
Abstract
The potential pathophysiological role of circulating microparticles (MPs) has been recognized in various conditions, such as cardiovascular and thrombotic diseases. Traumatic brain injury (TBI) has a complex pathophysiology that involves coagulopathy and inflammation. We investigated endothelial-, platelet-, and leukocyte-derived microparticles (EMPs, PMPs, and LMPs, respectively) in 16 patients with severe isolated TBI. Arterial and cerebrovenous samples were taken repeatedly, during 1–72 h after injury. Subpopulations of MPs, exposing tissue factor (TF) and P-selectin, were also studied. MP counts in cerebrovenous samples, irrespective of cellular origin, were higher in TBI cases, compared to healthy controls (peak levels of EMPs were approximately 7 times higher, PMPs 1.4 times higher, and LMPs 2 times higher, respectively; p<0.001 for all). MP counts declined sharply from high levels shortly after the trauma toward slightly elevated levels 72 h later. EMPs and PMPs exposing TF, as well as PMPs exposing P-selectin, showed a transcranial gradient with higher concentration in cerebrovenous, compared to arterial, samples. In contrast, LMPs exposing TF were higher in arterial samples, suggesting accumulation of LMPs in the brain. We conclude that the pattern of circulating MPs is altered after TBI. PMPs exposing P-selectin and EMPs exposing TF seem to be generated in the injured brain, whereas LMPs exposing TF are accumulated. The pathophysiological significance of these changes in MP pattern in TBI should be further investigated. Including MPs exposing brain-specific antigens in the assessment of brain injury would give further information of origin and likely give additional information of the size of the injury, given that the MP phenotypes investigated in the present study are not brain-specific markers.
Introduction
T
Recently, circulating cellular microvesicles or microparticles (MPs) have attracted a vast interest within various areas of medicine. Cells may release MPs upon processes of activation and cell death. 5 Increased circulating levels of MPs have been observed in various conditions, such as arterial thrombosis, 6 cancer, 7 subarachnoid hemorrhage, 8 neuroinflammation, 9 and perhaps also in schizophrenia. 10 Elevated levels of MPs have also been reported in cerebrospinal fluid of patients with neuroinflammatory disease 11 or after TBI. 12
Accumulating data indicate that MPs play an active physio- and pathophysiological role in the triggering of processes such as coagulation and inflammation 13 and that they may boost vascular repair. 14 They may also have a role in cancer progression and mediate metastasis. 15
The aim of the present study was to investigate the presence and temporal profile of circulating MPs in TBI, as a pathophysiological marker of underlying events, not as a brain-specific marker. We determined their different phenotypes in order to get insights in the pathophysiology of severe isolated TBI, with a focus on microcirculatory disturbances, inflammation, and coagulopathy. Because MPs descend from their “parent cell,” they expose the same cell-specific antigens on their surface and thus have the potential to provide valuable information on the functional status of the original cell. Thus, we studied endothelial-derived, platelet-derived, and leukocyte-derived MPs (EMPs, PMPs, and LMPs, respectively) in plasma. We investigated MP exposure of P-selectin (CD62P), an adhesion molecule expressed on MPs from activated platelets, and tissue factor (TF), a central molecule in the activation of coagulation. The key role of TF in triggering coagulopathy in TBI patients was postulated already during the 1970s, based on several theoretical arguments and indirect observations, 16 but the role of “blood-borne” or “soluble TF” is still controversial. Current data are, however, in favor of TF on circulating cells or MPs as important mediators in hemostasis and thrombosis, 17 but the presence of circulating TF in human cerebrovenous blood has, according to our knowledge, never been reliably detected.
Methods
Study design
MPs were studied in plasma obtained from cerebrovenous and arterial blood samples of 16 patients with severe isolated TBI during the period 1–72 h after the injury. The study was conducted at the neurointensive care unit (NICU) of an urban, academic level 1 trauma center with a catchment area of approximately 2.5 million inhabitants (Karolinska University Hospital, Stockholm, Sweden). It is a prospective study of nonconsecutive included patients suffering from isolated severe TBIs (sTBIs). Patients were enrolled as a convenience sample based on availability of study physicians. The study was approved by the local ethics committee, and informed consent was obtained from the patient's next of kin.
Patient population
Patients subjected to blunt head trauma were included during the time period May 2009 to December 2010. The Glasgow Coma Scale (GCS) at admission was ≤8, and patients were sedated intravenously and mechanically ventilated. We used the Abbreviated Injury Score (AIS) to evaluate injuries. Patients with head AIS >3 and nonhead AIS ≤3 were considered to have severe isolated TBI. 18 All patients underwent a computed tomography scan of the brain and were included a mean of 6 h (range, 1–20) after trauma. Patients with known coagulation disorders, ongoing antithrombotic treatment with warfarin or platelet-inhibiting drugs, or patients suffering from multiple trauma were excluded.
Patient management
Patients were treated and monitored in accord with the local guidelines (based on Brain Trauma Foundation guidelines) at the NICU. 19 An arterial line in the radial artery was established at arrival to the emergency room (ER). A jugular venous catheter was inserted in the jugular bulb shortly after the patient arrived to the NICU, in order to monitor the jugulovenous oxygen saturation and collect cerebrovenous blood samples. In acute trauma situations, this procedure may be delayed by other therapeutic and/or diagnostic measures of higher priority. In our material, the first cerebrovenous blood samples were therefore taken 9 h (mean) after injury (range, 3–22).
Bleeding tendency was evaluated by a senior intensive care physician and a neurosurgeon, using a scoring system (no bleeding or moderate [+] and overt bleeding [++]). Hemostatic treatments were given indicatively (Table 1). Patients received thrombosis prophylaxis with enoxaparin 20 mg subcutaneously daily, starting from day 2 after injury. The investigators performing the present study were not involved in clinical decisions regarding the patients included in the study.
Main X-ray findings: SDH, subdural haematoma; SAH, subarachnoid haematoma; CC, cerebral contusions. Coagulative therapy: E, red blood cells concentrate; P, plasma; Plt, platelets concentrate; O, desmopressin; C, tranexamic acid; fib, fibrinogen concentrate.
GCS, Glasgow Coma Scale; M, male; F, female.
Measurements and blood sampling
Blood samples were taken repeatedly and simultaneously from the arterial line and from the jugular bulb line at approximately 6, 12, 24, 48, and 72 h after trauma, respectively. The initial samples obtained at admission were taken from the arterial line only.
All blood samples were drawn into a syringe containing citrate (pH 7.4; 1 part 0.129 M trisodium citrate+9 parts blood). The blood samples were centrifuged at 2500g in room temperature (RT) for 20 min immediately after sampling. The plasma obtained was divided into smaller tubes (0.4 mL) and kept frozen at –70° C until the analysis.
The MP analyses (see below) were performed in a two-step manner: First, “all MPs,” irrespective of cellular origin, were gated according to phosphatidylserine (PS) exposure and later phenotyped with monoclonal antibodies (mAbs) to detect cellular origin (EMPs, PMPs, and LMPs). Thereafter, we analyzed subpopulations of MPs, that is, those exposing TF, and in PMPs also the expression of the adhesion molecule P-selectin (CD62P).
Analysis of circulating microparticles
The previously frozen platelet-poor plasma was thawed and centrifuged at 2000g for 20 min at RT. The supernatant was recentrifuged at 13,000g for 2 min at RT. Twenty microliters of the supernatant were then incubated for 20 min in the dark with phalloidin/Alexa 660 (Invitrogen, Paisley, UK) lactadherin/fluorescein isothiocyanate (FITC; Haematologic Technologies, Essex Junction, VT), and mAbs to detect cell origin: anti-CD42a/phycoerythrin (PE; BD Biosciences, Franklin Lakes, NJ) to detect platelet origin; anti-CD144/allophycocyanin (AH Diagnostics, Stockholm, Sweden) to detect endothelial origin; and anti-CD45 (Beckman Coulter, Brea, CA) to detect leukocyte origin. P-selectin was detected with anti-CD62P/PE (BD Biosciences) and TF with anti-CD142/PE (clone HTF-1 IgG1κ; BD Biosciences). 20
MPs were measured by flow cytometry on a Beckman Gallios instrument (Beckman Coulter) and identified by size (<1.0 μm in size) and binding of fluorescent-labeled lactadherin and the labeled antibodies, as described above. The MP gate was determined using Megamix beads (BioCytex, Marseille, France), which is a mix of beads with diameters of 0.5, 0.9, and 3.0 μm, respectively. MPs were defined as particles less than 1.0 μm in size, negative to phalloidin (in order to exclude cell membrane fragments; see a previous work 21 ) and positive to lactadherin. Notably, lactadherin, instead of AnnexinV, was used to identify PS exposure on MPs because lactadherin is a better probe than Annexin V in detecting PS-expressing particles. 22 Conjugate isotype-matched immunoglobulin (IgG1-FITC, IgG1-PE, and IgG1-APC) with no reactivity against human antigens was used as a negative control to define the background noise of the cytometry analysis. The absolute number of MPs was calculated by means of the following formula: (MP counted×standard beads / L) / standard beads counted (FlowCount; Beckman Coulter).
Statistical analyses
Statistical analyses were performed using STATISTICA software (v.10; StatSoft, Inc., Tulsa, OK) and SPSS Statistics software (IBM Software, Armonk, NY). Data are presented as mean±standard deviations.
We analyzed the data for the presence of time trends between the sampling points (falling time trend) and for the difference between the arterial and cerebrovenous values (transcranial gradient). This was performed using the mixed-model analysis. This model accommodates missing data, assuming that data are missing at random.
Post-hoc tests were performed within the mixed-model analysis, using Sidak's correction for multiple comparisons.
Importantly, the results obtained with the mixed-model procedure were in agreement with those obtained with the traditional t-test procedure for repeated measurements of nonparametric data (Wilcoxon's test).
Data were considered to be normally distributed if –1<skewness<1. This was true for all MP data included in the mixed-models analysis, except at the 48-h measurement for CD42a/142 and CD42a/62P where skewness was marginally outside these limits (i.e., 1.08 and 1.05, respectively).
The GraphPad program (6.0; GraphPad Software, Inc., La Jolla, CA) was used for presentation of the results.
Results
Sixteen adult patients (mean age, 45 years; 4 women and 12 men) were studied. Reference values for MP counts in venous plasma were obtained from a control group of 15 healthy volunteers (7 men and 8 women; mean age, 44 years). Patient characteristics are shown in Table 1.
MPs of various cellular origins regardless of CD142 or CD62P exposure
Plasma levels of EMPs (CD144+ particles), PMPs (CD42a+ particles), and LMPs (CD45+ particles) in the first samples obtained were higher in TBI patients than in healthy controls (4.1±1.8×109 vs. 0.5±0.3×109/L, 15.3±9.8×109 vs. 9.9±2.6×109/L, and 4.9±1.4×109 vs. 2.1±0.6×109/L, respectively; p<0.001 for all; Fig. 1). The mean plasma counts of EMPs, PMPs, and LMPs when all sampling time points were included in the calculations were 3.6±1.2×109/L, 13.7±2.9×109/L, and 4.2±1.6×109/L, respectively. When we compared arterial and cerebrovenous samples, we found a significant transcranial gradient in total MPs irrespective of cellular origin (p<0.001; cerebrovenous vs. arterial samples) and in the subpopulation of CD42a+ MPs, especially in early samples (PMPs; p<0.05). Regarding CD144+ MPs (i.e., EMPs), there was only a tendency of a transcranial gradient (p<0.06). There was no transcranial gradient in LMPs, but a significant declining trend in LMPs in both cerebrovenous and arterial samples.
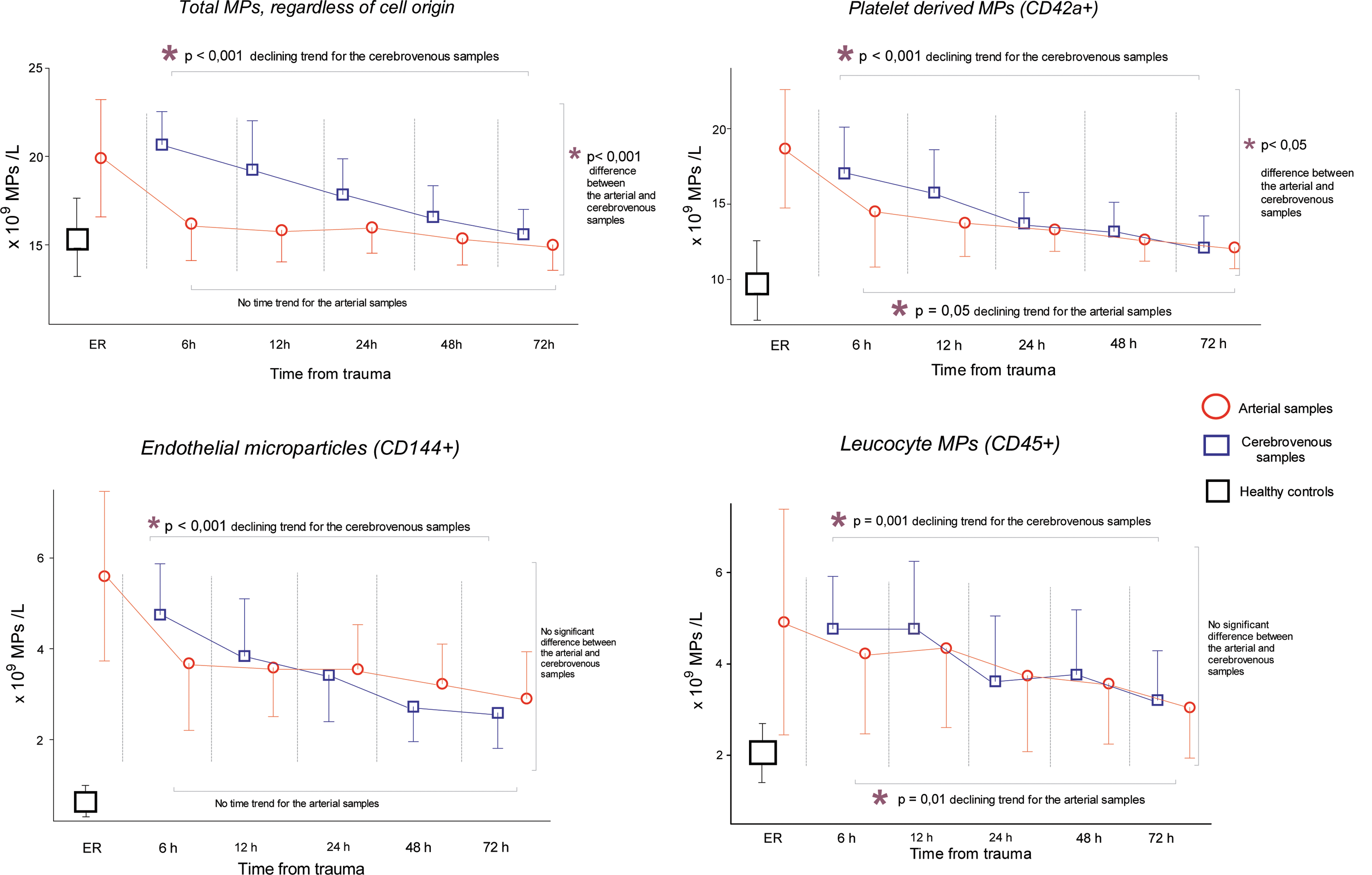
Microparticle (MP) counts in arterial (red) and cerebrovenous (blue) plasma samples in 16 patients with traumatic brain injury, collected in the emergency room (ER) and 6–72 h after trauma. Values are mean±standard deviations. The mixed-models procedure was used to accommodate for missing samples. The following numbers of samples were available at each time point: at 6 h, n=10; 12 h, n=11; 24 h, n=14; 48 and 72 h, n=13. p values refer to changes over time for arterial and cerebrovenous samples, respectively, as well as between arterial and cerebrovenous samples (mixed-model analysis; see Methods section for further details). MP counts for healthy controls (n=15) are also shown for comparison (left). Pair-wise post-hoc tests detected significant arteriovenous gradients at time points 6, 12, and 24 h for total MPs (p<0.001 for all three time points), at time point 6 h for endothelial MPs (p=0.001), and at time points 6 and 12 h for platelet-derived MPs (p=0.016 and p=0.013, respectively).
MPs exposing TF (CD142) or P-selectin (CD62P)
Data on TF+ and P-selectin+ MPs are shown in Figure 2. The declines in TF+ and P-selectin+ PMP counts over time were more pronounced, compared to the total PMP population. On average, TF+ and P-selectin+ PMPs decreased by approximately 4- to 7-fold during the 72-h measurement period, respectively, in comparison to an approximate 50% decrease in total MPs. Notably, the TF+ and P-selectin+ MPs constituted approximately 10–15% each of the total MP population.

Microparticles (MPs) of platelet, endothelial, and leukocyte origin exposing tissue factor (TF) or P-selectin (CD62P) in arterial (red) and cerebrovenous (blue) plasma samples in 16 patients with traumatic brain injury. Values are mean±standard deviation. Samples were collected in the emergency room (ER) and 6–72 h after trauma, as indicated (number of samples as in Fig. 1). PMPs, platelet-derived MPs; EMPs, endothelial MPs; LMPs, leukocyte MPs.
The temporal profile of TF+ EMPs was similar to the profile of all EMPs (see Figs. 1 and 2), but a significant transcranial gradient was only observed in TF+ EMPs (p<0.01 for cerebrovenous vs. arterial samples). Notably, TF+ LMPs showed a different—and, in fact, a reversed—pattern, compared to the other TF+ MP types. Thus, TF+ LMPs concentrations were lower in cerebrovenous samples than in arterial samples at all sampling points.
Discussion
The present study was designed to study MPs in TBI reflecting pathophysiological events in the microcirculation of areas within the injured brain. Several previous investigations have demonstrated elevated concentrations of circulating MPs in various pathological conditions in vascular medicine, 23 attributed to processes of microthrombosis, inflammation, and apoptosis. Our data show that sTBI also results in increased circulating MPs. The study design allowed comparison of MPs in cerebrovenous versus arterial samples at several time points after the injury. This made it possible to study the temporal profile of changes in the transcranial gradients of several populations of MP during the first 3 days after the injury.
The maximal levels of PMPs were approximately 1.7 times higher and the TF+EMPs more than 2 times higher in the TBI patients, compared to patients with prothrombotic antiphospholipid syndrome. 24 In addition, the maximal levels of circulating PMPs in the present study were approximately 35% lower than in patients with acute coronary syndrome, 6 but in the latter study, we defined PMPs as MPs exposing CD61 (i.e., as the platelet-specific antigen, instead of CD42a as done in the present study). Thus, the comparison of circulating PMP levels in patients with ACS versus patients with TBI should be interpreted with caution.
The greatest increase in circulating MPs was observed for EMPs, which were 7-fold higher than in healthy controls in the first samples collected. EMPs were defined as MPs expressing PS and CD144. Notably, CD144 (also known as VE-cadherin) is an endothelial-specific molecule located at the junction of endothelial cells (ECs) and of vital importance for EC contact. 25 Detecting elevated levels of MPs exposing this molecule together with PS is highly suggestive to reflect damage of ECs of brain vessels. We thus interpret our finding as a reflection of microvascular thrombosis and vessel injury in the penumbra area of the brain, similar to processes observed earlier in animal TBI models using in vivo fluorescent microscopy 26 and in postmortem electron microscopic examinations of baboon brain preparations. 27
As shown in Figure 1, transcranial gradients in MP counts were demonstrated in samples collected early after TBI. In all MPs, a gradient was detectable until 24 h after injury, whereas in EMP and PMP subpopulations, gradients were only detectable up to 6 and 12 h after trauma, respectively. Thus, the pathophysiological events, as detected by circulating MPs, unfold rather rapidly after trauma. It is conceivable that we only observe “the tail” of TBI-induced increments in MP formation.
Taking into consideration the methodological difficulties collecting cerebrovenous blood in TBI patients early after the trauma, and the potential significance of MPs for further understanding TBI pathophysiology, studies in animal TBI models would be likely to provide additional important information on these issues, including mechanisms behind MP changes in relation to pathophysiological events. In addition, animal experiments could also be of help in elucidating relationships between brain trauma severity and MP patterns and MP plasma levels. Given that a larger injury would lead to more-pronounced cell death and a stronger inflammatory response, it is likely that this would show as increased levels of circulating MPs from the cell types involved in the injury, as well as from those cells that would participate in the inflammatory response. Notably, we have previously found that circulating MPs exposing damage-associated molecular pattern molecules, such as high-mobility group protein B1, are significantly elevated in response to strong inflammatory stimuli. 28 The present study is unfortunately too small to investigate associations between TBI severity and MP patterns and plasma levels.
In the present study, 48–72 h after injury, we noted that MPs were still markedly elevated, compared to the samples from healthy controls, testifying to the “diseased state” of TBI patients. These elevations may also include complications to TBI, such as infection or systemic inflammatory response syndrome (SIRS).
Comparisons of MP subpopulations to the “general” MP population revealed that there was a significant transcranial gradient in TF+ EMPs, but not in the total circulating EMP population. The brain is very rich in TF, and the blood–brain barrier is damaged in TBI. Our data suggest that MPs released from ECs carry TF out in the circulation. It is possible that TF exposed in this way may influence coagulation in the microvasculature of the brain, but other effects, such as proinflammatory actions of TF, may also be exerted. Our study shows that TF can be detected in the cerebrovenous blood after TBI.
Also, the transcranial gradient of P-selectin+ PMPs was obvious in both early and late samples. Platelet activation is characterized by formation of MPs exposing P-selectin. 6,29 Thus, our finding indicates activation of platelets within the damaged brain, possibly leading to a further increase in the risk of microthrombotic complications.
Regarding LMPs, we found a reversed transcranial gradient, that is, the concentration of TF+ LMPs was lower in cerebrovenous, compared to arterial, samples. The significance of this finding is unclear, but we hypothesize that leukocytes and LMPs accumulate in the damaged brain. Animal experiments show infiltration of polymorphonuclear cells in penumbra areas early after TBI, whereas infiltration of mononuclear cells is observed at days 2–14 after the injury. 30 Recruitment of LMPs to the penumbra area may be involved in initiating/propagation of microvascular thrombotic events and contribute to inflammation within the damaged brain, as reported previously by us 31 and others. 32 The MP pattern observed in plasma, with signs of brain accumulation of LMPs, in combination with formation of PMPs exposing CD62P and TF over the injured brain, could reflect—and be a part of—the cellular response to inflammation caused by the brain injury. It would be of interest to characterize this response further by measuring MPs from other inflammatory cells, such as lymphocytes and/or MPs exposing cytokines 28 or proteins of the complement system.
We consider detection of transcranial gradients of MPs in humans suffering TBI as the novel and most interesting finding in the present study.
Limitations of the study
The patients received procoagulative treatment during the study period, which is a limitation of the study, but unavoidable for ethical reasons. In most cases, treatment was given after the initial samples were collected, that is, the first samples obtained in each case should have no traces of pharmacological intervention.
Our findings demonstrated statistically significant alterations in MPs; however, the small sample size is a very important limitation of this study. Larger studies are needed to confirm our results. Future studies will also have to include non-trauma-related conditions as control groups in order to compare values in these different conditions. The clinical utility of MPs in TBI is uncertain and will require further investigation. Clinical applications of MP analyses were recently proposed, such as monitoring of glioblastoma therapy. 33 The present study does not include detection of brain-specific antigens on the MPs and is not designed for monitoring of brain-specific biomarkers. However, it is theoretically possible that certain MPs carrying brain-specific antigens can be used in future as biomarkers of TBI, but this will require additional studies. At present, the value of MPs is mainly their contribution to increased understanding of the pathophysiology of TBI.
Conclusions
Early after TBI there is an increased formation of circulating PMPs, LMPs, and EMPs, which, however, are not brain-specific markers.
The transcranial gradients of these endothelial MPs suggest, however, that their origin is the damaged brain. In TBI patients, the presence of elevated TF in cerebrovenous blood has been demonstrated. Over time, the level of MPs declines in cerebrovenous and arterial blood. Levels of MPs remain, however, elevated, compared to healthy controls. This may reflect the diseased state of TBI patients, including various complications such as infection or SIRS. Levels of LMPs exposing TF were higher in arterial than in cerebrovenous samples, indicating accumulation of LMPs in the brain.
Footnotes
Acknowledgments
The authors thank Dr. Margareta Blombäck for being such a powerful source of energy for our team. The authors thank the nursing personnel at NICU for all the assistance and everlasting helpfulness during the study. The authors thank the medical statistics expert, Eva Hagel, Karolinska Institute, for professionally performed analyses of our data. The authors also thank the Carnegie Foundation, the Capio Foundation, and Karolinska Institute fund 245 for financial support.
Author Disclosure Statement
No competing financial interests exist.