
Abstract
Significant cardiovascular and autonomic dysfunction occurs after era spinal cord injury (SCI). Two major conditions arising from autonomic dysfunction are orthostatic hypotension and autonomic dysreflexia (i.e., severe acute hypertension). Effective regulation of cerebral blood flow (CBF) is essential to offset these drastic changes in cerebral perfusion pressure. In the context of orthostatic hypotension and autonomic dysreflexia, the purpose of this review is to critically examine the mechanisms underlying effective CBF after an SCI and propose future avenues for research. Although only 16 studies have examined CBF control in those with high-level SCI (above the sixth thoracic spinal segment), it appears that CBF regulation is markedly altered in this population. Cerebrovascular function comprises three major mechanisms: (1) cerebral autoregulation, (i.e., ΔCBF/Δ blood pressure); (2) cerebrovascular reactivity to changes in PaCO2 (i.e. ΔCBF/arterial gas concentration); and (3) neurovascular coupling (i.e., ΔCBF/Δ metabolic demand). While static cerebral autoregulation appears to be well maintained in high-level SCI, dynamic cerebral autoregulation, cerebrovascular reactivity, and neurovascular coupling appear to be markedly altered. Several adverse complications after high-level SCI may mediate the changes in CBF regulation including: systemic endothelial dysfunction, sleep apnea, dyslipidemia, decentralization of sympathetic control, and dominant parasympathetic activity. Future studies are needed to describe whether altered CBF responses after SCI aid or impede orthostatic tolerance. Further, simultaneous evaluation of extracranial and intracranial CBF, combined with modern structural and functional imaging, would allow for a more comprehensive evaluation of CBF regulatory processes. We are only beginning to understand the functional effects of dysfunctional CBF regulation on brain function on persons with SCI, which are likely to include increased risk of transient ischemic attacks, stroke, and cognitive dysfunction.
Introduction
It is now clear that cardiovascular diseases are among the most common causes of death in those with SCI. 10 Recently, epidemiological evidence shows stroke risk is two to three times greater in those with SCI, even after controlling for a number of cardiovascular risk factors such as hypertension, diabetes, arrhythmia, and coronary artery disease. 11 Brief disruptions of cerebral blood flow (CBF) caused by impaired vascular control may cause irreversible neuronal cell death. 12,13 Conversely, inadequate counter-regulation against excessive cerebral perfusion can cause intracranial hypertension and predispose to hemorrhagic stroke. 14,15
We have recently reviewed baroreflex control after SCI and highlighted that the function of this feedback system is significantly impaired in those with SCI. 9 Consequently, poor BP control in those with SCI makes appropriate regulation of CBF crucial for preventing stroke. As a negative relationship exists between baroreflex function and CBF regulation, a review of the current literature examining CBF control in those with SCI may shed light on the overall trends and underlying pathophysiology in this complex and diverse condition. 16 There is evidence that CBF dysfunctions contribute to the variety of clinical conditions that not only cloud present clinical challenges for management of individuals with SCI, but could even be life threatening in nature. For example, poor control over CBF during orthostatic hypotension can result in transient ischemic attack or even stroke. 17 Further, CBF regulatory dysfunction could also result in cognitive deficits in these persons and negatively affect quality of life. 18
We review the current understanding of CBF regulation in humans and the related alterations after SCI.
Regulation of CBF
It is often highlighted that the brain receives a disproportionate amount of blood relative to its mass. Specifically, the brain receives approximately 20% of cardiac output although it only makes up approximately 2% of body mass. 19,20 This relationship highlights the extremely elevated metabolic demands occurring in cerebral tissue. 20 The brain also uses more than 20% of resting oxygen and glucose. 19 Sufficient and appropriate matching of brain blood flow to metabolic demand is essential because cerebral ischemia and syncope can result after only 3 sec and neuronal death can occur after approximately 5 min of disrupted CBF. 20
Regulation of CBF is achieved through several factors including metabolic, myogenic, neurogenic, and systemic control (Fig. 1). 21 All these factors interact to regulate CBF through the adjustment of cerebrovascular resistance. 22 Together, cerebral autoregulation (CA), cerebrovascular reactivity, and neurovascular coupling are commonly used parameters to reflect the complex interplay of regulatory factors.

Illustration of primary pathways relevant to human cerebral blood flow control. Cardiac output (Qc) and total peripheral resistance (TPR) together generate mean arterial pressure. Cerebral perfusion pressure (1; CPP) is the difference between mean arterial pressure and intracranial pressure (2; ICP) when central venous pressure (3; CVP) is lower than ICP. Neurogenic control over cerebral vascular tone (4) is widely disputed; however, there is some limited evidence suggesting an autonomic influence on dynamic cerebral autoregulation, 34,38,39 as well as potentially cerebrovascular reactivity (Jordan et al. 2000). There is also controversial evidence that Qc alters cerebral blood flow independently of CPP (5). When CVP and/or ICP are elevated, venous outflow from the brain is likely also partially “regulated” by a Starling resistor, because of the relatively static dimensions and enclosed nature of the cranium (6). Cerebral blood flow is altered primarily in response to changes in pH (7; also metabolism and other factors), using both endothelial-dependent, and endothelial-independent pathways (8) to provide redundant regulation of cerebral [H+] to maintain optimal enzymatic pH within the brain. Endothelial function appears to partially mediate cerebrovascular reactivity to hypercapnia, 100,110 possibly hypocapnia, 98,101 as well as neurovascular coupling, 124 but not cerebral autoregulation. 37,60
CA
CA, which describes the ability of the brain to maintain CBF under a variety of perfusion pressures, can be divided into static and dynamic components. Static CA (sCA) is used to describe CBF during steady-state conditions 23 and is traditionally considered to be held relatively constant over a wide range of perfusion pressures (i.e., mean arterial pressures (MAP) from 60 to 150 mm Hg). 24, 25 Lucas and associates, 26 however, recently reported a relatively “pressure passive” CBF response to both increased and decreased BP induced pharmacologically. The assumption that diameter remained constant during the infusion of vasoactive substances was made based on the results of Giller and colleagues, 27 who measured the outer diameter during craniotomy and found minimal change in diameter of 4%. Volumetric blood flow through the internal carotid artery during steady state phenylephrine infusions, however, showed increases in middle cerebral artery blood velocity, but apparently no measurable change in internal carotid artery flow, 28 raising the possibility that elevations in flow velocity may reflect increase in arterial vascular tone, potentially of the insonated vessel itself. Such changes would invalidate the use of transcranial Doppler as an accurate surrogate of CBF. Discrepancies between these studies have not been resolved. Nevertheless, cerebral autoregulation has been shown to be reduced in acute ischemic stroke, 29 traumatic brain injury, 30 carotid artery stenosis, 31 and autonomic failure. 32
Dynamic CA (dCA) on the other hand, describes the ability of the cerebrovascular system to oppose short-term changes in perfusion pressure over a period of less than 5 sec. 23,33 Both sCA and dCA are thought to be regulated by a combination of myogenic, metabolic, neurogenic control mechanisms (for a review, see Paulson and coworkers, 21 ); however, sCA and dCA and may rely on relatively different influences from the various regulatory factors. 34 For example, it has been suggested that neural control over cerebral vascular tissue more heavily influences dCA than sCA, 34 while endothelial function has been shown to not relate to dCA. 35 Myogenic control, on the other hand, is thought to play a role primarily in passive, compliance-based dCA (i.e., transfer function analysis phase) and less of a role in active, resistance-based dCA (i.e., gain and coherence). 36,37 Further, although on a continuum, dysfunction in dCA is thought to occur before sCA in some pathological scenarios. 23
Very recently, Pukayastha and colleagues 38 showed, using prazosin (α1a antagonist), that sympathetic control is important to dCA (namely, low-frequency [LF] gain) at rest and during exercise. 38 Ogoh and associates 39 also showed that the middle cerebral artery velocity (MCAv) responsiveness to thigh cuff deflation was reduced after generalized α-adrenergic blockade. 39 Further, evidence showing increased transfer function analysis gain after cholinergic blockade through glycopyrrolate in the same frequencies as sympathetic control (i.e., >0.05 Hz) suggests that both the parasympathetic and sympathetic system are active in dCA that may predominantly oppose rising and falling perfusion pressures, respectively. 40 Recent evidence in both humans 16,41,42 and animals, 43 however, supports the idea of hysteresis in CA. In other words, the brain can more effectively buffer hypertension compared with hypotension. Whether this is the case for SCI remains unknown, but if comparable to healthy humans would provide a means to defend against autonomic dysreflexia.
Cerebrovascular reactivity
The index of cerebrovascular reactivity (i.e., relative change in CBF in response to altered blood gas concentration) allows for quantification of the sensitivity of the intracranial vessels to a given stimulus. Most commonly, stimuli include either pharmacological or ventilatory driven alterations of arterial blood gas content (i.e., oxygen or carbon dioxide). A reduced CBF response to hypocapnia and hypercapnia has been shown to be related to obstructive and central sleep apnea, 44,45 hypertension, 46 carotid artery stenosis, 47 congestive heart failure, 48 and have independent predictive value for future ischemic stroke. 49 –51
The overarching control of CBF is via PaCO2. Specifically, CBF adjusts roughly 3.8%·mmHg−1 change in PaCO2 partial pressure, with a slightly more robust response to hypercapnia in comparison with hypocapnia. 52 –56 Briefly, CBF is thought to preferentially alter CO2 and [H+] at the level of the brain stem to maintain pH within a narrow range. 52 Maintenance of cerebral pH helps stabilize respiratory and chemoreceptor centers, which are also sensitive to [H+]. The mechanisms through which this is achieved are only partially understood but thought to be the result of activated potassium channels in smooth muscle as well as rapid adjustment of several key vasoactive substances such as nitric oxide and prostaglandin. 52
Neurovascular coupling
The close temporal and regional linkage between neural activity and CBF response is termed neurovascular coupling. CBF responses in the MCA, posterior cerebral artery and anterior cerebral artery to visual, verbal, and cognitive tasks have been widely measured. 57 –61 Previous work shows altered neurovascular coupling in those with hypotension, 62 type I diabetes, 63 and Alzheimer disease 64 ; however, to our knowledge, no study has directly related the neurovascular coupling response to cognitive function. The metabolic portion of CBF regulation is related directly to increased oxygen availability and reduced carbon dioxide during periods of increased CBF. 65 Similarly to working muscle, a complex interplay occurs, whereas nitric oxide and other endothelial metabolites work to match blood flow provision to metabolic demand. 66,67 This functional hyperemia uses intimate interactions between cerebral vascular cells, such as endothelium and smooth muscle, with neurons and glia (i.e., astrocytes, microglia, oligodendrocytes) to match neural activation with CBF. 68
CBF after SCI
A total of 16 studies have examined CBF control in those with SCI. 7,69 –82 In the current section, we will highlight the literature examining these CBF regulatory indicators and discuss the potential underlying mechanisms (Table 1). Specifically, we will discuss how CA, cerebrovascular reactivity and neurovascular coupling are influenced by high-level SCI (HLSCI). The vast majority of these studies have examined CBF in those with HLSCI, with only one including a lower level SCI group (i.e., <T6). 80 As such, specific focus will be on studies examining those with HLSCI. Also, we have decided against discussing one of the studies because the researchers used a liquid-meal ingestion to alter BP, which did not alter BP in their HLSCI group, precluding the evaluation of a CBF response. 81,83 Several reports support the idea that those with HLSCI complain of orthostatic intolerance in the early stages after injury; however, these complaints reduce in frequency as duration of injury occurs, even with persistent and severe orthostatic hypotension. 72,84 –86 It has been speculated that CBF regulatory process may adapt to overcome profound orthostatic hypotension in chronic HLSCI. 77 Considering the neurogenic, vascular, metabolic, and respiratory changes known to occur after SCI, it is plausible that CA is altered in this population.
↑, significant increase compared with controls; ↓, significant decrease compared with controls; ≈, no difference compared with controls.
CBF, cerebral blood flow; CA, cerebral autoregulation; CBFRx, cerebrovascular reactivity; NVC, neurovascular coupling; SCI, spinal cord injury; AB, able-bodied; HLSCI, high-level SCI (≥T6); IOH, idiopathic orthostatic hypotension; sCA, static cerebral autoregulation; MCA, middle cerebral artery; LBNP; lower body negative pressure; ACE, angiotensin-converting enzyme; MCAv, midde cerebral artery velocity; VLF, very low frequency; dCA, dynamic cerebral autoregulation; PCAv, posterior cerebral artery velocity ; LLSCI, low-level SCI (<T6); EtCO2, end-tidal carbon dioxide.
Cardiovascular consequences of SCI
The autonomic nervous system is composed of both sympathetic and parasympathetic divisions. Parasympathetic nervous outflow occurs from cranial nerves III, VII, IX, and X superiorly and S2–4 inferiorly. Only cranial nerves IX and X participate in cardiovascular control, and as they exit above the spinal cord level; therefore any parasympathetic influence on cerebrovascular after SCI tone would be intact. 87 On the other hand, sympathetic nervous outflow occurs from the sympathetic preganglionic neurons (SPNs) localized within T1–L2 spinal segments of the spinal cord. These neurons synapse into the sympathetic paravertebral ganglia (ganglionic neurons in the sympathetic chain), 88 and finally postganglionic fibers innervate target organs in the periphery. After SCI, tonic supraspinal control to the spinal SPNs is lost, and sympathetic outflow from the spinal cord below the level of injury is severely disrupted. 4 Drastic changes in systemic cardiovascular regulation occur in those with lesion levels above the sixth thoracic spinal cord segment. Lesions above T6 are associated with a loss of supraspinal control over the heart and splanchnic blood vessels, 89 both of which are needed for effective long- and short-term BP regulation. 90 Further to this, sympathetic control of the cerebral blood vessels is transmitted through the superior cervical ganglion, which originates from spinal nerves T1–T4. 91 As such, any SCI occurring above T4 would be expected to result in partial or complete (>T1) abolishment of sympathetic cerebrovascular control.
Also highly relevant to cerebrovascular regulation, endothelial function, estimated by flow mediated dilatation, has been shown to be systemically reduced after SCI, 92 in some cases in as little as 6 weeks post-injury. 93 Nitric oxide is the primary substance causing vasodilatation in response to sheer-stress, 94 and leads to smooth muscle relaxation and increased blood flow. 95 The production of nitric oxide relevant to vascular control occurs in endothelial cells 96 and mediates the relationship between cerebral vascular tone and the majority of signaling messengers (i.e, acetylcholine, adenosine diphosphate, adenosine triphosphate, bradykinin, endothelin, sodium fluoride, oxytocin, vasopressin, etc.). 97 Reduced nitric oxide, however, does not appear to influence cerebral autoregulation but is associated with reduced cerebrovascular reactivity. 98 –101 After this, it could be suspected that systemic endothelial dysfunction influences the regulation of CBF (Fig. 2).

Illustration of theoretical mechanisms of cerebral blood flow control that would be impacted by high-level spinal cord injury (SCI). Briefly, decreased central sympathetic control of systemic blood vessels leads to greatly reduced cerebral perfusion pressure (CPP). Exacerbating the hypotension, those with SCI above T5 also have impaired central sympathetic control over the heart. Systemic endothelial (Endo) dysfunction may also lead to decreased CO2 reactivity, neurovascular coupling, and potentially cerebral autoregulation. As mentioned in Figure 1, direct neurogenic control of cerebral vascular tone is controversial, but cerebral vessels are innervated by sympathetic fibres originating from the T1–4 level (superior cervical ganglia). As such, SCI above T5 would result in impaired or absent central sympathetic control of cerebrovasculature. denotes where cerebrovascular dysfunction may occur after SCI (1-endothelial dysfunction, 2- altered direct neural cerebrovascular influence, 3- sympathetic neural control over total peripheral resistance (TPR), 4- sympathetic neural control over cardiac output [Qc]). *Note that when a complete injury occurs, sympathetic vasomotor control is disrupted below the lesion level. ICP, intracranial pressure; CVP, central venous pressure.
sCA
Early studies from Nanda and coworkers 72,73 that examined CBF using inhaled 133Xenon clearance rate during supine-to-seated and supine-to-raised leg/lower body negative pressure (LBNP) maneuver reported that CBF was well maintained during an average 18 mm Hg drop in MAP (Fig. 3). 72,73 Handrakis and colleagues 78 provided more insight by measuring MCAv after angiotensin-converting enzyme-inhibitor infusion combined with 45 degrees tilt; results revealed that MCAv decreased to a similar rate in chronic HLSCI patients compared with able bodied (AB), although the MAP drop in HLSCI patients was markedly higher (p=0.02). More recent work has shown that sCA (albeit using the relationship between MAP and MCA [diastolic]) was similar between HLSCI and AB patients in response to head-up tilt, although the HLSCI group reported greater decreases in end-tidal CO2. 80 These authors showed an interesting correlation between supine plasma noradrenaline concentrations (but not low frequency oscillations in systolic BP) and sCA, suggesting a relationship between sCA and autonomic completeness. 80
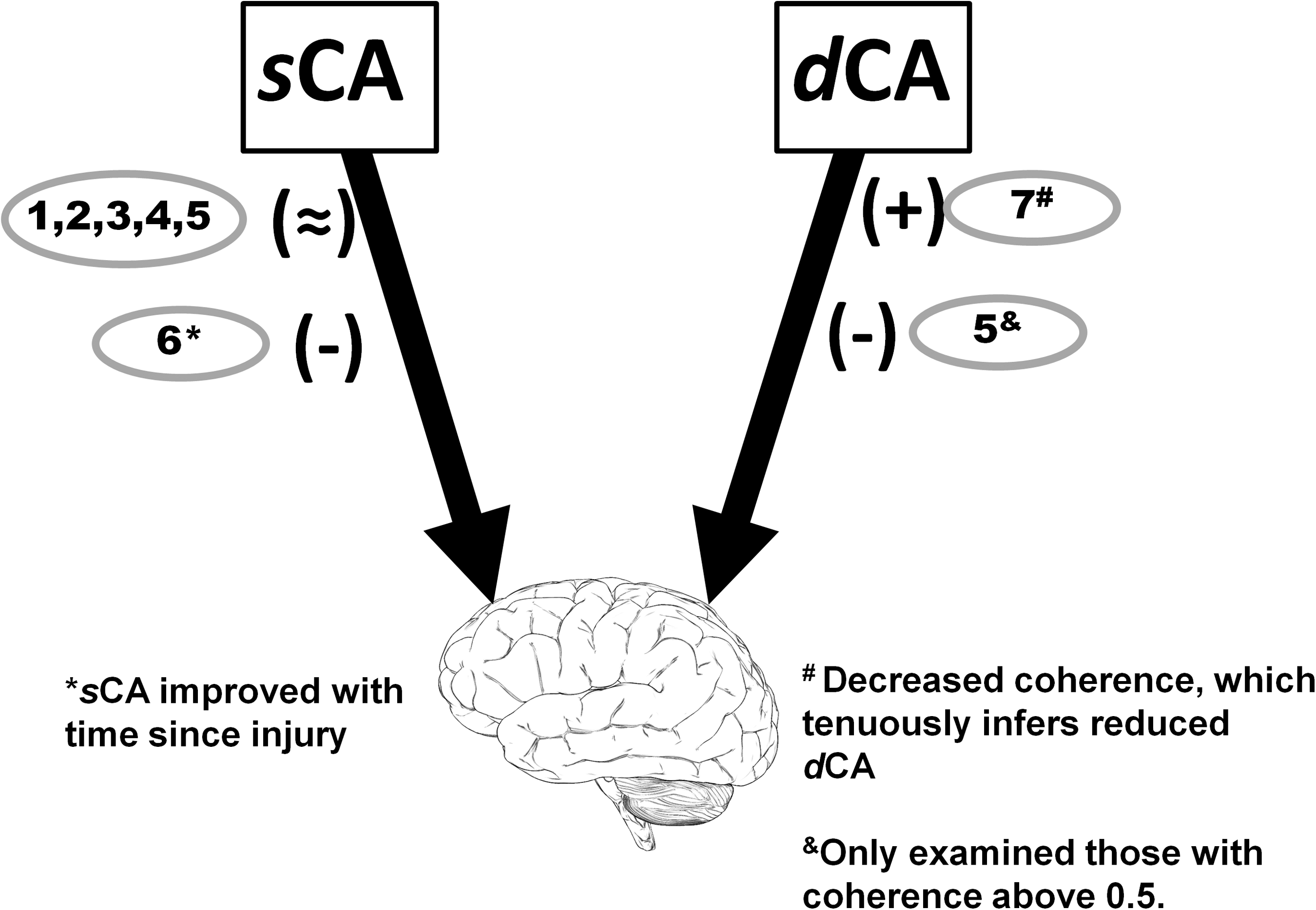
Using a similar 133Xenon technique in primarily patients with acute SCI (i.e., 92% <12 months since injury), Yamamoto and associates 74 showed that the sCA (=ΔCBF/ΔMAP) response to 30-degree tilt was impaired in acute HLSCI (i.e., greater changes in CBF for a given change in MAP); however, those who had injuries occurring within 6 months of examination had significantly impaired sCA compared with those closer to the chronic phase. Because CBF is the final common pathway before the development of syncope, it is not surprising that reduced static CBF control is a major factor related to the increased risk of orthostatic intolerance after SCI. In support of this, Gonzalez and colleagues 75 showed that orthostatically intolerant acute HLSCI had greater decreases in MCAv during steady-state 80 degrees tilt when compared with a tolerant chronic HLSCI group. 75
A landmark study by Houtman and coworkers 77 examined steady-state CBF velocity response to 5-min long bouts of progressive LBNP in a group of HLSCI patients. This study showed remarkably poor MAP control in HLSCI and large reductions in MCAv, but similar changes in cerebrovascular resistance (i.e., ΔMAP/ΔCBF) compared with AB indicating similar sCA. The large reductions in CBF velocity resulted in a trend for lower cerebral oxygenation in HLSCI. 77 The finding that CBF is reduced more than perfusion pressure during LBNP has long been shown in AB, and although compelling evidence now exists to the contrary, was originally thought to result from systemic sympathetic activation of both muscular and cerebral vasculature during central hypovolemia. 41,42,102 The majority (i.e., 8/10) of the HLSCI group in Houtman's the later work of Houtman and associates, 77 however, had a decentralized superior cervical ganglion (as the result of complete injury above T1) and likely would not be capable of sympathetically mediated increases in cerebral vascular resistance. 77 Because both HLSCI and AB persons reported similar changes in end-tidal CO2 (i.e., a surrogate marker of PaCO2), the findings of Houtman and colleagues 77 support the contention that cerebrovascular changes during orthostatic challenge are primarily related to changes in PaCO2. 103 –105
Finally, two interesting studies examined the CBF velocity response to increasing BP in those with HLSCI after 2 min of cold water immersion of the hand 69 or foot. 70 Although not originally interpreted in this manner, it is likely that both these studies induced autonomic dysreflexia in HLSCI. 106 As such, it is expected that in their HLSCI group (C4–C7), unopposed sympathetic outflow would have occurred not only in the systemic vascular bed, but also the cerebrovascular tissue. 87 Both of these studies reported similar average cerebrovascular resistance during autonomic dysreflexia in HLSCI when compared with AB. Further, these studies also included a group of participants with injuries ranging T4–T6, which would be expected to have preserved central sympathetic control over the cerebrovasculature 91 and showed that when the nociceptive stimulus is applied below the injury level (i.e., the foot), increases in cerebrovascular resistance do not occur during autonomic dysreflexia, 70 supporting the idea that sympathetic cerebrovascular control helps to mitigate flow increases during transient periods of increased perfusion pressure. 42,43
Together, the available literature suggests that sCA is well maintained in those with chronic HLSCI, but may be impaired in the acute phase of injury. Although the early studies showed that absolute CBF response is similar in HLSCI compared with AB, more recent work shows that dysfunctional BP control leads to larger fluctuations in CBF, but similar changes in resistance. 77 In one study, a small subset of three HLSCI patients suggests a strong systemic influence of sCA, because abdominal binders and compression stockings improved this metric. 74 It is likely that cerebrovascular regulatory factors other than sympathetic control allow for preserved sCA in response to decreasing BP in HLSCI with sympathetic decentralization and endothelial dysfunction (i.e., the myogenic influence). In response to autonomic dysreflexia, those with injuries above the superior cervical ganglion are able to increase cerebrovascular resistance to match BP increases, while those with lower level lesions (with preserved central sympathetic control over cerebrovasculature) do not, likely because of baroreflex mediated sympathetic withdrawal above the lesion level. 89
dCA
Only three studies have examined dCA in those with HLSCI, all of which used spontaneous transfer function analysis between MAP and MCAv and one with an additional analysis of MAP and posterior cerebral artery velocity (PCAv) (Fig. 3). 79,80,82 Recently, Wilson and associates 79 showed that supine coherence in the very low frequency (VLF) range was reduced in their group of six persons with HLSCI, while gain and phase were similar. 79 We later showed that upright MAP-MCAv coherence was reduced in both the range. 82 Similarly, we reported reduced PCAv coherence in the LF range. 82 Sahota and colleagues 80 showed that the supine LF gain was reduced and upright LF phase was increased, both indicative of reduced dCA. The latter study evaluated only the LF transfer function analysis for MAP-MCAv when coherence was >0.5. 80 To our knowledge, no existing studies examine dCA using a model of perturbed BP/CBF SCI to improve the reliability of transfer function analysis. 107
Considering the combined findings of these studies, spontaneous dCA is altered in those with HLSCI, and is possibly related to both sympathetic decentralization of the cerebrovasculature and increased parasympathetic tone after HLSCI. 108 A fundamental inverse relationship has recently been shown between both spontaneous and non-spontaneous dCA and cardiac baroreflex sensitivity (BRS) in healthy AB patients. 16 According to this relationship, and our recent review highlighting that cardiac BRS is markedly reduced in HLSCI, it could be expected that coherence and MAP-MCAv gain would be reduced while phase would be increased. 9 It is possible that cardiovascular and autonomic pathology after HLSCI leads to a loss of this fundamental compensatory relationship. 16 More work should be completed, however, using larger sample sizes, incorporating VLF ranges as well as driving BP changes using either oscillatory LBNP or tilt to increase confidence in transfer function analysis estimates. 109
Cerebrovascular reactivity
Early work using 133Xenon as well as more current work using transcranial Doppler shows that the CBF response to hypercapnia is preserved in those with HLSCI. 71,74,79 On the other hand, the response of CBF to hypocapnia does not appear as consistent. For example, Eidelman and colleagues 71 reported that the CBF decrease in response to hypocapnia was abolished after HLSCI. In contrast, other reports showed that the hypocapnic response of CBF is preserved. 72,73,79 The preliminary study in this group by Eidelman et al., 71 however, may be confounded as four of the nine HLSCI participants to have BP recorded during hyperventilation showed BP changes meeting the criteria for autonomic dysreflexia, 88 changes that would induce systemic, and more controversially, cerebral vasoconstriction. 89 The vasoconstriction induced by autonomic dysreflexia would thereby augment hypocapnie induced vasoconstriction. 110 Differences between Eidelman and associates 71 and Nanda and colleagues 72,73 in terms of hypocapnic CBF response in HLSCI are difficult to explain but may be because of different techniques for measuring CBF (i.e., modified vs. non-modified 133Xenon inhalation).
Although hampered by a low sample size, Wilson and coworkers 79 reported differences in subtle parameters of CBF between HLSCI and AB during both hypo- and hypercapnia. Pulsatile flow (pulsatility index) of MCAv decreased significantly more in response to hypercapnia in the HLSCI group, while the cerebrovascular resistance response to hypocapnia was reduced. These values suggest nuanced cerebrovascular changes occur after HLSCI, which influence the CBF response to PaCO2. In support of the idea that the hypocapnic CBF response is blunted in HLSCI, Sahota and colleagues 80 also showed that end-tidal CO2 decreased significantly more in HLSCI compared with AB during upright tilt, but diastolic CBF velocity changed similarly. Although end-tidal CO2 decreases may have been secondary to greater increases in dead space and/or decreases in cardiac output during head-up tilt in SCI compared with AB, 111,112 this differential response between HLSCI and AB suggests a reduced CBF sensitivity to hypocapnia.
Pulsatile flow patterns represent an area of increasing interest within the field of CBF regulation, which has been suggested as a primary mechanism through which the brain maintains perfusion during low mean perfusion pressure, such as severe hemorrhage, and orthostatic hypotention, the latter of which is widespread in HLSCI. 89,113 –115 Hypercapnia preferentially alters the small distal resistance vessels, which regulates pulsatile flow, 116 and the adaptation of this vascular bed in HLSCI, as shown by Wilson and associates, 79 may represent an adaptation to chronically altered hemodynamic and/or respiratory patterns in those with HLSCI as a means to maintain cerebral perfusion in the face of reduced perfusion pressure. 79 Reduced cerebrovascular reactivity, especially to hypocapnia, has been previously related in patients and healthy persons to sleep apnea. 48,117 Sleep apnea has been reported to occur in up to 40% of those with HLSCI, and cerebrovascular reactivity may be an important mediating factor. 118 Clearly, further examination of this relationship is warranted, especially considering the increased risk of stroke in the HLSCI population and the independent predictive value of cerebrovascular reactivity for stroke. 100
Neurovascular coupling
Neurovascular coupling describes the hyperemic response to cognition. To our knowledge, only two studies have examined neurovascular coupling in those with HLSCI. Wecht and colleagues 7 showed that mean MCAv and cerebrovascular resistance did not change when completing a Stroop test in either AB or HLSCI. This technique is quite different from the established neurovascular coupling procedures, because Wecht and colleagues 7 reported mean values from three repeated 45-sec long Stroop tests, and it is not clear if an eyes-closed resting period occurred, or if the peak response (as opposed to the average) was different between groups. In many other studies, albeit using more established protocols, MCA, anterior cerebral artery, and posterior cerebral artery CBF velocity have been shown to increase in healthy controls during cognitive tasks and visual motor stimuli while cerebrovascular resistance decrease. 57,59,119 Regardless, these authors reported a trend suggestive of a differing response in controls compared with combined HLSCI and low-level SCI (p=0.08), and a significant positive relationship between changes in cerebrovascular resistance during cognition and Stroop test performance in the AB group (indicating a relationship between neurovascular coupling and cognitive performance); this relationship did not occur in the HLSCI group. 7
Recently, we have revealed that a change in posterior cerebral artery CBF velocity during visual cortex stimulation is essentially absent in those with HLSCI (Phillips and associates, unpublished data). We also have shown that the hyperemic response to cognition improves after administration of an α1a agonist (midodrine hydrochloride, which does not cross the blood brain barrier), albeit still reduced compared with matched controls (unpublished observations). Together, the scant available literature suggests that the neurovascular coupling response is heavily dependent on the underlying BP and systemic vascular tone, with HLSCI and decentralized systemic sympathetic vascular control resulting in an inability to match CBF to increases in neuronal activity. These preliminary data seem to link the plethora of evidence showing cognitive function is declined in those with hypotension with and without SCI. 62,120 –123 Other than hypotension, it is possible that nitric oxide availability, 124 glucose intolerance, 125,126 and dyslipidemia 127 also mediate the relationship between altered neurovascular coupling and HLSCI. Interestingly, cessation of statin therapy, which acutely increases stroke risk, 128 rapidly reduced neurovascular coupling through what is thought to be endothelium dysfunction. 60
Future directions
Many interesting potential directions of future research exist to better evaluate CBF regulation in HLSCI patients who are at increased risk of stroke, have orthostatic hypoperfusion, and experience autonomic dysreflexia. Most apparent to us, true orthostatic tolerance in those with HLSCI has yet to be examined, although it has been suggested that this population has remarkable orthostatic tolerance given extremely reduced perfusion pressure. 77 The use of LBNP to presyncope in those with HLSCI would evaluate this contention, and concurrent measurement of PaCO2, BP, and CBF would shed light on potential underlying mechanisms.
Recently, the beat-by-beat measurement of diameter and blood flow velocity in the internal carotid and vertebral arteries has emerged as a viable technique to measure CBF regulation. These extracranial assessments of CBF are free of the assumption that the three main cerebral arteries do not change diameter under most conditions. 129 Further, because extracranial arteries provide CBF regulatory influence, 130,131 this technique, in addition of transcranial Doppler, allows for a more global and comprehensive assessment of CBF control. Also, magnetic resonance imaging (e.g., pulsed arterial spin labeling, blood-oxygen-level-dependent contrast) allows for more localized assessments of specific regions should also be used to evaluate CBF control in those with HLSCI. 132, 133 The use of LBNP (or tilt), applied at specific frequencies where dCA is thought to be active, should also be examined in those with HLSCI, because this would increase the confidence in the transfer function analysis metrics. It has recently been demonstrated that various measures of dCA are often poorly, or even contradictorily related; thus, it is paramount that a variety of dCA measures are used before concluding as to whether dCA is impaired, improved, or unchanged after HLSCI. 134
The evaluation of cerebrovascular reactivity presents an especially clinically relevant area of future research in the HLSCI population, considering the independent stroke risk value of this metric and the elevated stroke risk in the HLSCI population. 11,49 It should be investigated whether altered cerebrovascular reactivity is secondary to, or implicated in, the increased prevalence of sleep apnea in those with HLSCI or endothelial dysfunction. Evaluating cerebrovascular reactivity and endothelial function in HLSCI before and after continuous positive airway pressure therapy and/or statin administration may shed light on this relationship. 135 From a neurovascular coupling perspective, very little is currently known. Future directions should include using established neurovascular coupling procedures while evaluating the MCA and posterior cerebral artery during a variety of different stimuli. Clearly relating cognitive performance and neurovascular coupling parameters is also needed, not only in HLSCI patients but the AB population as well.
HLSCI offers a relevant model to evaluate the influence of sympathetic control on CBF control. For example, evaluating the influence of systemic sympathetic control and oscillatory CBF patterns on CBF regulation is possible using a model including only those with injuries above T6. On the other hand, specifically examining those with injuries at T3–4 allows for a model with theoretically preserved central sympathetic control of cerebral but not systemic blood vessels. 91
Finally, the result of long-term CBF regulatory dysfunction in SCI needs to be evaluated. The accumulated cerebrovascular trauma induced by the large swings in perfusion pressure may predispose to transient ischemic attacks, leading to cognitive deficits, depressive symptoms, and increased risk for stroke.
Conclusions
The available literature indicates that CBF control is altered in those with HLSCI (Fig. 2; Table 1), but may improve with recovery from injury. Specifically, neurogenic and vascular changes occurring after HLSCI are most consistently implicated for CBF regulatory dysfunction through both direct and mechanistic evidence. Although sCA appears to be preserved, spontaneous metrics of dCA are different in those with HLSCI compared with AB persons. Further to this, preliminary information suggests that the CBF response to changes in PaCO2 is also altered; however, this needs to be confirmed with a larger sample size and confirmed autonomic completeness of injury. Finally, although very limited evidence exists, significant reductions in the neurovascular coupling are apparent in those with HLSCI. Some potential causes of CBF regulatory change in HLSCI include sympathetic decentralization, vascular dysfunction, as well as sleep-related breathing problems.
Footnotes
Author Disclosure Statement
The authors report no disclosures relevant to this article.