
Abstract
The robustness of plasticity mechanisms during brain development is essential for synaptic formation and has a beneficial outcome after sensory deprivation. However, the role of plasticity in recovery after acute brain injury in children has not been well defined. Traumatic brain injury (TBI) is the leading cause of death and disability among children, and long-term disability from pediatric TBI can be particularly devastating. We investigated the altered cortical plasticity 2–3 weeks after injury in a pediatric rat model of TBI. Significant decreases in neurophysiological responses across the depth of the noninjured, primary somatosensory cortex (S1) in TBI rats, compared to age-matched controls, were detected with electrophysiological measurements of multi-unit activity (86.4% decrease), local field potential (75.3% decrease), and functional magnetic resonance imaging (77.6% decrease). Because the corpus callosum is a clinically important white matter tract that was shown to be consistently involved in post-traumatic axonal injury, we investigated its anatomical and functional characteristics after TBI. Indeed, corpus callosum abnormalities in TBI rats were detected with diffusion tensor imaging (9.3% decrease in fractional anisotropy) and histopathological analysis (14% myelination volume decreases). Whole-cell patch clamp recordings further revealed that TBI results in significant decreases in spontaneous firing rate (57% decrease) and the potential to induce long-term potentiation in neurons located in layer V of the noninjured S1 by stimulation of the corpus callosum (82% decrease). The results suggest that post-TBI plasticity can translate into inappropriate neuronal connections and dramatic changes in the function of neuronal networks.
Introduction
P
The neuronal and axonal loss associated with pediatric TBI is well documented in animal models. 8 –11 Whereas histological analysis is a reliable means to characterize TBI-induced pathology, it provides little insight into the plasticity mechanisms and neuronal pathways involved in reshaping the function of the surviving cells and circuits as well as how they may dictate degree of rehabilitation. Increases in cortical function measured by electrophysiology (EP) and functional magnetic resonance imaging (fMRI) signals have been shown to be tightly correlated to degree of recovery in stroke patients and experimental animal models of stroke. 12 –14 Therefore, changes in cortical neuronal function are believed to be a reliable indicator of plasticity and neurorehabilitation. In order to determine the neurophysiological responses and elucidate the altered plasticity mechanisms associated with injury to the developing brain, we studied cortical function in the controlled cortical impact (CCI) rat model with adaptation for the immature brain. The CCI model generates reproducible cortical lesions, mimics clinically relevant histopathological, neurophysiological, and behavioral characteristics of moderate-to-severe pediatric TBI, 9,15 –17 and is used for studying treatment strategies in head injuries. 15,18,19
Extracellular in vivo and intracellular whole-cell patch clamp EP techniques, fMRI, diffusion tensor imaging (DTI), and histology measurements were obtained 2–3 weeks postinjury. The results suggest that although there is a greater potential for plasticity during development, post-TBI plasticity can translate into inappropriate neuronal connections and dramatic changes in the function of neuronal networks, hence promoting increased brain vulnerability.
Methods
All animal procedures were conducted in accord with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Johns Hopkins University Animal Care and Use Committee (Baltimore, MD).
Animal model
TBI injury was performed using a CCI device (Pittsburgh Precision Instruments, Pittsburgh, PA). The injury was performed on 31 male Sprague-Dawley rats (∼30–45 g) at postnatal days 16–18. 17 This age range in rats is believed to be equivalent to human toddler age, 9,20 and thus this model is relevant to study brain injuries resulting from sports, playground activities, vehicle injuries, falls, and assault, which are the leading concern in young children. Anesthesia was maintained using a nose cone with 2% isoflurane and 30% oxygen for the duration of the surgical procedure. The rat's head was secured in a stereotaxic frame, and a rectal probe was placed. After a mid-line scalp incision, a left parietal craniotomy was performed. After a 20-min period of temperature stabilization, CCI was induced using a 6-mm flat metal impactor tip at 5.5 m/sec, duration of 50 msec, and depth of 1.5 mm. The craniotomy was then resealed with acrylic mixture, and the scalp incision was closed with interrupted sutures. This focal injury results in damage in the cortical sensory representations of the limbs, which allowed us to utilize established techniques and stimulation paradigms to investigate altered sensory processing associated with injury. Twenty-six aged-matched rats were used as a control group. All experimental assays were performed 2–3 weeks post-TBI. The functional responses of each individual rat were evaluated by a single technique, that is, extra- or intracellular electrophysiology recordings, fMRI, or DTI.
Electrophysiology
Extracellular in vivo recordings
The urethane anesthetized rat's head was fixed in a stereotaxic frame. A 1 mm2 craniotomy window was made over the right S1 centered at the AP (–0.5 mm) and ML (3.6 mm). Two needle electrodes were inserted into each forepaw to deliver electrical stimulation. An axial array microelectrode (FHC, Inc., Bowdoin, ME), with 12 sites spaced at 150 μm along the shank, was inserted into the center of the S1 cortex until reaching the depth of 1800 μm below pia mater to cover the whole depth of the rat cortex (starting at 150 μm). Multi-unit activity (MUA) and local field potential (LFP) were collected with a Cambridge Electronic Design interface and Spike2 data acquisition and analysis software (Cambridge Electronic Design, Cambridge, UK) and sampled at 11 and 1 KHz, respectively, and band-pass filtered between 500 and 5 KHz and 0.1 and 300 Hz, respectively. To identify MUA, the standard deviation (SD) of the MUA signals was calculated for 500 ms. MUA signals with amplitude greater than 4 times the SD were defined as spiking activity. To define stimulus-evoked spiking activity, poststimulus time histogram (PSTH) analysis was performed. PSTH was obtained for MUA in each trial by event correlation analysis of the spiking with the stimulation using 5-ms bins. The SD of neuronal firing rates was calculated for the last 150 ms of the interstimulus interval of each trial. MUA that showed increased (>2 SDs) activity in one 5-ms bin during the first 40 ms after onset of stimulation were considered to show stimulus-evoked response. MUAs were summed for each layer for every individual rat and averaged across the group. LFP waveforms were averaged with respect to the stimulation trigger. The mean amplitude of the negative deflection and the time to pick were calculated for each train of stimuli.
Intracellular whole-cell patch clamp recordings
Rats were anesthetized with isoflurane before decapitation. The brain was then rapidly removed and transferred to ice-cold, oxygenated cutting solution containing (in mM) 76 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 14.3 MgSO4, 25 glucose, 75 sucrose, and 0.66 CaCl2 (pH 7.4). Four coronal slices (350 μm thick) around bregma 0.0–1.5 containing S1 and corpus callosum were cut with a vibrating blade microtome (Leica Biosystems, Wetzlar, Germany) and incubated at 34°C for 30 min. Subsequently, the selected slices were transferred to a holding chamber with artificial cerebrospinal fluid (ACSF) containing (in mM) 125 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 0.4 ascorbic acid, 2 pyruvic acid, 4 L-lactic acid, 20 glucose, and 2 CaCl2 (pH 7.4) at room temperature (∼22°C). Whole-cell patch clamp recordings were made from visually identified pyramidal neurons in layer V (1000–1400 μm below the cortical surface) using infrared differential interference contrast microscopy equipped with a CCD camera (Carl Zeiss AG, Oberkochen, Germany). During recordings, slices were perfused with ACSF at room temperature. Recordings were obtained using a glass patch pipette (resistance, 4–6 MΩ) filled with an intracellular solution containing (in mM) 130 potassium gluconate, 10 KCl, 1 CaCl2, 6 NaCl, 20 HEPES, 10 ethylene glycol tetra acetic acid, 3 Mg adenosine triphosphate, 0.5 Na guanosine 5’-triphosphate, and 5 QX-314, and 0.2% biocytin, adjusted to pH 7.3–7.4 with KOH, at a holding potential of −70 mV. Electrical stimulation (0.1 ms in duration) was delivered at a frequency of 0.033 Hz to obtain baseline amplitude (10 min) approximately at the half-maximal response. A wire electrode was inserted into the corpus callosum (3.2–3.4 mm lateral to the mid-line) in the noninjured hemisphere. Long-term potential (LTP) was induced by high-frequency stimulation (HFS; 100 Hz in 1 sec, repeated 3 times with 10-sec interstimulus intervals) with the same stimulation intensity used for baseline recordings. All recordings were made in the presence of 100 μM of picrotoxin to block the gamma-aminobutyric acid (GABA)A receptor-mediated currents. Whole-cell recordings were collected using Multiclamp 700B amplifiers/Digidata 1440A (Molecular Devices, LLC, Sunnyvale, CA). Data were filtered and digitized at 10 kHz. Clampex (Molecular Devices) was used for both acquisition and analysis, and miniature excitatory postsynaptic current (mEPSC) was analyzed by Mini Analysis (Synaptosoft Inc., Fort Lee, NJ). Morphology reconstruction of the recorded neurons filled with biocytin was performed at the end of the experiment for several neurons. LTP of evoked EPSC responses to transcallosal stimulation were defined by a prolonged increase in synaptic transmission. Neurons that the series resistance changed more than ∼20% during recording were discarded. Mean values of potentiation±standard error of the mean (SEM) were calculated from the average values of the last 5 min of each recording.
Functional magnetic resonance imaging
Images were acquired on an 11.7 Tesla/16 cm horizontal bore small-animal scanner (Bruker Corporation, Billerica, MA), permitting high spatial and excellent temporal resolution. A 72-mm quadrature volume coil and a 15-mm-diameter surface coil were used to transmit and receive magnetic resonance signals, respectively. For blood-oxygenation-level–dependent (BOLD) fMRI, gradient echo, echo planar imaging was used with a resolution of 150×150×1000 μm. Five coronal slices with a 1-mm thickness were acquired with the following parameters: effective echo time (TE)=11 ms; repetition time (TR)=1000 ms; bandwidth=250 KHz; field of view (FOV)=1.92×1.92 cm; and matrix size=128×128. A T2-weighted RARE sequence was used to acquire high-resolution anatomical images with the following parameters: TE=10 ms; TR=5000 ms; bandwidth=250 KHz; FOV=1.92×1.92 cm; and matrix size=256×256. During fMRI measurements, rats were anesthetized with dexmedetomidine. Respiration rate, rectal temperature, PO2, and heart rate were continuously monitored throughout all measurements (Starr Life Sciences Corp., Oakmont, PA). fMRI block design was used, and stimulation of the right or left forepaw was conducted in a similar way as described above. The FMRIB Software Library (FSL 4.1.9) software was used for analysis and spatial normalization. 21 Activation maps were obtained using the general linear model. Z-score statistics were cluster-size thresholded for effective significance of p<0.05. The activation threshold was set at 2.3.
Diffusion tensor imaging
Rats were perfused with 4% paraformaldehyde (PFA), and brains were further immersed in PFA for 12 more hours at 4°C before they were transferred to phosphate-buffered saline (PBS). Brains were then kept in PBS for 2–3 weeks at 4°C to wash out PFA. Before DTI, brains were placed in a 15-mL tube filed with Fomblin. DTI was performed on an 11.7T scanner (Bruker) using a 20-mm-diameter volume coil. Diffusion tensor data were acquired using the diffusion-weighted three-dimensional (3D) GRASE sequence with the following parameters: TE=33 ms; TR=800 ms; bandwidth=100 kHz; FOV=17.0×14.0×25.8 mm; matrix size=128×96×192 (zero-padded to 256×192×384); and number of averages=4.
22
For DTI, six diffusion-weighted images (b-value, 1700 s/mm2) and two non-diffusion-weighted images were acquired with δ=3 ms and Δ=15 ms. Diffusion tensors were calculated using a log-linear fitting method using DTIStudio (
Histopathology
At the end of the neurophysiological measurements, TBI and control rats were perfused with 4% PFA. Brains were cut at 50 μm in a freezing microtome. Brain slices were stained with Luxol fast blue (for myelin), eosin (for neutrophils), and cresyl violet (for cells). Stained sections were imaged using the Zeiss microscope Imager M2 with a 5× objective. Four 50-μm stained sections corresponding to bregma −1, 0, 1, and 2 were analyzed using stereological approaches to determine corpus callosum volume. First, stained slices were coregistered to sections from the rat brain atlas using rotation, shifting, and scaling using reference points that included the corpus callosum, outer brain boundary, lateral vestibular nucleus, and the anterior commissure. ImageJ (NIH, Bethesda, MD) was used to segment the corpus callosum based on signal intensity corresponding to myelin staining. Then, the area of the corpus callosum in the noninjured hemisphere was selected from the mid-line to the border of S1 forepaw representation, and the number of pixels within the segmented area was calculated.
Statistical analysis
Sample-size calculations for each measurement were based on the power analysis (80% power) using the hypothesis test of noninferiority on group means for the average amplitude of evoked neurophysiological responses. A two-tailed Student's t-test or two-factor analysis of variance (ANOVA) with replication were performed between the different conditions and groups. Results and figures show the mean±SEM.
Results
The CCI was performed over the left S1 in 16- to 18-day-old rats, and measurements of neurophysiological responses were performed 2–3 weeks after the CCI procedure. The CCI caused a significant architectural damage to the left S1, as shown in the cresyl violet histology (Fig. 1A). At this time point, neurophysiological responses, as a consequence of tactile stimulation in the left, injured S1, were completely abolished (Fig. 2A). Thus, we focused on characterizing the neurophysiological responses in remote and anatomically intact cortical areas, specifically, S1 contralateral to the injury, that is, noninjured (right) S1 (Fig. 1B).
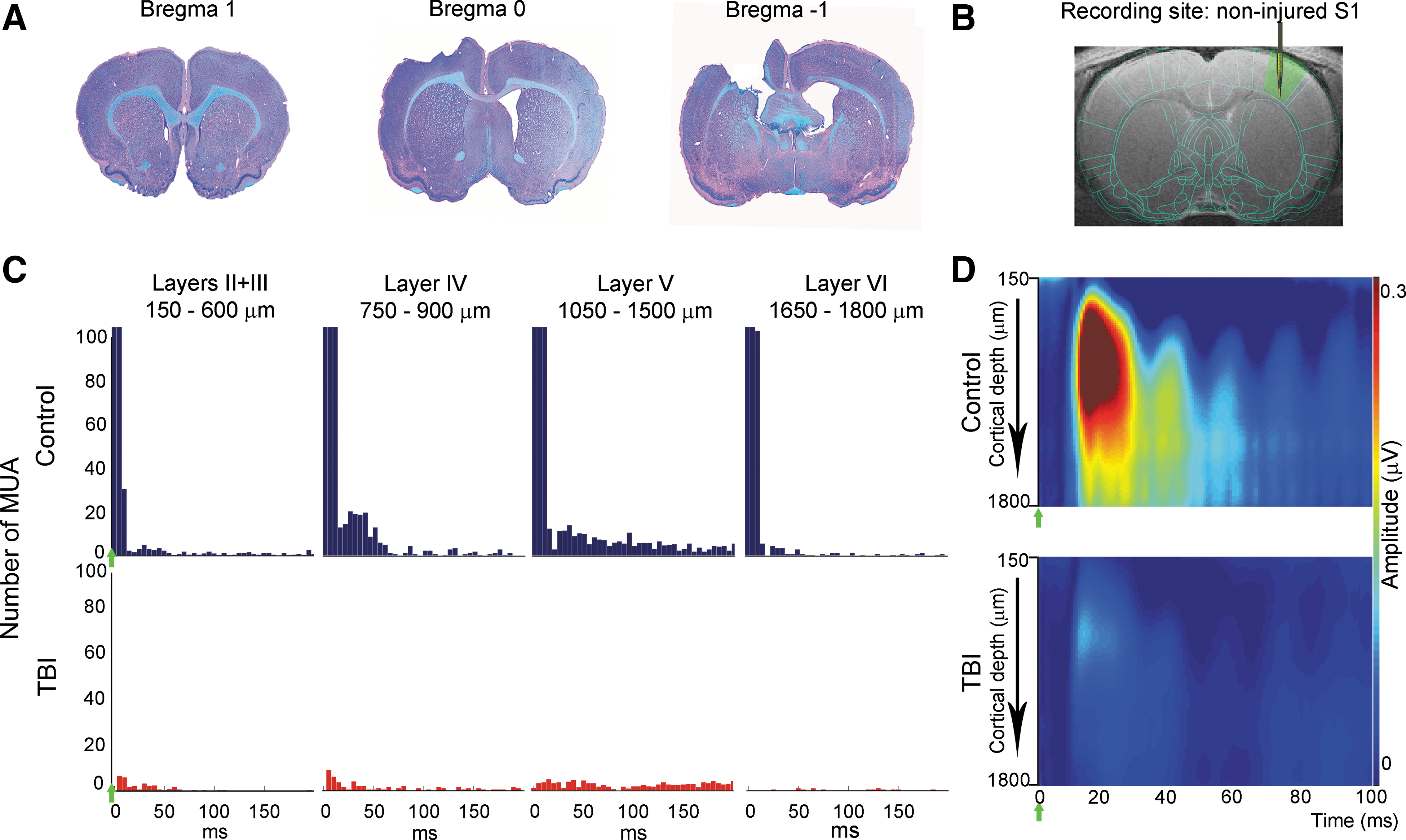
Decreases in stimulus-evoked electrophysiology responses in the noninjured S1 in TBI rats 2 weeks after injury. (
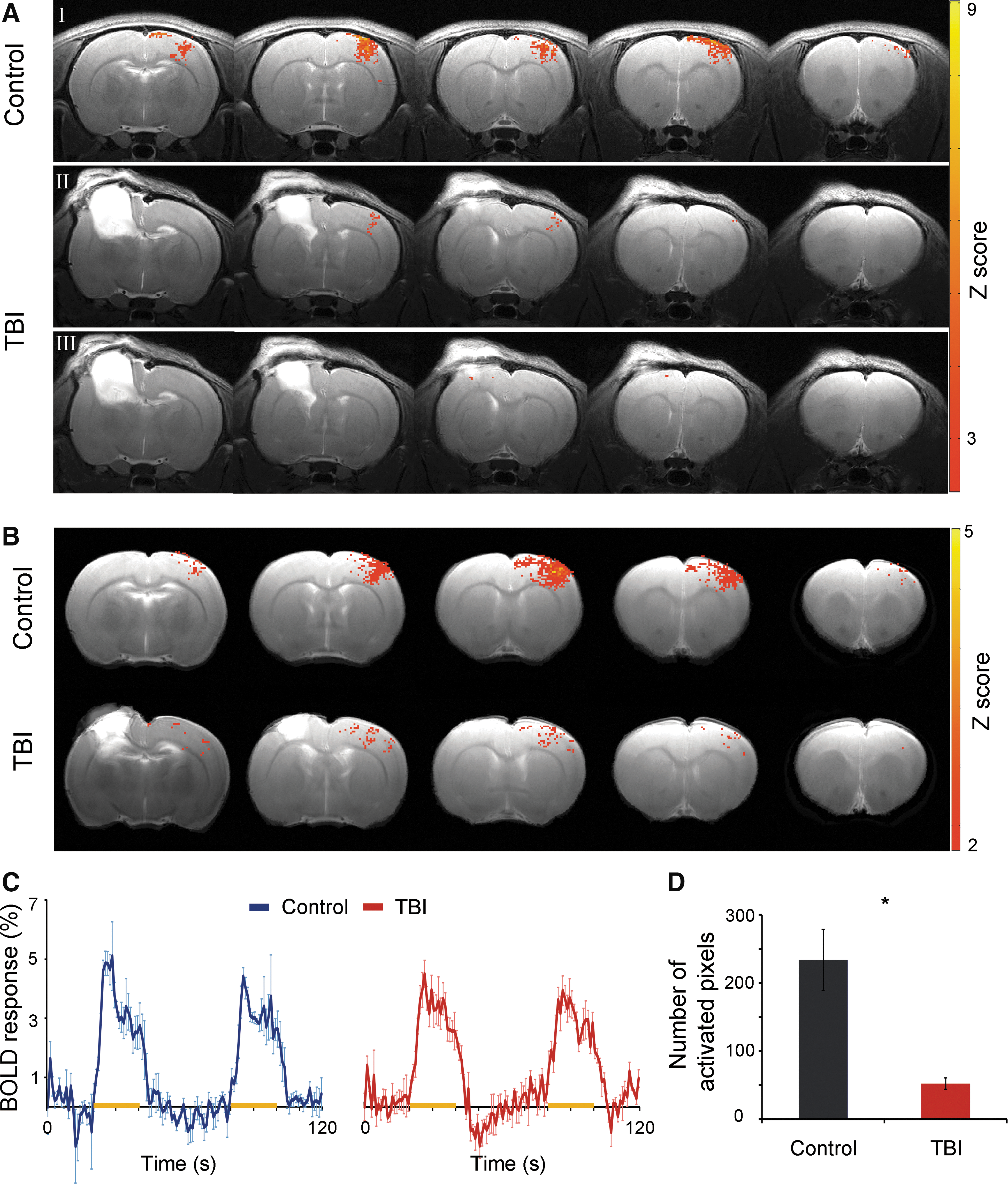
Decreases in BOLD fMRI responses in the noninjured S1 in TBI rats 2 weeks after injury. (
MUAs reflecting the spiking activity of neurons in the vicinity of the electrode were measured across the depth of the noninjured S1. As expected, in control rats (n=5), increases in average number of MUA responses across the cortical depth of S1 contralateral to forepaw stimulation were observed (layers II+III, 203.9±47.9; layer IV, 293.9±65.2; layer V, 261.1±71.9; layer VI, 169.6±62.9). However, in TBI rats (n=5), an average of an 86.4% decrease in number of MUA responses across the cortical depth, compared to controls, was observed. Decreases in number of MUA responses were found across all cortical layers (layers II+III, 29.1±4.1, 85.4%, p<0.05; layer IV, 41.5±11, 85.8%, p<0.05; layer V, 36±9.6, 86.1%, p<0.05; layer VI, 19.1±10.3, 88.7%). Figure 1C shows representative examples of MUA responses from individual rats in the different cortical layers in the noninjured S1.
LFP reflecting the averaged synaptic activity in the vicinity of the electrode was measured simultaneously with the MUA. Consistent with MUA results, TBI rats showed an average of a 75.3% decrease in LFP magnitude across the cortical layers in the noninjured S1, compared to controls (controls: layers II+III, 0.31±0.04 μV; layer IV, 0.44±0.08 μV; layer V, 0.31±0.06 μV; layer VI, 0.22±0.04 μV; TBI: layers II+III, 0.06±0.01 μV, 78.0%, p<0.05; layer IV, 0.09±0.02 μV, 77.7%, p<0.05; layer V, 0.08±0.01 μV, 74.1%, p<0.05; layer VI, 0.06±0.01 μV, 68.6%, p<0.05). In addition, the time when the peak of the LFP responses occurred after stimulation, reflecting response latency, was calculated. A significant delay in response latency was found only in layer V in TBI rats, compared to control rats (controls, 14.9±1.0 ms; TBI, 23.3±1.1 ms; p<0.05). Figure 1D shows individuals and LFP responses across the cortical depth in the noninjured S1.
We then tested whether the altered neuronal activity in the noninjured S1 observed with extracellular in vivo EP can be visualized with noninvasive imaging modalities. This technique is particularly appealing because it could be clinically translated into detailed investigation of postinjury plasticity. BOLD fMRI responses in the noninjured S1, as a consequence of contralateral forepaw stimulation, were assessed in control (n=5) and TBI (n=5) rats. Figure 2A and 2B show individual and group averaged BOLD fMRI activation Z-maps (p<0.05), respectively, of control and TBI rats overlaid on high-resolution anatomical MRI images (positioned at bregma −1.5–2.5 mm according to Paxinos and Watson 24 ) and the corresponding BOLD fMRI time course within S1 area (Fig. 2C). The number of activated pixels within the S1 forepaw representation, as was defined by the rat brain atlas, was calculated across the MRI slices covering the non-injured S1. Indeed, the BOLD fMRI responses were significantly decreased in the non-injured S1 of the TBI rats (52.2±8.2 pixels) compared to controls (233.8±44.9 pixels, p<0.05, Fig. 2D). The average BOLD fMRI amplitude of the activated pixels in the two groups was similar, suggesting that the injury resulted in significant changes in neuronal function of the non-injured hemisphere, but not in its vascular architecture.
The neurophysiological measurement results indicate that TBI in the developing brain causes significant decreases in neuronal function in the injured S1 as well as in remote, contralateral, cortical areas (noninjured S1). Elucidating the neuronal basis of these decreases would be advantageous in terms of improving recovery strategies based on pharmacological interventions and cortical manipulations. Thus, we sought to examine potential neuronal pathway(s) that may facilitate these decreases in neuronal responses. The corpus callosum is a clinically important white matter (WM) tract because it is consistently involved in post-traumatic axonal injury. 10,25 –27 We also showed previously that cortical plasticity associated with peripheral nerve injury is mediated by the corpus callosum. 28 –30 DTI has been shown to provide useful information regarding changes in WM integrity associated with injury. We measured the FA, which is the normalized SD of measured diffusivities, 31 across the corpus callosum of control (n=5) and TBI (n=5) rats. The results demonstrated a significant decrease in FA values across the corpus callosum in bregma 2.28–1.31 (total thickness, 0.97 mm; control, 0.43±0.0 mm2/sec; TBI, 0.40±0.0 mm2/sec; p<0.05, two- factor ANOVA with replication) of TBI rats, compared to control, that is suggestive of WM damage (Fig. 3A). Decreases in callosal FA were also significant when calculated only for the noninjured S1 (control, 0.43±0.0 mm2/sec; TBI, 0.39±0.0 mm2/sec; p<0.05, two-factor ANOVA with replication). In addition, at the end of the neurophysiological measurements, detailed histopathological examination of the corpus callosum area across S1 within the noninjured hemisphere were performed on control (n=5) and TBI (n=5) rats. The results demonstrate a significant decrease (p<0.05) in the corpus callosum area averaged across the noninjured S1 in TBI rats, compared to control rats (control, bregma 2, 1.94±0.1 mm2; bregma 1, 1.41±0.0 mm2; bregma 0, 1.75±0.0 mm2; bregma −1, 1.51±0.1 mm2, TBI, bregma 2, 1.63±0.2 mm2, 15.8%; bregma 1, 1.48±0.0 mm2, −5.0 %; bregma 0, 1.34±0.1 mm2, 23.9%; bregma −1, 1.26±0.1 mm2, 16.2%). Corpus callosum volume was 1.99±0.08 and 2.32±0.1 mm3 in TBI and control rats, respectively, as calculated using Cavalieri's estimator of volume. 32 Examples of corpus callosum staining of control and TBI rats are shown in Figure 3B.
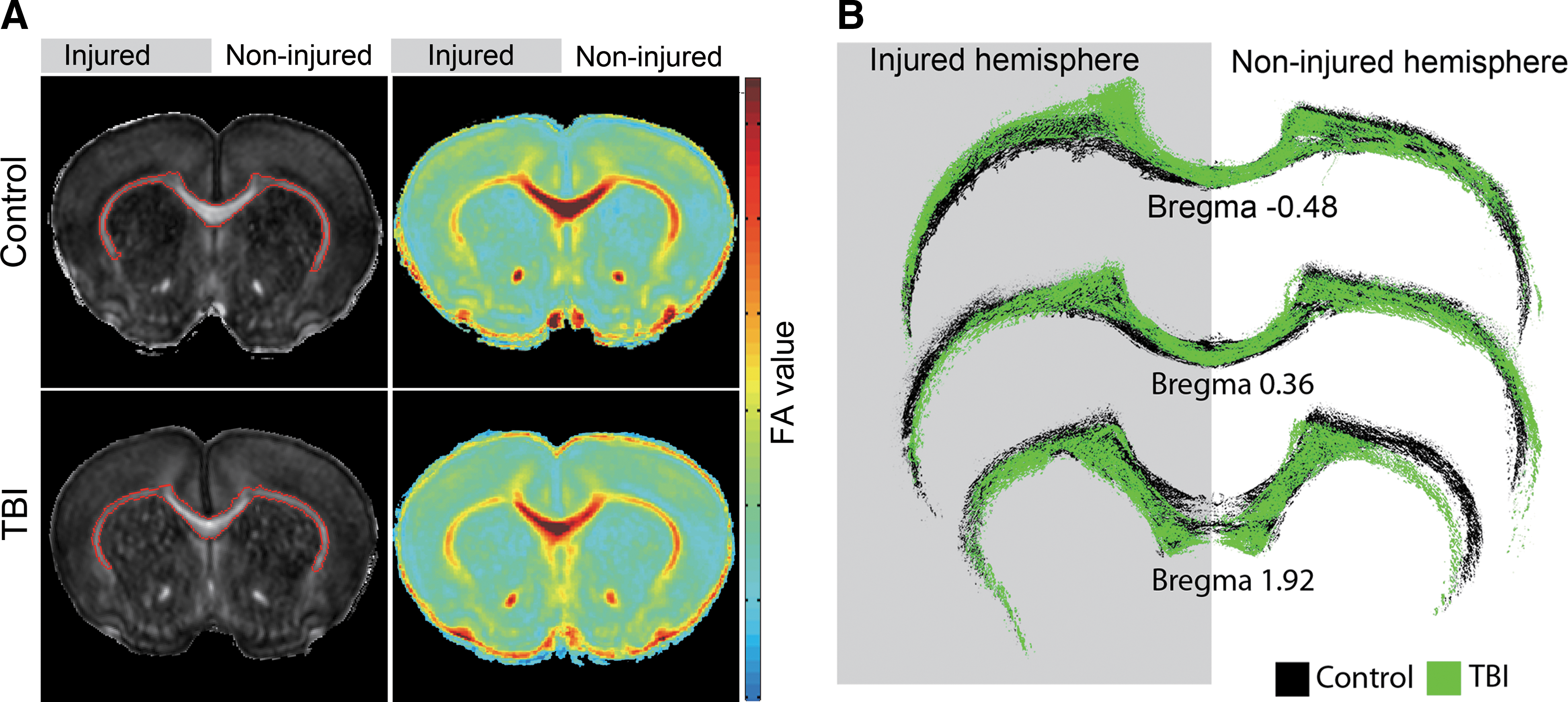
Corpus callosum abnormalities in TBI rats 2 weeks after injury. (
To investigate whether the corpus callosum damage may account for the altered neurophysiological responses observed in the noninjured S1, S1 brain slices were prepared from control (n=6) and TBI (n=11) rats 2–3 weeks after injury. Because one of the main inputs into cortical layer V is transcallosal fibers, whole-cell patch clamp recordings from identified pyramidal neurons in layer V in the noninjured S1 were collected with and without stimulation of the transacallosal projections. Amplitude and frequency of spontaneous mEPSC, reflecting random spontaneous release of presynaptic vesicles, showed that whereas the mEPSC amplitude was similar between the groups (controls, 13.63±1.8 pA, n=18 neurons; TBI, 13.43±0.7 pA, n=32 neurons), TBI resulted in significant decreases in mean mEPSC frequency (controls, 4.11±1.6 Hz; TBI, 1.8±0.3 Hz; p<0.05; Fig. 4A). LTP, which is one of the major cellular mechanisms that underlies learning and memory, was measured 40 min after high-frequency (100-Hz) stimulation of transcallosal fibers. The results demonstrated that stimulation of the transcallosal fibers induced 42.74±1.5% amplitude increase in control (n=11 neurons), but only 8.02±0.7% amplitude increase in TBI rats (n=8 neurons; Fig. 4B). In addition, the amplitudes of the evoked EPSC (eEPSC) that were measured in the presence of GABAA antagonist, were higher in TBI, compared to control, rats (controls, 208.94±66.5 pA, n=13 neurons; TBI, 1078.21±227.3 pA, n=17 neurons; p<0.005; Fig. 4B). Thus, the intracellular recording findings suggest decreased spontaneous neuronal activity, decreased ability to induce LTP, and alterations in GABA-mediated inhibition in layer V neurons in the noninjured S1 after TBI to the developing brain.

Altered synaptic transmission in the noninjured S1 in TBI rats 2–3 weeks after injury. (
Discussion
Plasticity mechanisms associated with learning and memory in the developing brain are often accompanied with architectural and functional modifications of neuronal networks. These plasticity changes occur within seconds (short term) and could last for days, weeks, and a lifetime (long term). As the occurrence of some of these mechanisms attenuates during adulthood, it was viewed that injury during development will activate these mechanisms, which, in turn, will maximize the functional outcome. Indeed, a great amount of work on the rodent barrel cortex suggests such plasticity. Removal of whiskers in young animals drastically changes the morphology of the barrel cortex. 1,33 In these cases, the barrels destined to represent the whisker that has been removed did not develop, and the neighboring barrels enlarged into the vacant territory. However, direct damage to the brain during development appears to produce unfavorable outcomes, even when recovery has been achieved. 5 –7,34
As a result of the cortical damage, as was expected, the neurophysiological responses in the injured S1 were significantly attenuated in TBI rats. Thus, we focused on characterizing the altered neuronal function and connectivity that occur after TBI in the developing brain of the remote, anatomically intact, noninjured hemisphere. Assessments of functional responses were performed 2–3 weeks postinjury, because, in this time frame, long-term plasticity mechanisms might be activated that, consequently, could affect behavioral outcome. 35 Postmortem studies also demonstrated ongoing injury progression 9 as well as abnormal trophic responses of neuronal fibers triggered by the injury 36 occurring at this time point, which could potentially affect postinjury plasticity. Another consideration is that different anesthetics agents have been used in this study. The agents were chosen according to their known mechanism of action and the specific requirements of the measurement. For example, urethane was used for in vivo extracellular recordings because it provides long-lasting anesthesia and its effect on evoked neuronal responses has been previously characterized. 37 On the other hand, fMRI measurements require anesthetic agents that are known to preserve neurovascular functions, such as dexmedetomidine. 28,38 Finally, isoflurane, which provides deep levels of anesthesia, was used only during the TBI procedure.
In TBI rats, dramatic decreases in MUA, LFP, and fMRI responses were observed throughout the depth of the noninjured S1. Similar to the control rats, the highest MUA and LFP responses to contralateral forepaw stimulation in TBI rats were observed in layer IV, as compared to other layers. However, a significant delay in S1 response latency was found only in layer V. This observation, which insinuates that neurons located in this layer may be particularly susceptible to the contralateral injury, requires further experimentation and analysis. Indeed, whole-cell patch clamp recordings revealed that neurons in layer V in the noninjured cortex show significantly reduced spontaneous firing rate and reduced potential to induce LTP by transcallosal stimulation after TBI. The former can be the result of decreased transmitter release probability, number of functional synaptic contacts, and quantal size originating in local, intracortical as well as remote, intercortical presynaptic neurons. The decreased spontaneous neuronal firing rate associated with the injury may also underlie the decreased ability to induce LTP, because the correlation between mEPSC frequency and LTP has been shown previously. 39 In addition, the decreased mEPSC frequency can be manifested by the decreases in the neurophysiological responses observed with extracellular EP and fMRI. We hypothesized that the decreases in LTP and neuronal activity account, at least partially, for the long-term physical, cognitive, psychological, and emotional impairments that survivors of pediatric TBI suffer from. Further, the mean amplitude of the eEPSC responses to transcallosal stimulation in layer V pyramidal neurons was greater in TBI, compared to control rats. Because the eEPSC responses were recorded in the presence of GABAA antagonist, the results suggest that TBI causes a shift in balance between excitatory and inhibitory neurotransmission. Possible mechanisms that can lead to this finding can include postsynaptic decrease in GABAA receptor sensitivity and/or expression, changes in the ratio between postsynaptic expression of GABAA/GABAB receptors, and decrease in presynaptic inhibitory interneuron activity. Alterations in GABA-receptor–mediated postsynaptic inhibition have been shown to be involved in epileptogenesis. 40,41 Thus, this putative attenuation in GABAA-mediated inhibition may contribute to post-TBI-induced seizures that are a common complication in the pediatric population. 42,43 Taken together, it appears that TBI in the developing brain leads to impaired cortical plasticity.
A study measuring the topography of the cortex and hippocampal circuitry after TBI in the immature brain with retrograde pseudorabies virus neuronal labeling showed that these connections are preserved in the noninjured hemisphere. 44 However, the number of infected (retrogradely labeled) neurons in the noninjured cortex was dependent on the degree to which transcallosal projections were affected by the injury. Other studies also report post-traumatic atrophy of the corpus callosum resulting from the direct injury and/or to the secondary insults related to hypoxic-ischemic injury and metabolic changes. For example, DTI revealed decreased transcallosal fiber density and disrupted transcallosal microstructure in children that suffered TBI. 25,26 Transcallosal dysfunction was also reported in immature 10,27 and adult rats after TBI. 45,46 In line with these observations, our DTI and histopathological findings suggest that TBI in the developing brain results in WM damage in the corpus callosum. Indeed, transcallosal-mediated plasticity has been shown to play a significant role in recovery and rehabilitation after stroke and peripheral nerve injuries. 47 –51 In an animal model of peripheral nerve injury, altered transcallosal communication was observed to occur within minutes after injury 52 and persist for weeks. 28 –30 In these cases, the postinjury changes in transcallosal connections resulted in increased cortical inhibition. Increase in inhibition and decrease in excitatory neurotransmission can affect LTP and long-term depression and are associated with impaired plasticity and synaptogenesis in the developing brain. 53 Thus, transcallosal-mediated plasticity can adversely influence the recovery and rehabilitation process. In an animal model of nerve injury, optogenetics manipulation of transcallosal communication caused an immediate increase in neurophysiological responses in infragranular layers of the deprived S1. 28 Strategies to reverse the altered transcallosal communication, such as constraint-induced therapy, 47 nerve blocking, 54 and transcranial magnetic stimulation (TMS), 55 –57 have been shown to maximize functional outcome in stroke and nerve injury patients. An improvement in hand function in children who suffered from stroke was also demonstrated after repetitive TMS treatment. 58
We suggest the impaired, potentially maladaptive plasticity after TBI in the immature brain as one of the potential neuronal mechanisms accounting for the long-term physical, cognitive, psychological, and emotional impairments that affect survivors of pediatric TBI. It emerges that the increased potential for plasticity during development may cause the brain to be more vulnerable to injury. This impaired plasticity in the developing brain could be attributed to several mechanisms (reviewed by Giza and Prins 59 ), such as critical windows that were missed during the recovery process, ongoing injury progression that disturbed the circuit maturation process, 9 and abnormal trophic responses of neuronal fibers triggered by the injury. 36 An additional consideration is that whereas postinjury recovery in the adult brain can be defined by the return of function to the premorbid level, the functional responses in the developing brain are dynamic. Postinjury recovery in the developing brain may reach premorbid levels, but may not match, at the end of the recovery period, age-matched peers. Understanding the plasticity mechanisms after TBI and how they are translated into recovery and behavior may benefit the development of appropriate therapeutic strategies based on pharmacological interventions and brain stimulation, such as TMS. Moreover, it would elucidate neuroplasticity mechanisms associated with normal brain development.
Footnotes
Acknowledgments
This work was supported by Johns Hopkins Brain Science Institute and the National Institutes of Health/National Institute of Neurological Disorders and Stroke (grant no. R01NS072171).
Author Disclosure Statement
No competing financial interests exist.