activator; NMDA-R,
Numeric scoring from 0 to 5.
Closed cranial window and brain injury procedures
Newborn pigs (1–5 days old, 1.0–1.4 kg) of both sexes were studied. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Animals were anesthetized with isoflurane (1–2 MAC) and maintained with a-chloralose (50–80 mg/ kg. supplemented with 5 mg/ kg/h intravenously [i.v.]).
The closed cranial window technique for measuring pial artery diameter and collection of cerebrospinal fluid (CSF) for enzyme-linked immunosorbent assay (ELISA) analysis has been described. 6 This window consisted of three parts: a stainless steel ring, a circular glass cover-slip, and three ports consisting of 17-gauge hypodermic needles attached to three precut holes in the stainless steel ring. For placement, the dura was cut and retracted over the cut bone edge. The cranial window was placed in the opening and cemented in place with dental acrylic. The volume under the window was filled with a solution, similar to CSF, of the following composition (in mM): 3.0 KCl, 1.5 MgCl2, 1.5 CaCl2, 132 NaCl, 6.6 urea, 3.7 dextrose, and 24.6 NaHCO3. This artificial CSF was warmed to 37° C and had the following chemistry: pH 7.33, pCO2 46 mm Hg, and pO2 43 mm Hg, which was similar to that of endogenous CSF. Pial arterial vessel diameter was measured with a microscope, a camera, a video output screen and a video microscaler.
The method used to induce brain FPI has been described. 15 A device designed by the Medical College of Virginia was used. A small opening was made in the frontoparietal skull contralateral to the cranial window. A metal shaft was sealed into the opening on top of intact dura and fluid coupled to the brain injury device; i.e, the shaft was connected to the transducer housing, which was in turn connected to the fluid percussion device. The device itself consisted of an acrylic plastic cylindrical reservoir 60 cm long, 4.5 cm in diameter, and 0.5 cm thick. One end of the device was connected to the transducer housing, whereas the other end had an acrylic plastic piston mounted on O-rings. The exposed end of the piston was covered with a rubber pad. The entire system was filled with 0.9 % saline. The percussion device was supported by two brackets mounted on a platform.
FPI was induced by striking the piston with a 4.8 kg pendulum. The intensity of the injury (usually 1.9–2.3 atm. with a constant duration of 19–23 ms) was controlled by varying the height from which the pendulum was allowed to fall. The pressure pulse of the injury was recorded on a storage oscilloscope triggered concurrently with the fall of the pendulum. The amplitude of the pressure pulse was used to determine the intensity of the injury.
Protocol
Pial small arteries (resting diameter, 120–160 μm) were examined. Typically, 2–3 mL of artificial CSF were flushed through the window over 30 sec, and excess CSF was allowed to run off through one of the needle ports. For sample collection, 300 μL from the total cranial window volume of 500 μL was collected by slowly infusing artificial CSF into one side of the window and allowing the CSF to drip freely into a collection tube on the opposite side.
Seven experimental groups were studied (all n=5): (1) sham control, (2) FPI, vehicle treated, (3) FPI treated with tPA (1 mg/kg i.v.), (4) FPI treated with tPA-S481A (1 mg/kg i.v.), (5) FPI treated with tPA-A296–299 (1 mg/kg i.v.), (6) FPI treated tPA-A296–299S481A (1 mg/kg i.v.), and (7) FPI treated with tPA-S481A+the p38 MAPK antagonist SB 203580 (1 mg/kg i.v.). Hypotension was induced by the rapid withdrawal of either 5–8 or 10–15 mL blood/kg to induce moderate or severe hypotension (decreases in mean arterial blood pressure of 25 and 45%, respectively). 15 Such decreases in blood pressure were maintained constant for 10 min by titration of additional blood withdrawal or blood reinfusion. 15 The vehicle for all agents was 0.9% saline, except for the MAPK inhibitor, which used dimethyl sulfoxide (100 μL) diluted with 9.9 mL 0.9% saline. In sham control animals, responses to hypotension (moderate, severe), papaverine, NMDA (10−8, 10−6 M), and PGE2 (1, 10 ng/mL) were obtained initially and then again 4 h later in the presence of the agent vehicle. In animals treated post-FPI, tPA variants were administered 3 h post-injury, and responses to vasoactive stimuli were measured both before and 4 h post-injury.
CBF was measured in the cerebral cortex and hippocampus using radioactively labeled microspheres. 15 Briefly, neutron activated microspheres (Biopal; 15μm diameter; 300,000–800,000 spheres) were injected into the left ventricle. After each experiment, the piglet was sacrificed and the brain removed and weighed. The energy from each nuclide was separated by differential spectroscopy. Aliquots of the actual microsphere solutions injected were used for overlap calculations. The count in each milliliter per min of blood flow was determined by dividing the counts in the reference withdrawal by the rate of reference withdrawal. Thus, blood flow can be calculated as Q=C×R×CR−1, where Q is brain blood flow (in mL/min), C is counts per min (cpm) in the tissue sample, R is the rate of withdrawal of reference blood sample (in mL/min), and CR is the total counts in the reference blood sample. CBF so determined reflects flow to the cerebral cortex and hippocampus both ipsilateral and contralateral to the injury site. Previous studies have shown that multiple injections of microspheres in similar quantities (300,000–800,000 spheres per nuclide) do not cause significant detectable microvessel plugging, thereby limiting this as an experimental caveat for data interepretation. 6,15
ELISA and radioimmunoassay (RIA)
Commercially available ELISA kits were used to quantity CSF p38 MAPK (Assay Designs) concentration, while RIA kits were used to quantify ET-1 (Phoenix Pharmaceuticals). Values for phosphorylated p38 MAPK enzyme were normalized to total form and then expressed as percent of the control condition.
Statistical analysis
Pial artery diameter, CSF p38 MAPK, and ET-1 values were analyzed using analysis of variance for repeated measures. If the value was significant, the data were then analyzed by Fisher protected least significant difference test. An α level of p<0.05 was considered significant in all statistical tests. Values are represented as mean±standard error of the mean of the absolute value or as percentage changes from control value.
Results
Wild type tPA aggravates, tPA- S481A mitigates, and tPA-A296–299 and tPA-A296–299S481A have little effect on impaired cerebral autoregulation during hypotension post-FPI
CBF was unchanged during hypotension (moderate 25%, severe 45% reduction in mean arterial pressure) in sham control animals, indicating cerebral autoregulation remained intact (Fig. 1A, 1B). CBF was reduced after FPI and reduced further during hypotension, which was aggravated by administration of exogenous wild type (wt) tPA (1 mg/kg iv) 3 h post-injury (Fig. 1A, 1B). Similarly, FPI reduced pial artery diameter, which was reduced further by wt tPA (Fig. 1C). In contrast, administration of tPA-S481A (1 mg/kg i.v.) 3 h post-FPI, prevented injury-induced pial artery constriction and hypoperfusion in the parietal cortex and hippocampus monitored 1 h later (4 h post-injury) (Fig. 1). Further reductions in CBF during hypotension (at 4 h post-injury) were also blunted by tPA-S481A (1 mg/kg i.v.), administered at 3 h post-FPI (Table 1), similar to the effect of tPA-S481A administered 30 min post-injury. 21 Aggravation of hypoperfusion, impairment of cerebral autoregulation during hypotension, and reductions in pial artery diameter by exogenous wt tPA after FPI was blocked by co-administration of tPA-S481A (Fig. 1). Neither tPA-A296–299 nor tPA-A296–299S481A, however, caused a significant reduction in pial artery vasoconstriction, hypoperfusion, or impaired cerebral autoregulation after FPI (Fig. 1).

Effect of fluid percussion injury (FPI) on cerebral blood flow (CBF) in the parietal cortex (
FPI blunts pial artery dilation during hypotension. 15 This response was reversed to vasoconstriction when wt tPA was administered after FPI (Fig. 2). In contrast to wt tPA, administration of tPA-S481A (1 mg/kg i.v.) 3 h post-FPI blocked injury-induced pial artery constriction and almost fully restored pial artery dilation monitored 1 h later (4 h post-injury) (Fig. 2). In contrast, neither tPA-A296–299 nor tPA-A296–299S481 protected pial artery dilation during hypotension (Fig. 2). Papaverine-induced pial artery dilation was unchanged after FPI, 15 and none of the tPA variants significantly affected its cerebrovasodilator effect either before or after brain injury (Fig. 2). Amelioration of pial artery vasoconstriction associated with FPI by tPA- S481A occurred within minutes and was similar when administered 30 min or 3 h post-injury (Fig. 3).

Influence of A. hypotension (moderate, severe) and papaverine ((10−8,10−6 M) on pial artery diameter before (control), 4 h after FPI, and after FPI in animals post-treated (3 h) with tPA, tPA-S481A, tPA-A296–299, and tPA-A296–299S481A (1 mg/kg i.v.), n=5. Baseline pial artery diameters were: 130±14, 102±10, 94±8, 122±13, 111±10, and 108±8 μm, respectively for the six animal groups, control, FPI, FPI+tPA, FPI+tPA-S481A, FPI+tPA-A296–299, and FPI+tPA-A296-–99S481A. *p<0.05 compared with corresponding control; + p<0.05 compared with FPI alone.
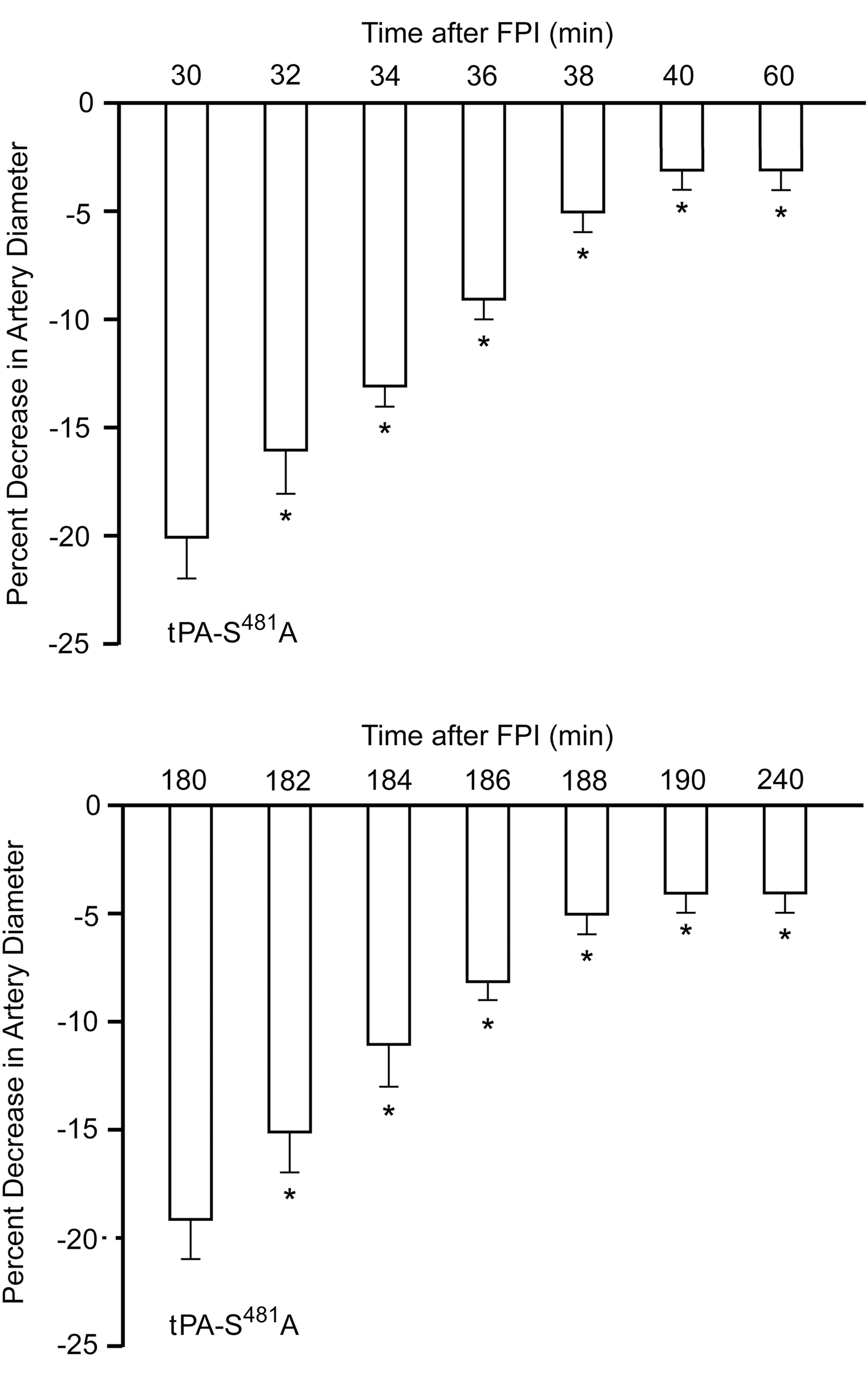
Influence of tPA-S481A (1 mg/kg iv) on pial artery diameter as a function of time when administered either at 30 min (
Wild type tPA aggravates, tPA- S481A mitigates, and tPA-A296–299 and tPA-A296–299S481A have no effect on impaired cerebrovasodilation mediated by activation of NMDA-Rs and PGE2
NMDA-R–mediated pial artery dilation was reversed to vasoconstriction after FPI (Fig. 4A). 20 Wild type tPA (1 mg/kg i.v.) administered 3 h post-insult aggravated NMDA-R–mediated pial artery vasoconstriction (Fig. 4A), whereas tPA-S481A (1 mg/kg i.v., 3 h post-insult) did not. Rather, vasodilation was observed (Fig. 4A), similar to that observed when tPA-S481A was administered at 30 post-injury. 21 tPA-S481A (1 mg/kg i.v., 3 h post-insult) also prevented impairment of PGE2-mediated cerebrovasodilation (Fig. 4B), similar to the protection observed when the tPA variant was administered 30 min post-injury. 21 Neither tPA-A296–299 nor tPA-A296–299S481A, however, prevented the reversal of NMDA-R–mediated dilation to vasoconstriction after FPI (Fig. 4A), consistent with their inability to bind to NMDA-Rs. Similarly, in piglets treated with tPA-A296–299 and tPA-A296–299S481, impairment of PGE2 dilation was no different in animals given vehicle post-FPI (Fig. 4B).
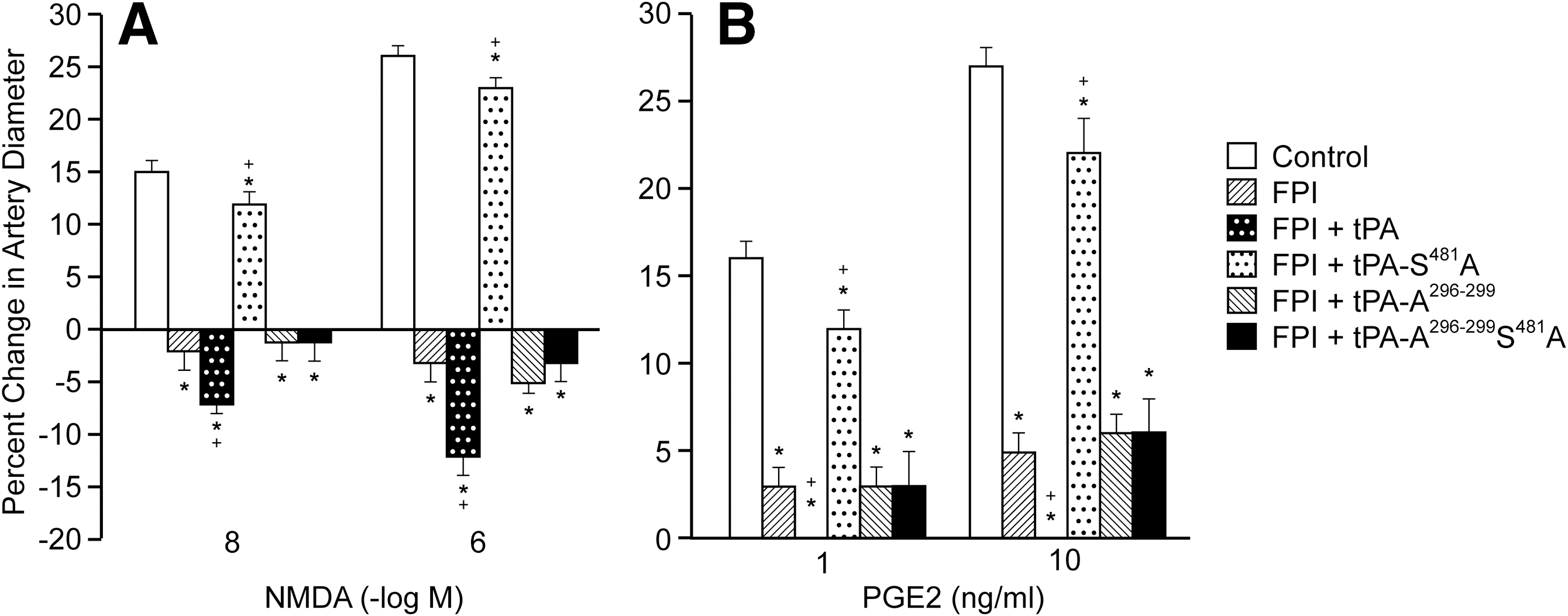
Influence of (
tPA-S481A prevents impaired autoregulation and NMDA-R–mediated pial artery dilation after FPI by upregulating p38 MAPK and downregulating ET-1
tPA augments reversal of NMDA-R–mediated dilation to vasoconstriction and contributes to impairment of autoregulation after FPI by upregulating ERK and JNK MAPK. 18,20 As upregulation of p38 MAPK by wt tPA appears to limit impairment of NMDA-R–mediated pial artery dilation after FPI, 18 we speculated that a change in the MAPK isoform profile toward further increasing p38, concomitant with diminished ERK might serve as the mechanism underlying the vasoprotective effect of tPA-S481A after TBI. Indeed, the increase in CSF p38 MAPK post-FPI was augmented mildly by wt tPA but was elevated more robustly by tPA-S481A (Fig. 5A).
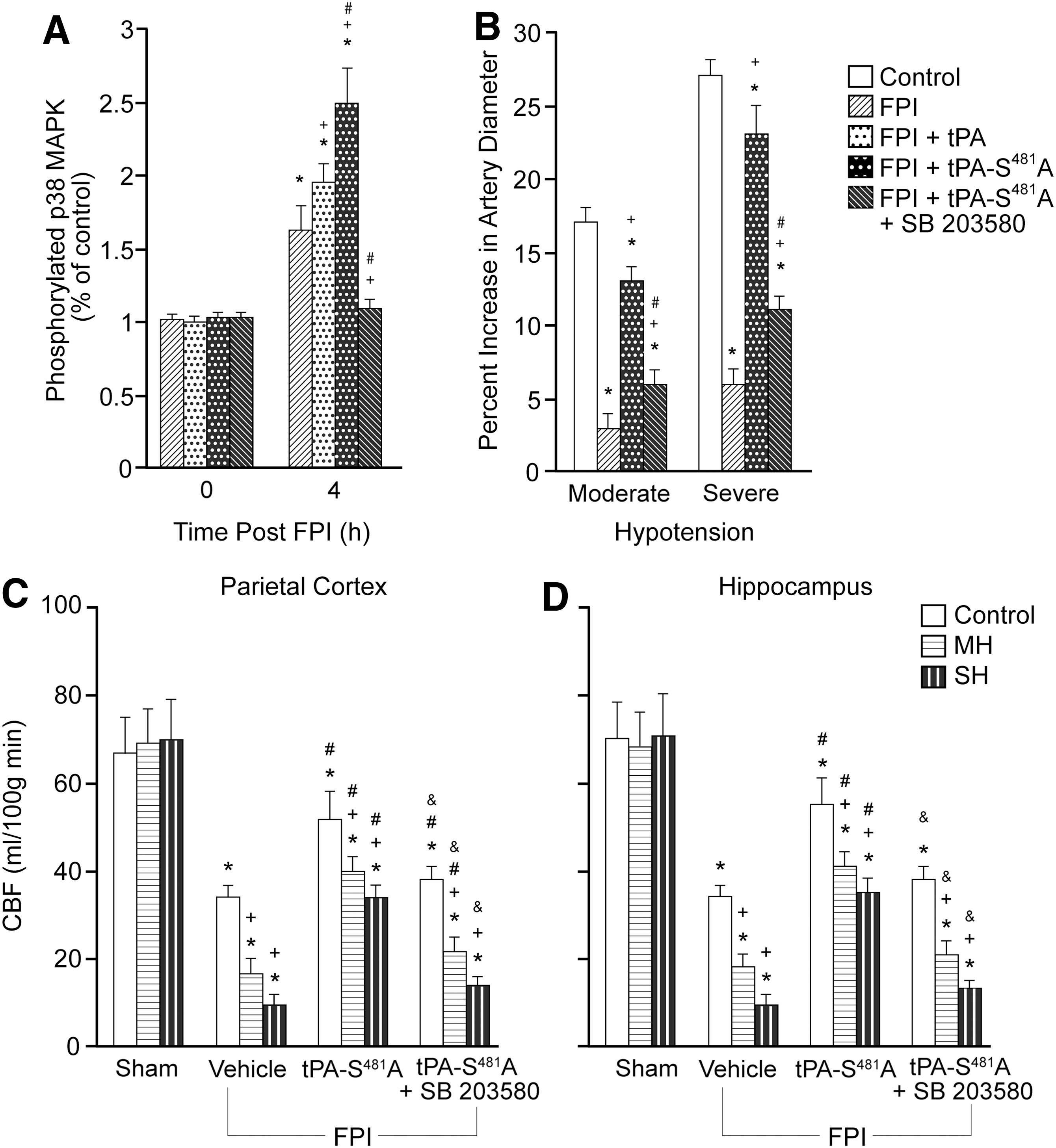
A. Influence of fluid percussion injury (FPI) (4 h) on (
We previously observed that tPA-S481A blocked the rise in phosphorylated ERK in the CSF seen after FPI. 21 Co-administration of the p38 MAPK antagonist SB 203580 (1 mg/kg i.v.) with the tPA- S481A 3 h post-FPI blocked upregulation of phosphorylated p38 MAPK (Fig. 5A) and the ability of the variant to prevent hypoperfusion and the further reduction in CBF during hypotension (Fig. 5B, 5C). Together, these biochemical and pharmacological data suggest that tPA- S481A prevents impairment of cerebral autoregulation after FPI both by augmenting p38 and by inhibiting upregulation of ERK MAPK. 21
We also observed that ET-1 is upregulated and antagonizes NMDA-R–mediated dilation after FPI. 19 Therefore, we examined the effect of wt tPA and tPA-S481A on ET-1 levels in the CSF after FPI. CSF ET-1 was elevated after FPI and was increased further by exogenous wt tPA, but both the total upregulation and that portion augmented by wild type tPA were both blocked by tPA-S481A administered at 3 h post-FPI (Fig. 6). Co-administration of SB 203580 with tPA-S481A modestly reversed the total blockade of ET-1 upregulation observed with tPA-S481A alone (Fig. 6). These data suggest that tPA-S481A-mediated protection of autoregulation after TBI may result from the capacity of p38 to block upregulation of ET-1 post TBI.
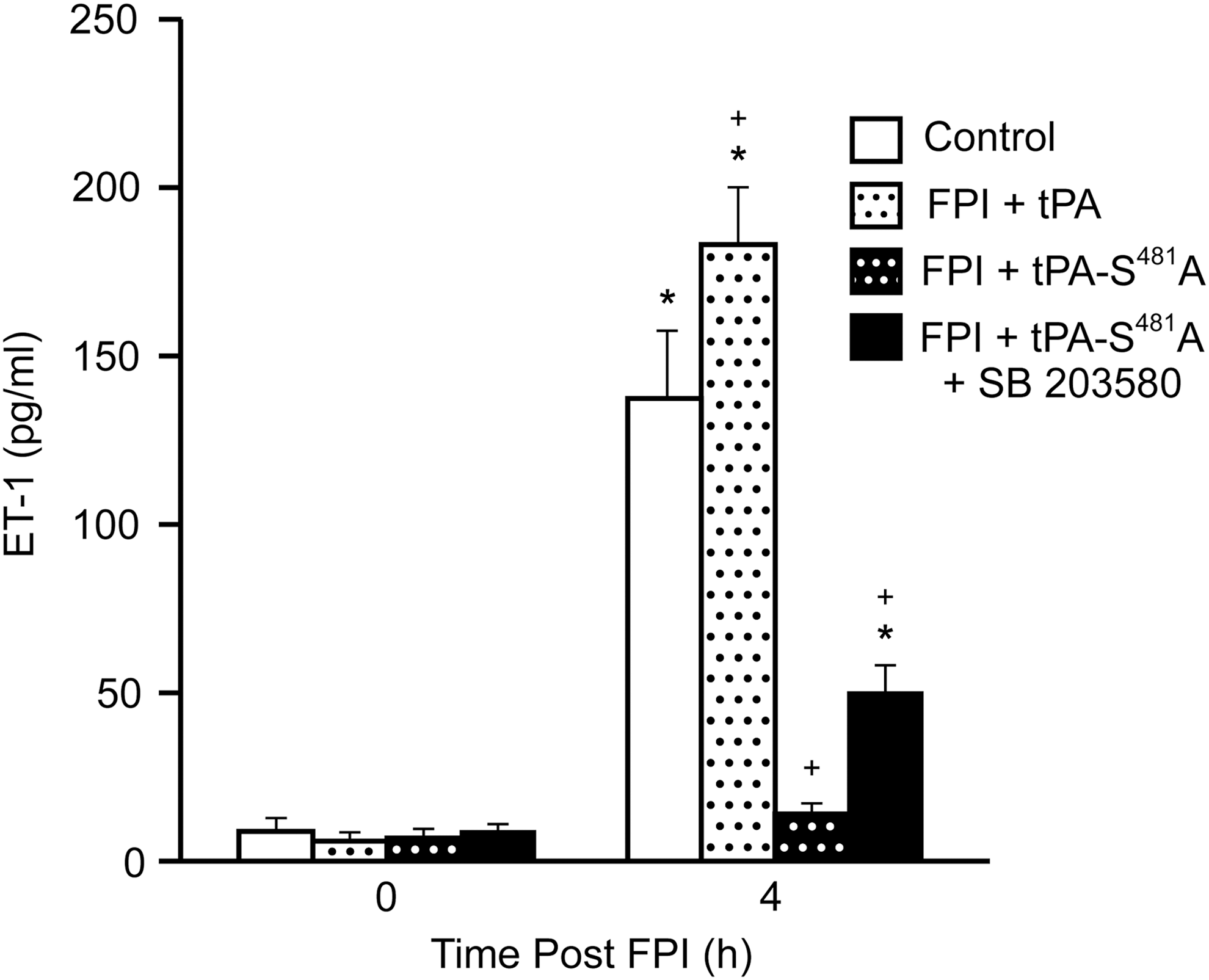
Influence of fluid percussion injury (FPI) (4 h) on endothelin-1 (ET-1) (pg/mL) in FPI, FPI+tPA, FPI+tPA-S481A, and FPI+tPA-S481A+SB 203580 (1 mg/ kg i.v.) treated (3 h) pigs, n=5. *p<0.05 compared with FPI; + p<0.05 compared with FPI+tPA.
Blood chemistry and ICH
Blood chemistry values were collected before and after all experiments. There were no statistically significant differences between sham control, FPI, and FPI and drug-treated animals. FPI is reported to produce little ICH, 26 and we did not observe hemorrhage after FPI in the present studies, consistent with previous studies using this model in the piglet. 14,15,17
Discussion
The toxicity caused by increased endogenous tPA has been well studied in animal models of stroke and in humans. TBI is not stroke, however: the etiology, pathophysiology, and the critical care pathways for treatment are related, but differ in important ways, including the role of endogenous wt tPA and the risk of hemorrhage. Endogenous wt tPA appears to be especially important in determining the outcome of TBI, whereas in stroke, the concentration of exogenous tPA administered as a therapeutic predominates. Excessive endogenous tPA released after TBI can cause tissue damage by over-activating NMDA-Rs or by exacerbating ICH, depending in part on the nature and force of the impact. To delineate the role of each pathway in TBI and to block whichever is/are operative in this model, the approach we used is based on our recently published finding that the docking site in tPA is required to bind and cleave and thereby activate NMDA-Rs. 22 –24 Previous work by others indicates that tPA cleaves the NR-1 subunit of NMDA-Rs, which contributes to excitoxicity by increasing the influx of calcium, 12,27 by signaling through an interaction with the NR2B subunit. 28
Human TBI is heterogenous, limiting the ability of animal models to mimic the complexity of the human situation. FPI is thought to be a good mimic of TBI caused by shaken impact and motor vehicle accidents. 26 We also chose to study FPI to isolate the effect of NMDA-R occupancy/cleavage/excitotoxicity by endogenous tPA from its role in causing ICH. FPI is reported to produce little ICH, 26 and we did not observe hemorrhage after FPI in the piglet. 14,15,17 This allowed us to explore the role of NMDA-R in tPA-mediated excitoxicity post-FPI and to use visible ICH as an adverse end-point from intervention.
We recently reported that administration of tPA-S481A 30 min post-injury prevents loss of cerebrovascular autoregulation during hypotension and limits histopathology produced by FPI in the piglet. 21 We therefore hypothesized that tPA-S481A provides a novel approach toward limiting neuronal injury by blocking excessive activation of NMDA-Rs caused by the robust increase in glutamate and tPA within the brain after TBI (Table 1). Whether tPA-S481A works by blocking NMDA-R–mediated signaling through non-proteolytic pathways and/or prevents diffuse microvascular bleeds in this model, however, could not be determined using this single variant because it acted on both pathways. Comparative study of the three mutants in the present study helped to definez the contribution of each pathway to negative outcome after TBI.
Results of the present study show that FPI blunts pial artery dilation during hypotension, 15 and this response is reversed to pial artery vasoconstriction when wt tPA was administered. Although our previous studies had suggested that wt tPA inhibits autoregulation, 21 these data are the first direct demonstration. In contrast, administration of tPA-S481A 3 h post-FPI blocked injury-induced pial artery constriction and hypoperfusion in the parietal cortex and hippocampus monitored 1 h later (4 h post-TBI). These data are very similar to that reported for tPA-S481A administered at 30 min post-injury, with hemodynamics monitored at 1 h post-insult. 21 Amelioration of pial artery vasoconstriction associated with FPI by tPA-S481A occurred within minutes and was similar when administered 30 min or 3 h post-injury. This is important because it means that tPA-S481A acts on its target tissues within a realistic therapeutic time frame. Previously, we observed that the NMDA-R antagonist MK801 prevented impairment of cerebral autoregulatory pial artery dilation after FPI. 20 This outcome suggested that the protection of cerebral autoregulation by tPA-S481A post-TBI likely results from ameliorating toxicity induced by the joint action of tPA and glutamate signaling through NMDA-Rs. In support of this, papaverine-mediated vasodilation was unchanged after FPI and tPA-S481A, indicating the specificity of the variant on endogenous tPA as a mediator.
Our previous studies did not exclude the possibility that tPA-S481A prevents microhemorrhaging, which in turn impairs autoregulation and CBF. We did not, however, observe any hemorrhages in our animals, 14,15,17 consistent with the previous experience of others using this model. 26 Moreover, neither tPA-A296–299, which maintains proteolytic activity, nor tPA-A296–299S481, which does not, caused a significant reduction of pial artery vasoconstriction and hypoperfusion after FPI. In addition, pial artery dilation during hypotension was protected only modestly after FPI by tPA-A296–299 and tPA-A296–299S481A and neither variant prevented the loss in autoregulation and reduction in CBF that results from hypotension after FPI. Together, these findings focus attention on neurotoxicity mediated through NMDA-Rs.
Activation of NMDA-Rs elicits cerebrovasodilation and may represent one mechanism by which local metabolism is coupled to blood flow. 8 More recently, it was observed that tPA is critical for the full expression of the flow increase evoked by activation of the mouse whisker barrel cortex. 29 Specifically, tPA promoted nitric oxide (NO) synthesis during NMDA-R activation by modulating phosphorylation of nNOS. 29 These findings suggest that tPA is a key factor linking NMDA-R activation to NO synthesis and functional hyperemia.
NMDA-R induced pial artery vasodilation is reversed to vasoconstriction by FPI in the newborn pig. 11 Release of endogenous tPA in the setting of FPI aggravates NMDA-R–mediated vasoconstriction. 11,18 The PAI-1 derived hexapeptide EEIIMD blocked reversal of NMDA-R induced dilation to vasoconstriction as well as reductions in baseline pial artery diameter after FPI, 11 indicating that the interaction between tPA and NMDA-Rs is altered in the setting of TBI. Because the NMDA-R antagonist MK801 prevents impairment of pial artery dilation during hypotension after FPI, 20 these data indicate that reversal of NMDA-R–mediated dilation to vasoconstriction and its aggravation by tPA likely contributes to cerebral dysregulation in the setting of TBI.
Pial artery dilation during hypotension is associated with release of dilator prostaglandins (PGs), such as PGE2, while the cyclooxygenase inhibitor indomethacin blocks the vascular response, indicating that PGs contribute to autoregulation. 30 FPI impairs PGE2-mediated pial artery dilation, 31 which probably contributes to disturbed cerebral autoregulation after TBI. This might be better explained by increased superoxide production after FPI, 32 which together with increased NO will produce excessive peroxynitrite. Once formed, peroxynitrite might impair dilation of the cerebral vasculature. We, therefore, investigated the relationship between NMDA-R and tPA in the context of impaired PG-mediated vasodilation after TBI.
tPA-S481A (3 h post-insult) did not cause NMDA-R mediated vasoconstriction. Instead, vasodilation was observed, similar to what we observed when tPA- S481A was administered 30 min. 21 tPA-S481A also significantly prevented impairment of PGE2-mediated cerebrovasodilation, similar to our findings when that tPA variant was administered 30 min post-injury. 21 Taken together, these data suggest that tPA-S481A protects against cerebrovascular dysregulation after TBI in part by preventing impairment of NMDA-R and PGE2-induced pial artery vasodilation. Neither tPA-A296–299 nor tPA-A296–299S481A prevented the reversal of NMDA-R–mediated dilation to vasoconstriction after FPI, consistent with their inability to bind to NMDA-Rs. Similarly, in piglets treated with tPA-A296–299 and tPA-A296–299S481, impairment of PGE2 dilation was no different than in animals given vehicle post-FPI. The findings that tPA-A296–299 and tPA-A296–299S481 did not enhance inhibition of responses to PGE2 and activation of NMDA-Rs like wt tPA further argues against an important role for excessive bleeding in impairing cerebral hemodynamics in this model (Table 1B). Bleeding may contribute to dysregulation in the clinical setting, however, when TBI is more severe.
We next investigated the mechanism by which tPA-S481A prevented impairment of autoregulation post-TBI. MAPK, a family of kinases (ERK, p38, and JNK), is a post-receptor signaling system that contributes to setting vascular tone and is upregulated after TBI. 14 Release of ERK MAPK after FPI in the piglet contributes to reductions in CBF and worsens histopathology. 14 wt tPA aggravates reversal of NMDA-R–mediated dilation to vasoconstriction and contributes to impairment of autoregulation after FPI by upregulating ERK and JNK MAPK. 18,21 As upregulation of p38 MAPK by wt tPA appears to limit impairment of NMDA-R–mediated pial artery dilation after FPI, 18 we speculated that increasing p38 might serve as a mechanism for vasoprotection by tPA-S481A. In partial support of this hypothesis, the increase in CSF p38 MAPK post-FPI was mildly augmented by wt tPA but more so by tPA-S481A. We previously observed that tPA-S481A blocked the increase in phosphorylated ERK in the CSF that occurs after FPI. 21 The ability of tPA-S481A to prevent hypoperfusion and further reduction of CBF during hypotension while protecting pial artery dilation after FPI was superimposed was blocked by the p38 MAPK antagonist SB 203580 co-administered with the variant at 3 h post-FPI. SB 203580 blocked the upregulation of phosphorylated p38 MAPK after FPI, demonstrating efficacy of action. These biochemical data support the pharmacological data and suggest that tPA-S481A prevents impairment of cerebral autoregulation after FPI both by augmenting p38 and by inhibiting upregulation of ERK MAPK. 21
The severity of constriction after FPI in the presence of exogenous tPA is substantial and probably not the simple result of loss of a dilator, such as NO scavenging by superoxide, but also production of a vasoconstrictor. We previously observed that the spasmogen ET-1 is upregulated and antagonizes NMDA-R–mediated dilation after FPI. 19 Therefore, we investigated the influence of wt tPA and tPA-S481A on CSF ET-1 levels after FPI. CSF ET-1 was elevated after FPI and increased further by exogenous wt tPA, but upregegulation by both wt tPA and FPI was blocked by tPA-S481A administered at 3 h post-FPI. These data suggest that tPA-S481A-mediated protection of autoregulation after TBI may result in part from blocking induction of ET-1 post-TBI by endogenous tPA released as a result of injury.
ERK MAPK and ET-1 may be linked mechanistically in mediating inhibition of cerebral autoregulation after TBI. ET-1 promotes ERK MAPK upregulation, which contributes to impaired autoregulation after FPI in the piglet, 33 and inhibition of ERK MAPK prevents upregulation of ET receptors and reduction of CBF in rat models of subarachnoid hemorrhage and middle cerebral artery occlusive ischemic stroke. 34 –36 Therefore, it is possible that ERK MAPK may both upregulate ET-1 and contribute to vasoconstriction by this peptide in the setting of TBI, resulting in a feed forward cycle. TBI is not stroke. However, the etiology, pathophysiology, and the critical care pathways for treatment are related, but differ in important ways. In the stroke model used by the Edvinsson group, the ET-B receptor was observed to be the primary ET receptor contributory to vasoconstriction. 34 –36 In contrast, our data show that it is the ET-A receptor that is key to vasoconstriction after FPI in the piglet. 19,33 Because the p38 inhibitor SB 203580 modestly reversed the total blockade of ET-1 upregulation observed with tPA-S481A alone, protection of autoregulation by this tPA variant after TBI may result in part from p38 blocking signaling pathways that lead to the induction of ET-1 post-insult. The concomitant block of ERK MAPK and ET-1 may act in concert with the augmentation of p38 MAPK by tPA-S481A to protect cerebral autoregulation after FPI.
There are several potential clinical implications of the observed relationship between tPA and NMDA-R induced vascular activity. For example, hypotension is an important predictor of poor outcome after TBI in children and adults. 37,38 Hypotension may be more detrimental to immature than to mature brain. 39 Cerebral autoregulation during hypotension is known to be impaired after TBI in the pediatric population. 40 When autoregulation is impaired, hypotension decreases cerebral perfusion pressure (CPP) and CBF. Decreases in CPP exacerbate NMDA-R induced vasoconstriction, which leads to ischemia, cell swelling, and edema. The latter leads to increased intracranial pressure (ICP). Increases in ICP decrease CPP further, initiating a vicious cycle. 41
There are also several experimental caveats that may limit data interpretation. First, the focus of this study was on the role of NMDA-R over-activation in impaired cerebral hemodynamics and autoregulation and not histopathology. We reported that tPA-S481A prevented neuronal cell necrosis in CA1 and CA3 hippocampus when administered 30 min post-TBI. 21 Future studies will be needed to determine if prevention of disturbed autoregulation when tPA-S481A is administered at 3 h post-injury (as in this study) prevents histopathology from developing to the same extent. Later time points and varying the dose of tPA-S481A post-TBI will also need to be investigated to further refine the clinical therapeutic window. Second, the experimental design did not allow us to determine the cellular origin of the MAPK isoforms we detect in CSF, which include neurons, glia, vascular smooth muscle, and endothelial cells. While we were initially surprised to detect the products of an intracellular signaling system in the CSF under sham control conditions, we make the point that this does not reflect damage or pathology. In response to peer review concerns over the years, we have reproducibly detected MAPK in CSF under control conditions and monitored its change in response to diverse stimuli. 14,18 In particular, we documented parallel changes in tissue and CSF in response to FPI and hypoxia/ischemia using qualitative immunohistochemistry, semi-quantitative Western, and quantitative ELISA of CSF. 14,42 Therefore, changes in CSF MAPK reflect changes occurring within the tissue. Finally, the use of three tPA mutants does focus attention on glutamate toxicity but did not exclude the possibility that tPA-S481A also prevents microhemorrhaging. It is unlikely, however, that we overlooked sufficient hemorrhage to explain the changes in autoregulation and CBF we observed.
Conclusion
These studies extend our previous observations that tPA-S481A provides neuroprotection when given 30 min after TBI in several ways. 21 First, we found that tPA-S481A protects cerebrohemodynamics when administered 3 h post-trauma, entering a clinically relevant therapeutic window. Second, it appears likely that a concerted change in MAPK isoform profile contributes to outcome. In addition to blocking upregulation of ERK MAPK, tPA-S481A upregulates p38 MAPK, which in turn inhibits the upregulation of ET-1 that occurs after TBI, which is known to impair cerebral autoregulation. Third, the use of three tPA variants with separate but overlapping functions focuses attention on glutamate/NMDA-R toxicity as an important mechanism underlying the impairment of cerebral autoregulation after TBI and suggests a potential therapeutic. Additional studies will be needed to delineate the efficacy and safety of tPA-S481A in the prevention of neurotoxicity post-TBI.
Footnotes
Acknowledgments
Sources of financial support: This research was supported by grants from the National Institutes of Health, HD57355 (WMA), HL77760 and HL82545 (AARH), HL76406, CA83121, HL76206, HL07971, and HL81864 (DBC).
Author Disclosure Statement
No competing financial interests exist.