
Abstract
Traumatic brain injury (TBI) is a significant risk factor for chronic traumatic encephalopathy (CTE), Alzheimer's disease (AD), and Parkinson's disease (PD). Cerebral microbleeds, focal inflammation, and white matter damage are associated with many neurological and neurodegenerative disorders including CTE, AD, PD, vascular dementia, stroke, and TBI. This study evaluates microvascular abnormalities observed at acute and chronic stages following TBI in rats, and examines pathological processes associated with these abnormalities. TBI in adult rats was induced by controlled cortical impact (CCI) of two magnitudes. Brain pathology was assessed in white matter of the corpus callosum for 24 h to 3 months following injury using immunohistochemistry (IHC). TBI resulted in focal microbleeds that were related to the magnitude of injury. At the lower magnitude of injury, microbleeds gradually increased over the 3 month duration of the study. IHC revealed TBI-induced focal abnormalities including blood–brain barrier (BBB) damage (IgG), endothelial damage (intercellular adhesion molecule 1 [ICAM-1]), activation of reactive microglia (ionized calcium binding adaptor molecule 1 [Iba1]), gliosis (glial fibrillary acidic protein [GFAP]) and macrophage-mediated inflammation (cluster of differentiation 68 [CD68]), all showing different temporal profiles. At chronic stages (up to 3 months), apparent myelin loss (Luxol fast blue) and scattered deposition of microbleeds were observed. Microbleeds were surrounded by glial scars and co-localized with CD68 and IgG puncta stainings, suggesting that localized BBB breakdown and inflammation were associated with vascular damage. Our results indicate that evolving white matter degeneration following experimental TBI is associated with significantly delayed microvascular damage and focal microbleeds that are temporally and regionally associated with development of punctate BBB breakdown and progressive inflammatory responses. Increased understanding of mechanisms underlying delayed microvascular damage following TBI could provide novel insights into chronic pathological responses to TBI and potential common mechanisms underlying TBI and neurodegenerative diseases.
Introduction
T
Microvascular damage detected in later periods after TBI is associated with diffuse axonal injury (DAI), considered to be one of the major chronic pathological complications following TBI that could directly affect clinical outcome. 5 –8 Post-traumatic microvascular damage results in the genesis of focal microbleeds associated with microscopic areas of hemosiderin deposits, which can be detected by advanced magnetic resonance imaging (MRI) protocols such as susceptibility weighted imaging (SWI). 5,9 Studies have demonstrated a characteristic distribution of traumatic microbleeds after TBI that differ from other conditions such as intracranial hemorrhage (ICH), and predominantly occur in the deep white matter. 10 Microbleeds have been detected in both the acute and chronic stages following TBI, 8,11,12 and their prevalence has been associated with the degree of injury severity. 13 These abnormal accumulations of ferritin/hemosiderin are known to be toxic to astrocytes, neurons, and endothelial cells, 14,15 and are frequently observed surrounded by macrophages, which can initiate inflammatory and neurodegenerative cascades. These cascades can include activation of microglia, stimulation of gliosis, late complement activation, and apoptosis; pathological processes that have all been reported to be associated with post-traumatic neurodegenerative sequelae including AD 16 and dementia. 9 Inflammation and white matter degeneration can persist for years after a single TBI. 17
In addition to TBI, 8,12,18,19 microvascular damage is associated with several neurological and neurodegenerative disorders in humans, including stroke, 20,21 AD, 22 –24 and different types of dementia. 25 Moreover, recent studies suggest that temporal lobe microbleeds may play roles in the development of suicidality in stroke patients. 26 In patients with chronic traumatic encephalopathy (CTE) which is primarily reported in athletes who have sustained repeated brain injury, hemosiderin deposits and tau accumulation are frequently observed surrounding small cerebral vessels, suggesting microvascular involvement in the pathology. 27,28 Although, definitive diagnostic criteria for CTE are not yet established, 29 white matter abnormalities, including myelin loss in the corpus callosum and tau-positive fibrillary tangles, have been proposed as characteristics of CTE. 28,30 The presence of focal microbleeds has also been documented in a small number of studies employing animal models of cerebral small vessel disease causing stroke and dementia. 31,32
The etiology and mechanisms of cerebral microbleeds/microvascular damage, as well as their involvement in the neuropathological consequences of neurodegenerative diseases, are not well understood. The observation that focal brain microbleeds are a prominent feature of various neurodegenerative diseases, and that vascular damage with heme deposition is associated with cognitive impairment and dementia, 9,16,33 –35 suggest that focal microbleeds might play a role in both acute and prolonged pathological responses to TBI. 5,36 TBI has been reported to cause neurodegeneration and progressive brain atrophy that continues for at least 1 year after injury, and these processes are associated with neuronal and white matter degradation. 37,38 Nevertheless, few experimental studies have attempted to characterize the temporal and regional pattern of post-traumatic vascular changes and focal microbleed formation. 39 Equally important, no studies have systematically examined the temporal profile of the appearance and resolution of these microbleeds. Although the specific pathological pathways contributing to progressive white matter degeneration after TBI have not been well defined, several possible mechanisms have been proposed, including apoptotic cell death, 40 –43 inflammation 44,45 in white matter tracts, and prolonged acute and chronic regional hypoperfusion. 46 –48 Importantly, a recent review highlights the role of iron even in mild TBI, including focal deposition of heme-iron in the white matter as result of microhemorrhage. 49
In the present study, we hypothesized that TBI initiates delayed pathophysiological cascades, involving punctate opening of the blood–brain barrier (BBB), and increased inflammation. Activation of these cascades is associated with progressively increasing microvascular damage that spreads over time from the site of primary injury. These sites of damage can become foci for chronic inflammation and white matter degeneration. We utilized a well-characterized model of controlled cortical impact (CCI) in rats, 50 together with high resolution histopathological and immunohistochemical analyses, to determine whether prolonged white matter damage in the corpus callosum following TBI is temporally and regionally associated with microvascular damage, BBB breakdown, and/or neuroinflammation. The markers used in this study are routinely employed in postmortem studies in humans with white matter abnormalities and small vessel disease, 51 allowing us to further characterize the relationship between TBI and delayed chronic microvascular degenerative diseases.
Our results suggest that TBI results at least two discernible stages of microvascular damage. 1) A previously reported acute stage involving neuropathological processes initiated by mechanical injury primarily localized to the primary site of injury and persisting up to 1 week. This first stage includes biphasic widespread BBB opening, leucocyte translocation, and microglia activation. 2) A hitherto unreported subacute/chronic phase that involves delayed secondary pathologies, including focal microbleeds and toxic iron deposition that are regionally and temporally associated with delayed punctate BBB opening, inflammation, and white matter damage distal from the primary site of injury. These delayed and prolonged microvascular changes following TBI may represent an underappreciated component of cellular and molecular cascades that mediate evolving post-traumatic neurodegeneration and other neurodegenerative diseases for which TBI is a risk factor.
Methods
Animals
All experimental procedures of the study were approved by the University of Florida IACUC Committee. Male Sprague–Dawley rats weighing 230–300 g (Harlan, Inc. Indianapolis, IN) were used. The rats were allowed access to a normal laboratory diet and potable water ad libitum, and were acclimated to the housing facilities and diet for at least 1 week before being used in the study. The controls in the animal room were set to maintain a temperature of 20–24°C and 30–70% relative humidity. Rats were maintained on a 12 h light/dark cycle. Numbers of animals per experimental group were calculated based on power analysis and our previous experience, to obtain significant differences between measured outcomes. Three experimental groups (n=4–5 per group) were employed in the study: control, unilateral CCI (uCCI), and bilateral CCI (bCCI). The addition of the bCCI group was chosen to examine the effects of more severe bCCI on the corpus callosum. Following lower level uCCI, rats were euthanized and studied at 24 h, 1 week, and 1, 2, and 3 months following injury.
CCI
CCI brain injury 50,52 was induced using Benchmark™ Stereotaxic Impactor (MyNeurolab, USA). Animals were anesthetized with 4% isoflurane in oxygen/air mixture and maintained under anesthesia by maintenance anesthesia with 2.5% isoflurane in the same carrier gas. The surgeries were performed with the animals mounted in a stereotactic frame in a prone position and secured by ear and incisor bars. To produce uCCI, a 5 mm ipsilateral craniotomy was performed midway between bregma and lambda in the center of the right parietal bone with the dura mater remaining intact over the cortex. In case of bCCI, two equivalent craniotomies were performed symmetrically on both sides of midline (i.e., right and left parietal bones). Brain trauma in the uCCI model was produced by a single impact using a 4 mm impactor tip, velocity of 3.5 m/sec, dwell time 200 ms, and designed compression distance of 2.5 mm. To produce injury in the bCCI model, a single impact with the same parameter as in the uCCI model was performed sequentially on each hemisphere. Sham-injured control animals received identical anesthesia and surgical preparation including craniotomy, without impact injury. The following groups of animals with corresponding time points for brain tissue analyses were used: Sham control (n=4), uCCI 24 h (n=4), uCCI 1 week (n=4), uCCI 1 month (n=5), uCCI 2 months (n=4), and uCCI 3 months (n=5). Multiple bCCI animals were studied from 1 month to 3 months (n=3 per group). No quantitative analyses were performed on bCCI rats. In all animals, pre- and post-surgery monitoring and management was maintained to ensure compliance with guidelines set forth by the University of Florida Institutional Animal Care Committee.
Immuno- and histochemistry
At appropriate time points following TBI, animals were euthanized with a lethal dose of Beuthanasia®-D solution, transcardically perfused with 4% paraformaldehyde, and whole brains were removed, processed, and embedded in paraffin.
For the intracardiac perfusion, a commercially available perfusion system containing of two (50 cc) syringes with a valve and tubing elevated 100–120 cm above the animal was used according to manufacturer's protocol. Rats were anesthetized with intraperitoneal injection of Beuthanasia-D solution at a lethal dose of 200 mg/kg and perfused with washing solution (phosphate buffered saline [PBS], 100–150 mL) and fix solution (4% paraformaldehyde in PBS, 200–250 mL) at a rate of ∼2 mL/min. After perfusion, the brain was removed and placed in the same fixative solution for at least 24 h, and then processed and embedded in paraffin. Brain coronal sections (4 μM) used for all immuno- and histochemical analyses were cut between −3.8 mm and −2.8 mm from bregma, which was within the center of impact site, and then attached to slides
The coronal brain sections showing the core lesion and penumbra areas used for histopathological and IHC analyses were cut at the center of the circular craniotomy corresponding to the impact zone. These sections represented all important brain regions differently affected by focal CCI injury. Brain sections from stereotaxically corresponding regions were used in all animals. Based on the experimental data by us and others, these sections produced reliable and reproducible injury profiles in this well-characterized focal TBI model. 50,52 The selection of these brain sections was based on the major goal of this study: to investigate the temporal progression of brain pathology from injured regions and surrounded areas to more distal regions of the corpus callosum not directly impacted.
IHC procedures
To visualize myelin, a Luxol fast blue (NovaUltra) stain kit was used. Coronal brain sections were de-pariffinized and incubated with Luxol fast blue solution overnight at 56 C°. The following day, sections were rinsed with alcohol and distilled water and differentiated with lithium carbonate solution followed by rinsing with distilled water, and counterstained with cresyl violet solution. Iron Prussian blue staining, used for visualization of microbleeds (hemosiderin deposits), was performed using an Iron kit (Sigma-Aldrich). Previously de-paraffinized and hydrated 4 μm brain coronal sections were incubated with a mix of potassium ferrocyanide solution with hydrochloric acid solution for 10 min, rinsed with distilled water, and counterstained with pararosaniline solution to visualize morphology.
Immunohistochemical experiments
IHC analysis was performed on paraffin-embedded 4μm coronal brain sections. After de-paraffinization, the slides were incubated for 10 min at 95°C in Trilogy solution (Cell Marque, Hot Springs, AK) for antigen retrieval, and blocked for endogenous peroxides with 3% hydrogen peroxide. The sections were incubated overnight at 4C° with 1) one of the primary antibodies to rat IgG (Millipore) to assess BBB damage; 2) intercellular adhesion molecule 1 (ICAM-1, Abcam), an endothelial damage marker; 3) ionized calcium binding adaptor molecule 1 (Iba1, Abcam), a microglial marker; 4) glial fibrillary acidic protein (GFAP), cell signaling, (an astrocytic marker); and (5) cluster of differentiation 68 (CD68, Millipore) to assess inflammation. Treatment of the tissue with a primary antibody was followed by incubation with biotinylated anti-species-specific secondary antibodies and avidin-HRP conjugate (LSAB+kit, #K0679, Dako, Carpinteria, CA). The immunostaining was visualized with 3, 3′-diaminobenzidine (DAB) for brown color development, and sections were counterstained with hematoxylin (Dako, Carpinteria, CA). Negative controls were performed by treatment only with species-matched secondary antibodies. The staining was visualized by exposure to DAB (Dako, Carpinteria, CA). The sections were then counterstained with hematoxylin (Dako, Carpinteria, CA). For control experiments, primary antibodies were omitted. Sections were finally washed with PBS, mounted, air-dried, and cover-slipped with mounting medium Aquamount (Dako). The slides were scanned and examined using either Aperio ScanScope GL system using 20x objective or ScanScope software.
Quantitative analyses of immuno- and histochemistry
The number of microbleeds (Prussian blue positive hemosiderin deposits) was counted using ImageJ software in the ipsilateral and corresponding contralateral areas of corpus callosum (800×200 μm) with the same locations in all animals, and was expressed as counts per mm2. The number of punctate IgG staining was counted in the same areas (200×150 μm) of corpus callosum using ImageJ software, and was expressed as counts per mm2. To quantify the amount of a specific stain present in a scanned slide image, the Positive Pixel Count algorithm associated with the ImageScope (Aperio) digital image analysis software was used. The same areas (800×200 μm) of the corpus callosum within the interhemispheric fissure were used to measure immunopositivity for rat IgG, ICAM 1, Iba1, and CD 68 visualized with DAB staining using build-in Positive Pixel Count Algorithm (version 9.1) of ImageScope software (Aperio Technologies). The image analysis was performed with the default parameter setting, and visual inspection of markup images was performed to confirm that the algorithm results were sufficiently accurate for the purpose of specific biomarker quantification. The positivity value was expressed as a ratio of positive pixels detected (brown staining) to total area. Analysis was performed at full ×20 magnification.
Statistical analysis
For statistical analysis of specific immunostaining, one way ANOVA followed by Bonferroni's multiple comparison tests (Prism 5, Graphpad, La Jolla, CA) were used. Data were reported as mean±SEM. P values<0.05 were considered significant.
Results
Acute and chronic histopathological changes after CCI
Histopathology with Luxol fast blue staining demonstrated that uCCI caused delayed white matter lesions in the corpus callosum that expanded over time from 1 day to 3 months after experimental TBI when compared with sham-injured controls (Fig. 1).

Acute and chronic histopathological changes following unilateral controlled cortical impact (uCCI). Representative photograph of Luxol fast blue staining of myelin fibers in the corpus callosum of uCCI injured rat at 24 h, 1 month, 2 months, and 3 months following uCCI, and corresponding control. Low magnification images demonstrate regions of the corpus callosum. Higher magnification images demonstrate decreased myelin density, suggesting possible demyelination of white matter following uCCI. Color image is available online at
Association of delayed focal microbleeds and chronic BBB dysfunction after TBI
Detection of delayed microbleeds
No detectable signs of microbleeds were observed in the acute (24 h) post-injury period. However, Prussian blue iron staining revealed deposition of microbleeds with a range between 2 and 20 μm in diameter localized around the region of maximal white matter damage in the corpus callosum beginning at 1 week after injury following both uCCI and bCCI brain injury. The number of individual microbleeds continued to gradually increase for up to 3 months post-injury (Fig. 2A) when the number of microbleeds in the corpus callosum was found to be significantly increased when compared with control animals (p<0.05, Fig. 2B). The deposition of hemosiderin deposits/microbleeds was first evident in the white matter regions adjacent to the primary site of white matter damage, and expanded progressively over time into the white matter of the contralateral hemisphere (Fig. 2A, C). Both the number and relative size of microbleeds was higher following bCCI than those observed following uCCI (Fig. 2C). In addition, bCCI (but not uCCI) caused regional/focal deposition of microbleeds in the hippocampal areas adjacent to the injury site, suggesting a possibility of their involvement in hippocampal neurodegeneration (data not shown).
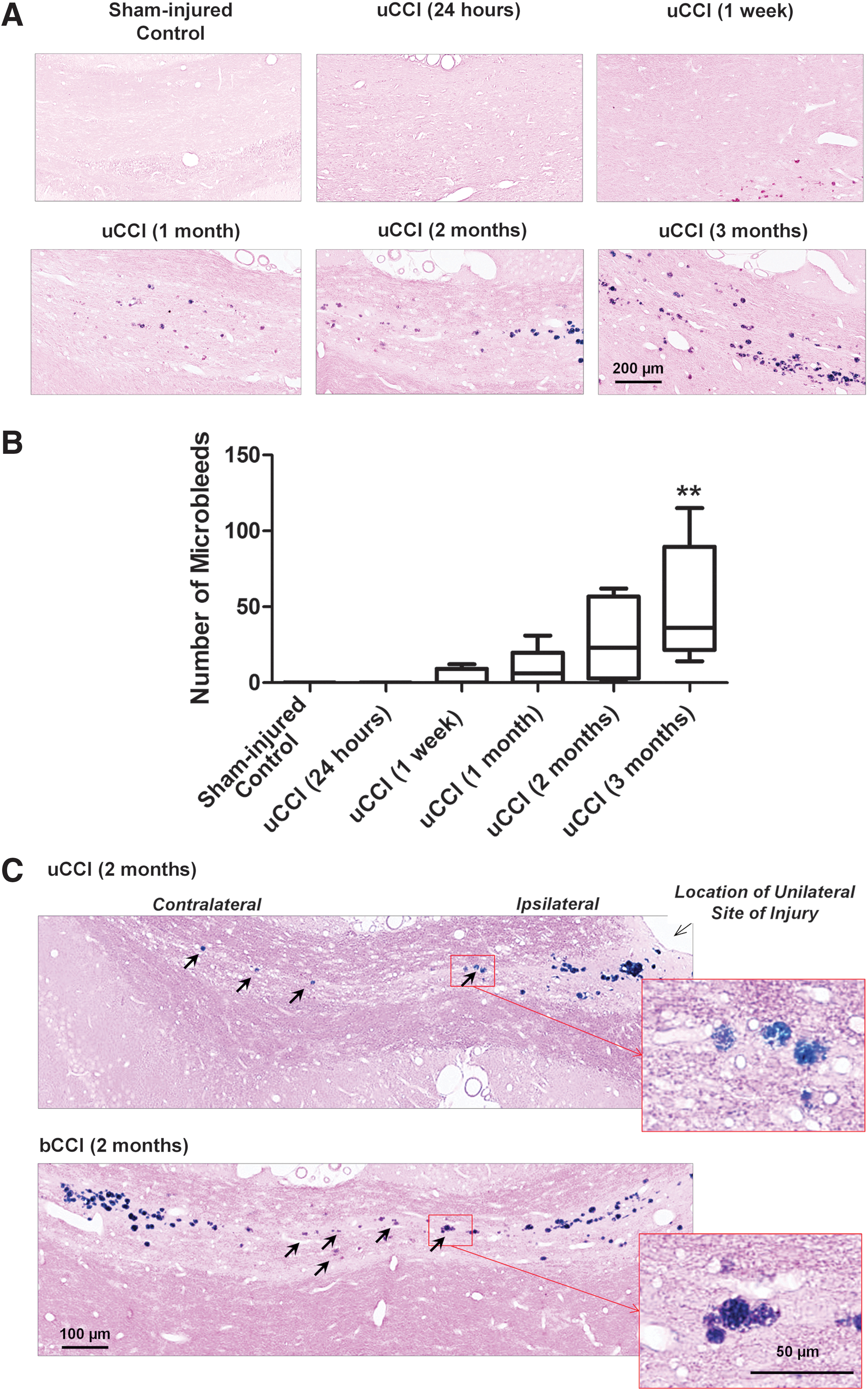
Temporal profile of increases in microbleeds in the corpus callosum following unilateral controlled cortical impact (uCCI)-induced injury: Comparison with more severe bilateral CCI (bCCI) induced injury. (
BBB breakdown
Immunohistochemical analysis with anti-rat IgG revealed prominent, diffuse rat IgG immunoreactivity surrounding the maximal sites of injury including regions of the ipsilateral cortex, ipsilateral hippocampus and corpus callosum (Fig. 3). A marked increase in diffuse IgG immunoreactivity was observed at early time points, reaching maximal staining at 24 h post-injury and remained increased for up to 1 week. Although diffuse IgG immunoreactivity decreased by 1 month post-injury, numerous focal ∼1 μm diameter IgG immunoreactive punctal stainings suggestive of localized BBB breakdown were observed within the corpus callosum at this delayed time point (Fig. 3A). The appearance of these delayed focal IgG-immunopositive puncta preceded microbleed formation and expanded over time to infiltrate into the contralateral site (Fig. 3B). However, by 3 months after injury, a prominent portion of punctate IgG-immunopositive staining co-localized with focal microbleeds. (Fig. 3C).
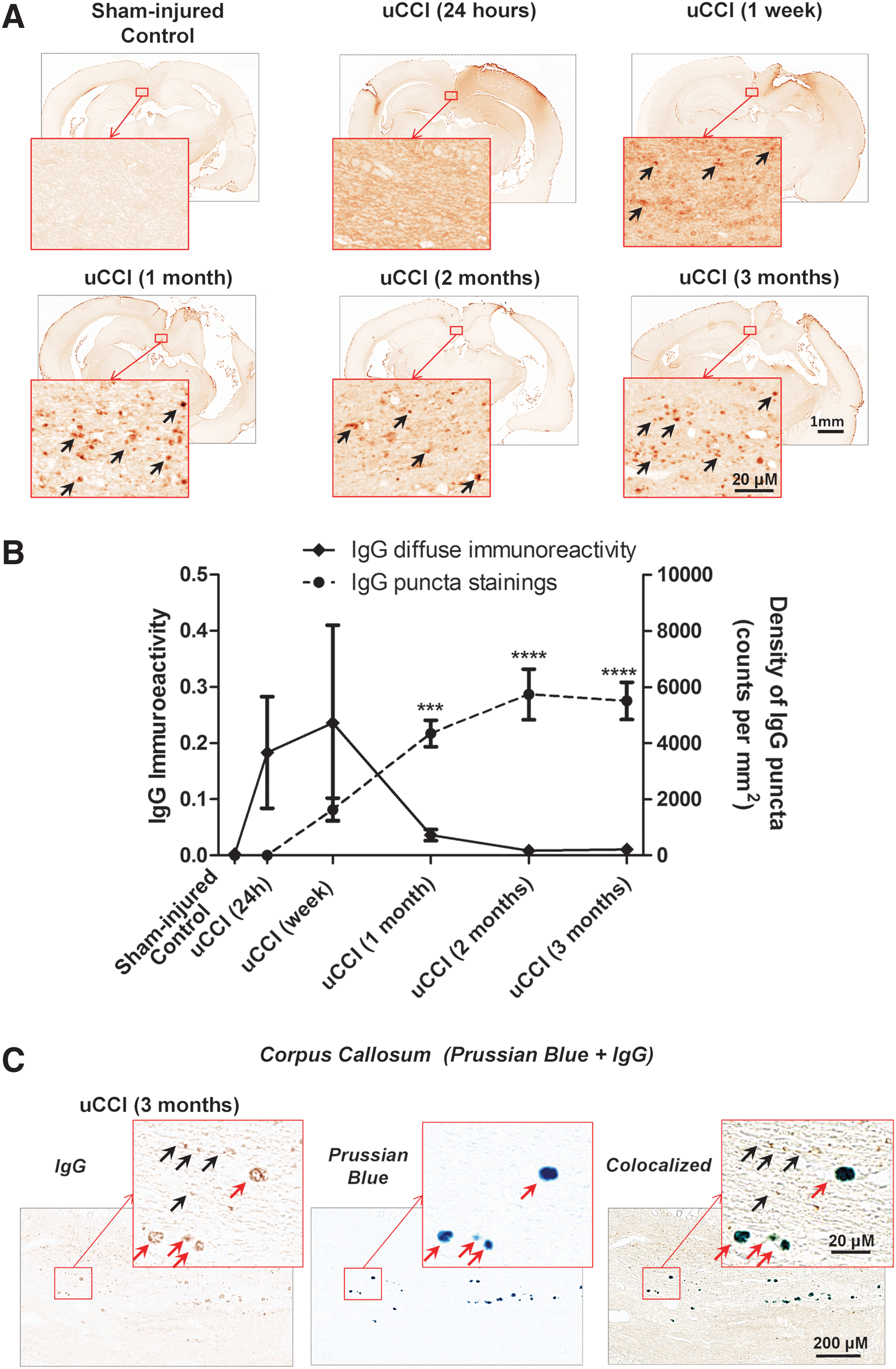
Temporal profile of changes in the blood–brain barrier (BBB) damage in the corpus callosum following unilateral controlled cortical impact (uCCI) and co-localization with microbleeds. (
Endothelial damage
TBI resulted in significantly increased ICAM-1 immunoreactivity at the injury site and the corpus callosum. At 3 months post-injury, the microbleeds were surrounded by increased ICAM-1 immunopositivity (Fig. 4A). Increased ICAM-1 immunoreactivity was biphasic, showing a significant increase at 1 week and 3 months following CCI (Fig. 4B).
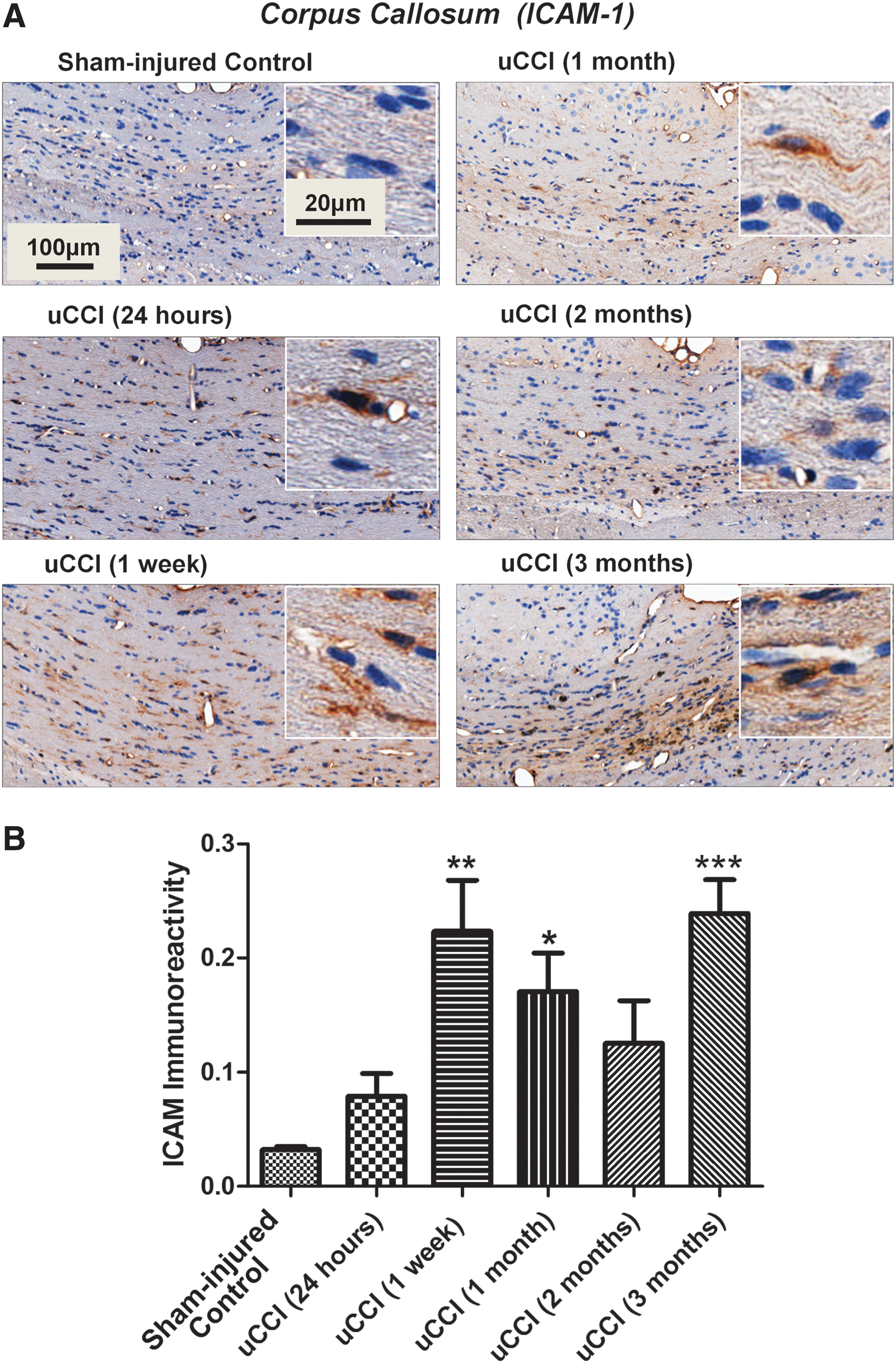
Time dependent changes in levels and localization of intercellular adhesion molecule 1 (ICAM-1) in rat brain following unilateral controlled cortical impact (uCCI)-induced injury. (
Association of delayed focal microbleeds with regional brain inflammation after TBI
Activated microglia (Iba1-immunostaining)
The onset of delayed focal microbleeds was temporally associated with increased Iba1 immunoreactivity in the corpus callosum, although Iba1 positive cells were evenly distributed in the tissue. The morphology of Iba1-positive cells in the corpus callosum changed over time and showed characteristics of reactive microglia with bush-like shapes at chronic time points following TBI (Fig. 5A). uCCI brain injury induced a significant increase in Iba1 immunoreactivity in white matter regions at 1 week post-injury (p<0.01 when compared with sham-injured control animals) that remained significantly increased for up to 3 months (p<0.01; p<0.05) (Fig. 5B), suggestive of persistent microglial activation in white matter following TBI.
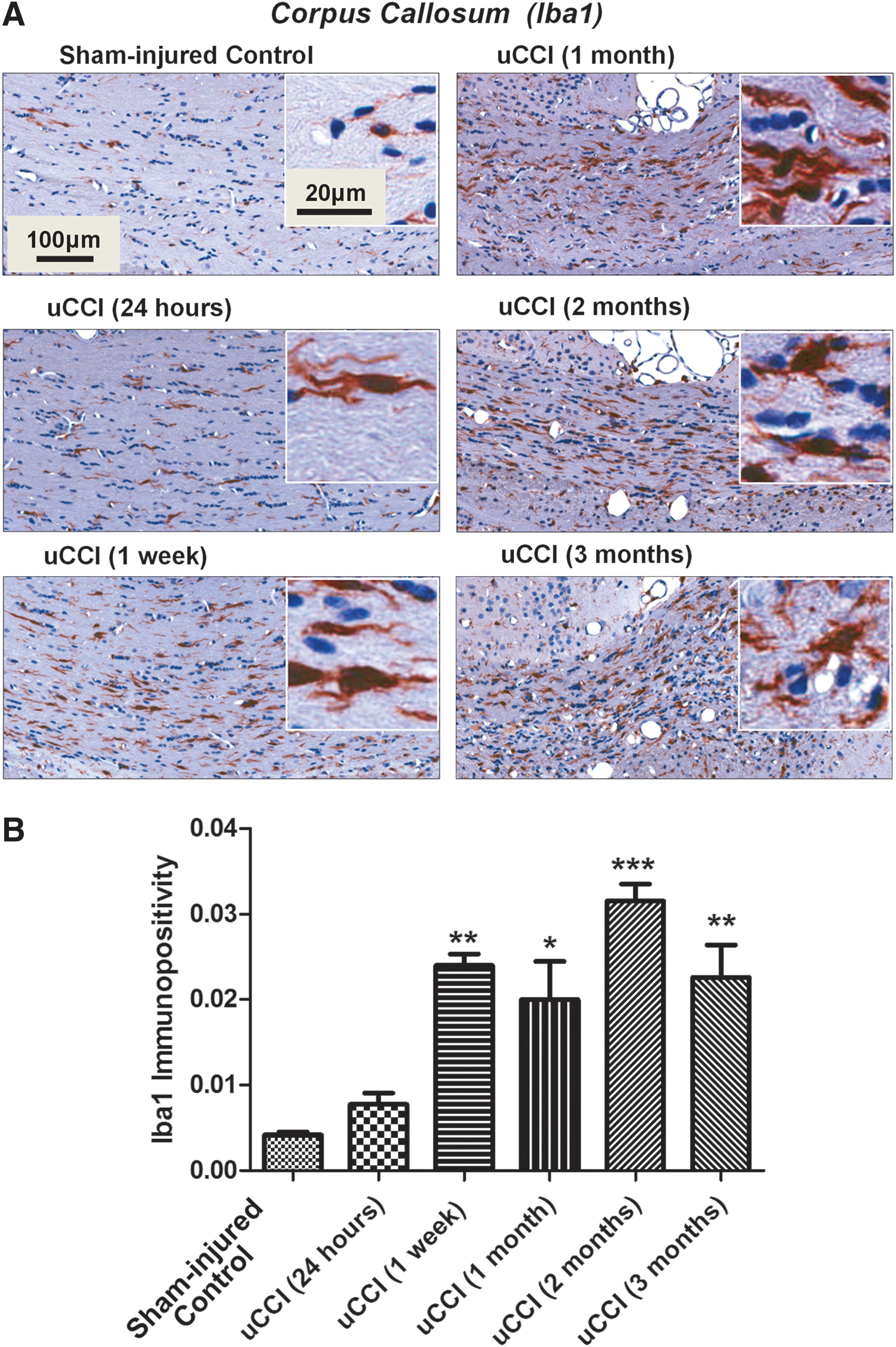
Temporal profile of changes in levels, morphology, and localization of ionized calcium binding adaptor molecule 1 (Iba1) positive cells in rat brain following unilateral controlled cortical impact (uCCI)-induced injury.
Brain inflammation
Immunohistochemical analysis revealed increased immunoreactivity for CD68, a lysosomal glycoprotein expressed by microglia/macrophages, at 1 week after uCCI, which gradually increased and reached maximal levels by 3 months post-injury (p<0.05 when compared with sham-injured controls) (Fig. 6A, B). In white matter regions including the corpus callosum, fimbria of hippocampus, and cerebral peduncles, increased levels of CD 68 immunoreactivity were detected in both ipsilateral and contralateral hemispheres following both uCCI and bCCI brain injury (data not shown).
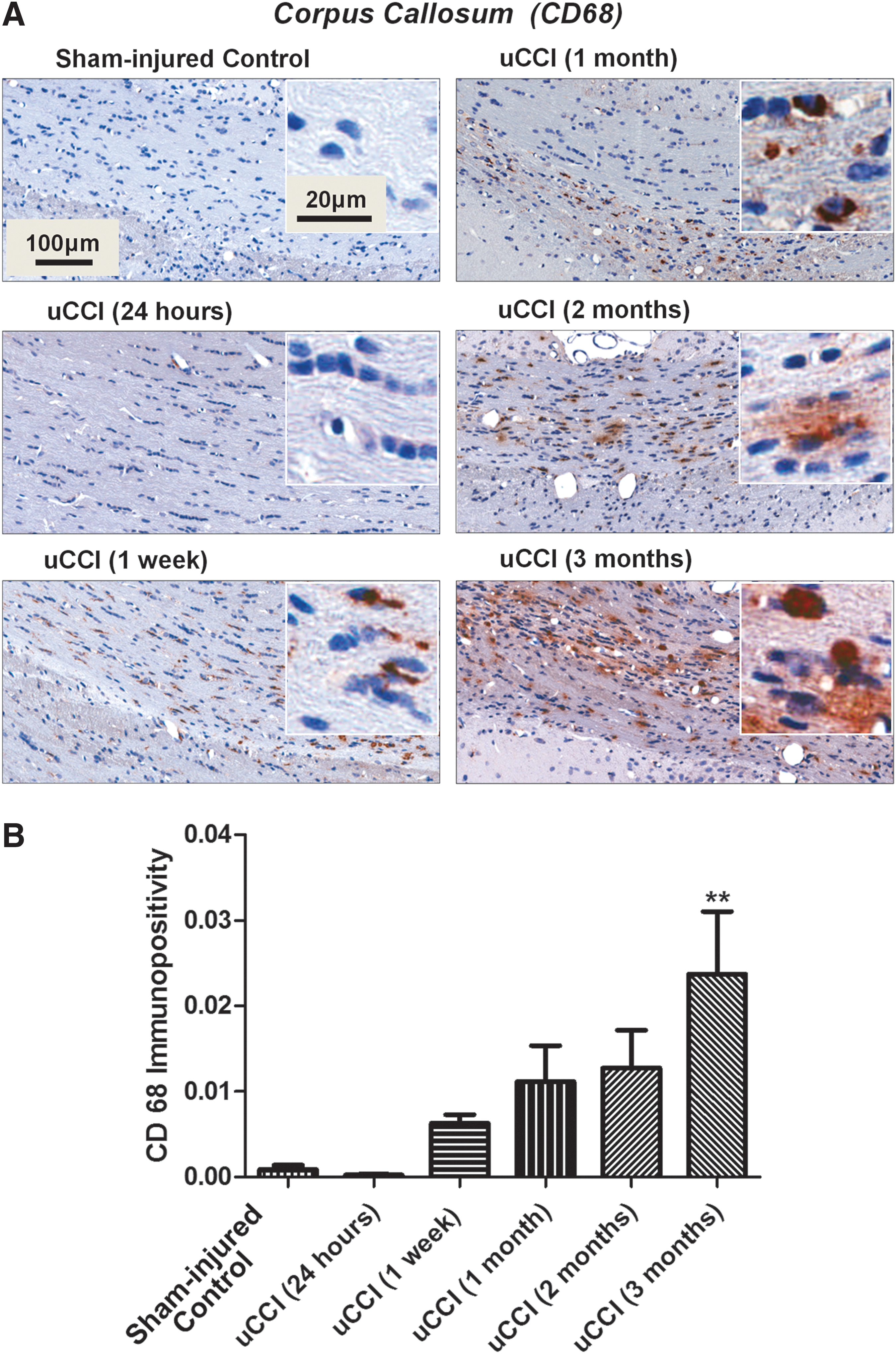
Temporal profile of changes in levels, morphology, and localization of cluster of differentiation 68 (CD68) positive cells in rat brain following unilateral controlled cortical impact (uCCI)-induced injury.
Association of microbleeds with abnormalities in white matter
Reactive astrogliosis (GFAP-immunostaining)
Increased GFAP reactivity following TBI showed a similar temporal and regional pattern to the upregulation of Iba1 immunopositivity. GFAP-immunoreactivity in the corpus callosum was found to be elevated at all time points up to 3 months post-injury (data not shown). Importantly, by 3 months post-injury, GFAP immunoreactivity in the corpus callosum was specifically localized to the formation of areas of glial scarring surrounding the discrete areas of focal microbleeds (Fig. 7A).
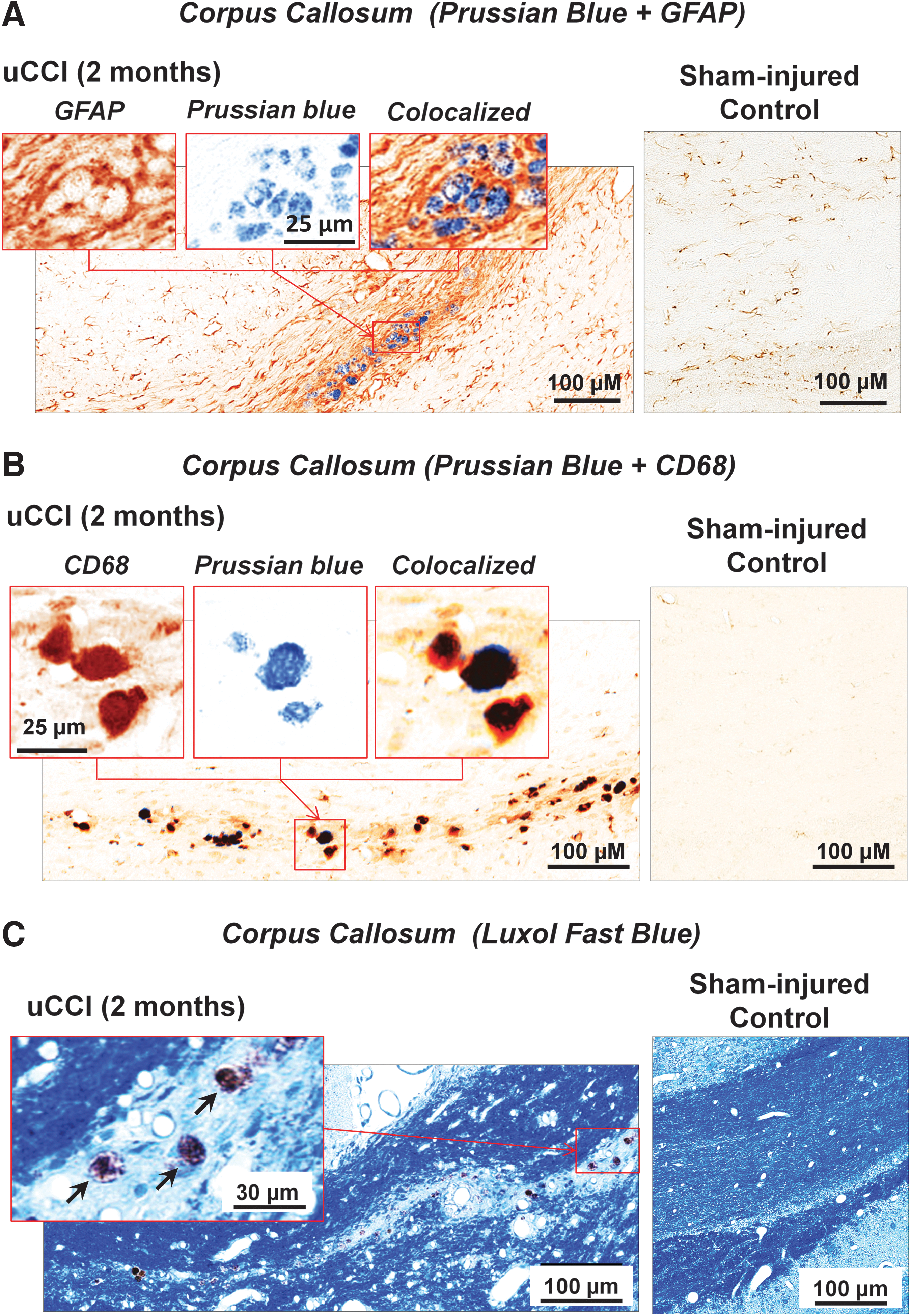
Pathological abnormalities in white matter associated with microbleeds.
Evolving white matter degeneration
Prussian blue positive staining, indicative of the presence of focal microbleeds in the corpus callosum, was observed at 1 week, and 1, 2, and 3 months after TBI, and co-localized with increased CD68 expression in double-labeled immunohistochemical experiments (Fig. 7B). bCCI caused higher CD68 expression with a concomitant increase in focal microbleeds from 1 to 3 months after injury when compared with uCCI (data not shown). Importantly, Luxol blue staining demonstrated that the density of myelin fibers was markedly decreased in areas surrounding focal microbleeds, suggestive of chronic degradation of white matter and myelin loss (Fig. 7C).
Discussion
Overview of results
Using the well-characterized model of CCI brain injury, our data suggest that chronic white matter degeneration following experimental TBI is associated with significantly delayed microvascular damage. A focal TBI model (i.e., CCI) was chosen specifically to investigate whether localized cortical injury and mechanical damage acutely trigger neurodegenerative and inflammatory cascades at later chronic stages, and whether the brain damage could spread over time to the brain regions not directly affected by the primary injury. The development of focal microbleeds was associated with secondary, delayed, localized BBB breakdown and progressive degeneration of white matter up to 3 months following injury. Moreover, delayed white matter damage and development of focal microbleeds were co-localized with cellular markers of regional inflammation. These data are the first to suggest that TBI is associated with focal microbleeds and BBB breakdown in white matter, which are delayed in onset, become foci of inflammation, and contribute to evolving post-traumatic white matter degeneration.
Acute and chronic inflammatory and morphopathological responses to TBI
Despite extensive studies and preclinical research, the precise molecular and biochemical mechanisms underlying chronic TBI pathology are poorly understood. Our data suggest that experimental brain injury causes abnormal changes within white matter in the corpus callosum, including demyelination, beginning at 1 month and continuing to evolve for at least 3 months after TBI. The breakdown of BBB during the acute period following TBI is associated with the infiltration of blood-borne cells including macrophages. 44,53,54 Moreover, intraparenchymal quiescent microglia have been reported to undergo acute morphological changes associated with activation following TBI, 55,56 and may contribute to the release of inflammatory cytokines. 57 Experimental studies have demonstrated that migration of activated microglia along the corticospinal tract occurs within 2 months after TBI. 58 Human studies using positron emission tomography (PET) imaging have demonstrated that activated microglia are present up to 17 years after TBI. 59 More recently, Johnson et al. (2013) have also reported that inflammation and white matter degeneration persist for years following a single TBI in humans. 17 Local cerebral inflammation is also believed to be a chronic and important component of neurodegenerative disease. 60
Previously published studies have demonstrated that microvascular abnormalities occur at acute and sub-chronic stages of TBI, and suggest their role in neurodegeneration. 61,62 Microvascular abnormalities associated with axonal changes have been reported up to 3 weeks after experimental TBI. 61 –64 Using ICAM-1 immunoreactivity, a marker of endothelial damage, we observed that TBI causes acute endothelial pathology spreading along the corpus callosum for at least 3 months after TBI. Elevated levels of ICAM-1 in acute and delayed TBI have been reported in both experimental and clinical human brain injury studies, and increased levels are known to be associated with BBB damage. 65 –68 Our staining with rat IgG showed microscopic IgG-positive puncta, which were localized in white matter beginning 1 week following TBI, and which persisted over time for at least 3 months. The sizes of punctate IgG immunostaining (diameter from 1 to 8 μm at initial stages) and Prussian blue staining (diameter from <10 to 25 μm at initial stages) are within the diameter ranges of microvessels in the rodent brain. 69 Importantly, the appearance of these punctate micro-tears in the BBB was associated with the appearance of focal microbleeds that also increased in number over time and expanded within the corpus callosum. These findings suggest that chronic endothelial and localized BBB damage may result in localized microvascular damage and the formation of focal microbleeds with the deposition of hemosiderin, which is neurotoxic to brain cells. 14 Moreover, delayed white matter damage and development of focal microbleeds were co-localized with cellular markers of reactive gliosis and regional inflammation. Taken together, these data suggest that the development of delayed, focal microbleeds resulting from delayed chronic microvascular damage after TBI can lead to toxic iron deposition, which could initiate abnormal changes in white matter such as gliosis, neuroinflammation, and demyelination of axons. Our findings confirm those from previous clinical studies that TBI microbleeds have a specific distribution in white matter, predominantly in the corpus callosum, 5,12 and that the number of microbleeds is associated with the severity of TBI. 13
Delayed focal BBB opening and early signs of localized inflammation preceded onset of microbleeds
We confirmed that BBB breakdown, previously reported in several studies 70,71 occurs acutely within the first 24 h following TBI. We also observed punctuate, secondary opening of the BBB in white matter at 1 week after injury, the same time that the first focal microbleeds were detected in the corpus callosum in the injured (ipsilateral) hemisphere. Regions of focal BBB breakdown expanded over time. By 3 months post-injury, BBB damage extended across the midline and into the contralateral hemisphere, including white matter.
Microscopic BBB damage preceded formation of microbleeds. Double immuno- and histochemical staining revealed that all focal microbleeds were surrounded by IgG-positive material, whereas not all IgG puncta are co-localized with microbleeds. These observations suggest that localized microvascular damage leads to the development of discrete microbleeds with subsequent accumulation of red blood cells and their breakdown products, potentially leading to toxic iron deposition. These micro-rents in the BBB co-localized with the presence of focal white matter microbleeds and upregulation of ICAM-1 immunoreactivity are suggestive of microscopic endothelial damage and transmigration of leucocytes. 65
Evolving white matter pathology surrounding microbleeds co-localized with inflammatory puncta and glial scarring
Our studies using Ibal1 immunohistochemistry demonstrated temporal changes in microglia activation and suggest that, in the chronic stages following TBI, the accumulation of reactive microglia is associated with focal neuronal and axonal damage. By 3 months post-injury, activated microglial cells were observed to migrate within the white matter.
Our findings both confirm previous reports
72
that CD68 positive microglia/macrophages accumulate acutely within injury site (e.g., ipsilateral hippocampus and cortex) and provide the first evidence that CD68-immunopositive cells expanded over time into white matter regions outside of the impact zone, including the corpus callosum (Fig. 6), as well as other regions even more distal from the site of impact, such as the cerebral peduncle and fimbria of the hippocampus (Supplementary Fig. S1) (see online supplementary material at
Reactive gliosis is another well-characterized hallmark of the histopathological response to human and experimental TBI. 73 –75 In the present study, we observed a significant increase in GFAP immunoreactivity in the acute post-injury period only at injured sites. However, the presence of significant clusters of GFAP-immunopositive cells and the appearance of localized glial scarring in the corpus callosum at 3 months following TBI indicate that sites of delayed white matter damage and decreased density of myelin fibers in areas surrounding focal microbleeds evolve into mature injury sites associated with classic glial profiles.
Potential pathological significance of delayed microbleeds in CTE and other neurodegenerative diseases
Microvascular changes and focal inflammation have been observed in neurological and neurodegenerative disorders and diseases including stroke and vascular dementia, 20,26,35 as well as CTE, AD, and PD, 22,28,25 suggesting that vascular damage BBB opening in microbleeds might be associated with chronic adverse neurological and cognitive outcomes. However, there are no systematic studies regarding the potential for chronic microvascular dysfunction following TBI to contribute to neurodegenerative diseases for which TBI is a risk factor.
A hypothetical injury cascade contributing to evolving and prolonged white matter degeneration after TBI
The precipitating events underlying delayed formation of punctuate the BBB opening remains unknown. It is possible that delayed axonal pathology, such as that observed during Wallerian degeneration, could contribute. Experimental studies (see review by Lingor et al. 76 ) have suggested that primary damage to cortical neurons induces a disruption of projecting axons that have not been primarily affected, which, in turn, show signs of degeneration after prolonged periods. This delayed axonal damage is accompanied by a local increase of Iba1-positive microglia, which is also thought to be involved in the progression of secondary damage. 76 Localized increases in barrier permeability produced by inflammation would allow molecules such as IgG and macrophages to additionally contribute to local inflammatory responses. 77 Further compromise of barrier permeability could ultimately lead to leakage of erythrocytes and subsequent elevation of potentially toxic iron and local deposition of its complexes with ferritin and hemosiderin. In combination, these vascularly mediated inflammatory events could synergize to promote evolving, substantial localized white matter damage. Compromised astrocytic morphology and function could additionally contribute to BBB dysfunction. This interpretation is consistent with an increasing awareness of the association of brain microvessels, astrocytes, and neurons to form functional “neurovascular” units and “gliovascular” units, 78 the integrity of which is critical to sustaining normal brain function.
Limitations and future studies
Although the current study provides a systematic IHC and morphological description of evolving changes in BBB disruption, inflammation, microbleeds, and delayed white matter damage, the data are subject to a number of interpretational limitations. CCI is not a pre-clinical model of mild TBI, and was originally developed as a model of moderate to severe TBI associated with mass lesions. 50 The study also did not employ repeated concussions, which are potentially important mediators of later neurodegeneration. In addition, this study did not present any data on the relationship of microvascular changes to accumulation of other proteins important to the diagnosis of neurodegeneration, such as tau. 28,79
Stereology is most frequently employed where quantitative three-dimensional information is required. The technique has been employed, for example, to calculate the total length of capillaries per unit volume of biological tissue. Although comprehensive stereological studies are not essential for the phenomenological observations of microvascular injury and the conclusions presented in this article, the technique can provide additional details regarding spreading secondary injury along the rostrocaudal axis. Therefore, we plan to incorporate stereological changes in future work. Future studies in humans employing noninvasive techniques, such as MRI and biomarkers, could also significantly enhance our understanding of the evolution of BBB dysfunction, inflammation, and neurodegenerative diseases.
Footnotes
Acknowledgments
The authors thank Marda Jorgensen, at the University of Florida, McKnight Brain Institute Cell and Tissue Analysis Care (MBI CTAC) and the University of Florida Molecular Pathology Core for assistance with histology. This study was supported in part by DoD Award number W81XWH-11-2-0002.
Author Disclosure Statement
Ronald L. Hayes owns stock, receives compensation from, and is an executive officer of Banyan Biomarkers, Inc. and, as such, may benefit financially as a result of the outcomes of this research or work reported in this publication. Olena Glushakova and Danny Johnson are employees of Banyan Biomarkers, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.