
Abstract
Although the mechanisms that contribute to the development of traumatic brain injury (TBI)-related deficits are not fully understood, it has been proposed that altered energy utilization may be a contributing factor. The tuberous sclerosis complex, a heterodimer composed of hamartin/Tsc-1 and tuberin/Tsc-2, is a critical regulatory node that integrates nutritional and growth signals to govern energy using processes by regulating the activity of mechanistic Target of Rapamycin complex 1 (mTORC1). mTORC1 activation results in enhanced protein synthesis, an energy consuming process. We show that mice that have a heterozygous deletion of Tsc2 exhibit elevated basal mTORC1 activity in the cortex and the hippocampus while still exhibiting normal motor and neurocognitive functions. In addition, a mild closed head injury (mCHI) that did not activate mTORC1 in wild-type mice resulted in a further increase in mTORC1 activity in Tsc2+/KO mice above the level of activity observed in uninjured Tsc2+/KO mice. This enhanced level of increased mTORC1 activity was associated with worsened cognitive function as assessed using the Morris water maze and context discrimination tasks. These results suggest that there is a threshold of increased mTORC1 activity after a TBI that is detrimental to neurobehavioral performance, and interventions to inhibit excessive mTORC1 activation may be beneficial to neurocognitive outcome.
Introduction
T
One of the major cellular pathways for sensing energy status involves signaling between the AMP-activated protein kinase (AMPK) and tuberous sclerosis complex (TSC, composed of Tsc1 and Tsc2) cascades. 13 When activated by signals such as a lowered adenosine triphosphate/adenosine monophosphate (ATP/AMP) ratio, AMPK enhances catabolic processes that produce ATP, while simultaneously inhibiting anabolic processes that consume energy (e.g., protein synthesis). The mechanistic Target of Rapamycin Complex 1 (mTORC1), a master regulator of protein translation, is negatively regulated by TSC, with mutations of either Tsc1 or Tsc2 resulting in enhanced mTORC1 activity. mTORC1 directly regulates protein synthesis via phosphorylation of its downstream targets S6 kinase (S6K) and eukaryotic translation initiation factor 4E-binding protein (4E-BP), resulting in increased synthesis. 14
Although mTORC1 activity has been reported to be increased after moderate-to-severe TBI, blocking its activity with the selective inhibitor rapamycin has yielded mixed results on behavioral outcomes. 15 –17 Given the conflicting literature, additional investigation is needed to more fully elucidate the role of mTORC1 in TBI pathobiology.
We used Tsc2 mutant mice to examine if altered regulation of mTORC1 after a TBI can influence cognitive outcome. To examine the consequences of activation of mTORC1, we used heterozygous Tsc2+/KO knockout mice. Deletion or inactivation of one Tsc2 allele is known to result in dysregulated mTORC1 by removing one of its negative regulators, while homozygous inactivation is embryonic lethal. 18,19 To help isolate the influence of altered mTORC1 activity after injury, we used a mild closed head injury (mCHI) paradigm that did not activate mTORC1 signaling in wild-type (WT) animals.
We observed that uninjured Tsc2+/KO mice exhibit increased mTORC1 activity in both cortical (CTX) and hippocampal (HIPP) protein extracts compared with WT siblings, as assessed using the surrogate marker phospho-S6. A single mCHI further enhanced acute mTORC1 activity in Tcs2+/KO mice, while WT mice showed no significant difference between uninjured and mCHI groups. When tested for cognitive function beginning 7 days post-injury, the Tsc2+/KO mice performed poorly compared with WT littermates receiving a mCHI. Our results suggest that increased mTORC1 activity has a deleterious effect on cognitive outcome after brain injury, suggesting that inhibiting excessive mTORC1 activity after a TBI may be beneficial.
Methods
Antibodies
The following antibodies were purchased from Cell Signaling Technology (Danvers, MA): phosphorylated ribosomal protein S6 (Serine240/244), total ribosomal protein S6, total Tsc2, and phosphorylated Tsc2 (Threonine1462). Antibodies directed against β-actin were purchased from Sigma-Aldrich (St. Louis, MO).
Animals
All protocols involving the use of animals were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee. Tsc2 +/KO were generated by crossing Tsc2 flox/flox mice to a mouse strain containing the Cre recombinase gene controlled by a Cytomegalovirus promoter. 19 The progeny (Tsc2 +/flox;Cre) undergo Cre recombinase-mediated deletion of Tsc2 exons 2–4 to produce heterozygous Tsc2+/KO;Cre mice. These progeny were intercrossed to generate the Tsc2+/KO and Tsc2 +/+ mice (homozygous Tsc2 KO/KO mice are embryonic lethal) used for these experiments (Fig. 1). A mixture of males and females (balanced across groups) were used in these studies.

Tsc2 genotyping scheme. (
Separate groups of animals were used to generate the western blot (a total of 32 mice) and behavioral (a total of 47 mice) data. The histology data (a total of nine animals) were generated from animals that had undergone behavioral assessments. For the western blot data in Figure 2a–c, uninjured Tsc2+/+ WT, and Tsc2+/KO group sizes were n=5 each, while in Figure 3, group sizes were n=6/condition for sham and injured Tsc2+/+ animals, and n=5/condition for sham and injured Tsc2+/KO . The behavioral data shown in Figure 2d–g were generated using uninjured Tsc2+/+ (n=9) and Tsc2+/KO (n=10) animals. The data from uninjured sham Tsc2+/+ (n=8), mCHI Tsc2+/+ (n=10), and mCHI Tsc2+/KO (n=10) animals were presented in Figures 4, 5, and 6.

Uninjured Tsc2+/KO
mice exhibit reduced tuberin expression, elevated phospho-S6 levels, and normal spatial learning. Brain tissues from 2 month-old Tsc2+/KO
mice (n=5) and wild-type control littermates (n=5) were collected for western blot analysis. (

Phosphorylated S6 levels are elevated in Tsc2+/KO
mouse brains 30 min after closed head injury (CHI). Tsc2+/+
(n=6) and Tsc2+/KO
(n=5) mice received a single mild CHI (mCHI), and brain tissues were dissected 30 min after injury. (

Acute neurological and motor performance assessments of mCHI Tsc2+/KO
and Tsc2+/+
mice. Cohorts of Tsc2+/+
(n=10) and Tsc2+/KO
(n=10) animals received a mild closed head injury (mCHI), and acute neurological assessments were performed beginning immediately after discontinuation of anesthesia. A representative group of sham animals (n=8) is shown for reference. There was no significant difference in either (

Tsc2+/KO
mice have impaired memory performance in the Morris water maze (MWM) task 7 days after mCHI. The performance of a sham group (n=8) is shown for reference on each panel. (
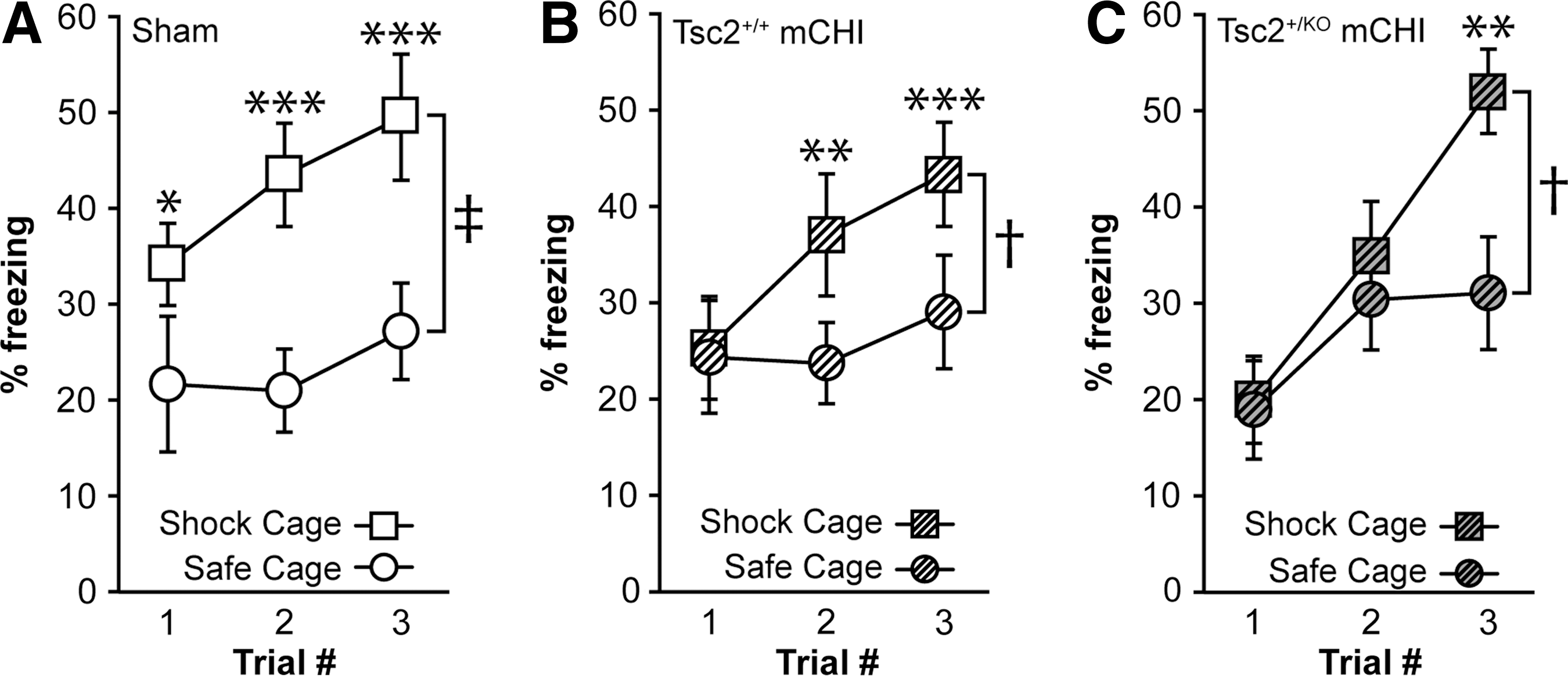
Tsc2+/KO mice have impaired context discrimination after mild closed head injury (mCHI). (
mCHI
A single mCHI was delivered to mice essentially as described previously. 20 Heterozygous knockout (Tsc2 +/KO) and WT (Tsc2 +/+) littermates were deeply anesthetized with 5% isofluorane in a 1:1 O2/air mixture, then maintained with 2.5% isofluorane in a 1:1 O2/air mixture via face mask. A midline incision was made to expose the skull, and anesthesia was discontinued for the remainder of the surgical manipulations. Immediately after discontinuation, mice were placed on a foam pad that kept their head level with the body. A pneumatically driven controlled CTX impact (CCI) device with a metal impactor tip (5 mm diameter) was used to deliver a single impact to the skull midway between lambda and bregma over the sagittal suture. The impactor was driven at 4 m/sec to a depth of 1 mm below the plane of the skull. Immediately after impact, the length of apnea was monitored. When spontaneous breathing returned to normal, the scalp was closed with sterile surgical staples. Sham animals received the same surgical treatment but did not receive an impact.
Western blots
Mice underwent the mCHI or sham surgery and their brains were dissected at 30 min post-injury. The brains were submerged in ice-cold artificial cerebrospinal fluid containing phosphatase inhibitors (2 mM NaF, 2 mM Na2MoO4, and 1 mM Na3VO4), and HIPP and CTX tissues were quickly dissected and snap frozen on dry ice. To prepare total protein extracts, tissues were thawed and disrupted in homogenization buffer (10 mM Tris, pH 7.4, 1 mM ethylene glycol tetraacetic acid, 1 mM ethylenediaminetetraaceticacid, 0.5 dithiothreitol, 0.1 μM okadaic acid and 1 mM Na3VO4, 1 mM phenylmethanesulfonylfluoride, and 10 μg/mL leupeptin). An equal volume of 2X Laemmli buffer was added, and the protein concentration was determined by NanoOrange Protein Quantification Assay (Invitrogen, Carlsbad, CA) using bovine serum albumin (BSA) as the standard.
Samples were resolved on Tris-glycine SDS-PAGE gels, and the proteins transferred to Immobilon-P membranes (Millipore, Bedford, MA). Membranes were blocked for 30 min in Tris-buffered saline (TBS) containing 5% BSA. Blots were incubated with primary antibodies overnight at 4°C in TBS/BSA, washed, and incubated for 1 h at room temperature with alkaline phosphatase-conjugated secondary antibodies (Vector Laboratories, Burlingame CA). Immunoreactivity was detected using the CDP-Star chemiluminescence system (New England Biolabs, Beverly, MA) and the blots exposed to Kodak XAR5 film (Rochester, NY). The detected immunoreactive band intensities were normalized against the signal obtained for β-actin using ImageJ (National Institutes of Health). Western blot data were compiled from at least five independent animals.
Assessment of acute neurological functions
Post-injury apnea, the duration of suppression of tail pinch, and righting response were monitored to assess injury severity across groups. After anesthesia was discontinued, the animal was quickly positioned underneath the impactor and the mCHI was delivered. A timer was started on impact, and the time to recovery of spontaneous breathing was recorded by an independent observer. Immediately after breathing resumed, the scalp was closed with sterile surgical staples and a topical ointment (Tritop, which contains neomycin sulfate, isoflupredone acetate, and tetracaine hydrochloride) was applied while the observer monitored recovery of the tail pinch response by applying firm pressure to the distal tip of the tail at regular intervals. Concurrently, the righting response was monitored by placing the animal on its back and recording the time needed to right itself on three consecutive trials.
Beam balance task
To assess vestibulomotor and motor functions of mCHI Tsc2+/KO heterozygous and Tsc2+/+ wild-type mice, the beam balance task was used. 21,22 On the day after injury, animals were placed at the farthest end of a metal beam (0.5 cm wide), and the time animals remained on the beam was recorded. A foam pad was placed beneath the animals to prevent injury if they fell. Animals were given three trials of 1 min each on the beam, and the trials were averaged to give a group mean for each day.
Foot fault task
Vestibulomotor function of Tsc2+/KO and WT injured mice was tested using the foot fault task. 21,22 For this task, a mouse was placed on a wire grid with an opening size 1×1 cm and allowed to walk freely until the experimenter counted 50 steps. Foot faults were recorded when either of the front paws passed below the plane of observation through a gap in the grid. Each animal was given three trials per day, and the results averaged to give a group mean, until all groups performed as well as sham controls.
Abbreviated water maze
Spatial learning and memory were assessed using a 1-d abbreviated version of the hidden platform water maze task as described previously. 23,24 On day 7 post-injury, mice were given 10 consecutive training trials with an intertrial interval of 4 min. Animals were allowed to search for a hidden escape platform for a period of 60 sec, and the time to find the hidden platform was recorded. Forty-eight hours later, animals were tested in a probe trial in which the escape platform was removed from the tank, and the mice were allowed to search for a period of 60 sec. Movement within the water maze was monitored using a video camera linked to tracking software (Ethovision, Noldus Information Technology, Leesburg, VA).
Context discrimination
Fear context discrimination was performed on days 14–16 post-injury by pre-exposing animals for 10 min to two similar, but not identical, contexts without shock. These contexts shared certain features (e.g., background noise, horizontal grid floor) but differed in others (differently spaced grids, distal cues, floor color, shape, and scent). Animals were given two trials a day, one in each of the two chambers. Half of the animals from each group were trained in the morning in the shock chamber where they stayed for a total of 3 min, with a 2 sec, 0.75 mA shock given at 178 sec. In the afternoon, they were placed in the safe chamber for a total of 3 min, and no shock was given. The other half of each group received the same treatment with reversed order and thus experienced a foot shock in the shock chamber in the afternoon.
On the following 2 days, an animal's ability to discriminate between the two contexts was assessed by monitoring freezing behavior in 2 sec intervals for 178 sec in each chamber, with the same presentation order. The animals received a shock during the last 2 sec in the shock chamber, and no shock in the safe chamber. Percent freezing in each chamber was calculated and analyzed to determine if animals were able to discriminate between the shock and no-shock (or safe) contexts. 25
Immunohistochemistry
At the conclusion of behavior testing, mice were killed with an overdose of sodium pentobarbital followed by transcardial perfusion with ice-cold phosphate-buffered-saline (PBS) followed by buffered 4% paraformaldehyde. Brains were removed and stored in buffered 4% paraformaldehyde for an additional 24 h. Brains were then cryoprotected in 30% sucrose and 40 μm sections cut on a cryostat in the coronal plane. Sections were incubated in primary antibody solutions (0.1–0.5 μg/mL antibody, 2.5% normal goat serum in PBS) overnight at room temperature. After extensive washing, sections were incubated for 1 h in PBS containing species-specific secondary antibodies linked to AlexaFluor dyes (Alexa-488 or Alexa-568; Invitrogen). Sections were mounted onto glass slides and coverslipped with Fluoromount-G to inhibit fading. Slides were examined using an upright microscope with epifluorescence capabilities, and images were captured with a MagnaFire camera using settings that remained constant across groups.
Statistics
In all experiments, data collected from the same animal with one or more factors were subjected to repeated measures (RM) analysis of variance (ANOVA). Data comparing only one factor between groups, such as probe trial data analysis and western blot data, were subjected to a two-tailed Student's t-test for unpaired (behavioral data) or paired variables (western blot data and contextual discrimination data). Data comparing more than two groups were subjected to one-way ANOVA. Data were considered significant at p<0.05.
Results
Tsc2+/KO heterozygous mice
To generate Tsc2+/KO mice, a floxed Tsc2 allele (Tsc2flox/flox ) was made using a targeting construct that contained loxP sites inserted in introns 1 and 4 of the Tsc2 gene (Fig. 1). 19 Homozygous Tsc2flox/flox mice were crossed with mice carrying the Cre recombinase gene under the control of a cytomegalovirus promoter. The F1 offspring of this cross produced heterozygous Tsc2+/flox;Cre mice, which subsequently underwent Cre-mediated deletion of the region of the Tsc2 gene between the two loxP sites in all tissues including germ cells. 19 The F1 generation were interbred to produce viable heterozygous Tsc2+/KO and WT Tsc2+/+ siblings, while the homozygous Tsc2KO/KO embryos died in utero.
Genotyping was performed on genomic DNA extracted from ear punch tissue from all surviving mice. Amplification reactions with primers 2F/1R produced a 390 bp PCR product from the WT Tsc2 allele, while the floxed allele produced a 434 bp product because of the introduction of the loxP sequences. After Cre-mediated deletion of Tsc2 exons 2–4, a 590 bp PCR product was generated by primers 1F/1R (Fig. 1), indicating loss of the floxed allele.
Tsc2+/KO mice exhibit decreased tuberin levels and increased S6 phosphorylation
Tsc2 (tuberin) binds to Tsc1 (hamartin) to form the TSC complex that functions as a negative regulator of mTORC1 activity. Thus, loss of a Tsc2 allele is anticipated to result in lower tuberin expression, less mTORC1 inhibition, and increased phosphorylation of mTORC1 substrates. To measure the levels of tuberin in the brains of Tsc2+/KO mice, CTX and HIPP tissue extracts were prepared for western blot analysis. Figure 2A shows representative western blots and summary results indicating an approximately 30% reduction of tuberin immunoreactivity in both CTX and HIPP protein extracts compared with extracts prepared from WT littermates. Total and phospho-S6 immunoreactivities were measured using the same CTX and HIPP protein extracts. Figure 2B shows there was no significant difference in total S6 immunoreactivity between Tsc2+/+ and Tsc2+/KO animals, while phospho-S6 immunoreactivity (Fig. 2C) was significantly elevated in Tsc2+/KO animals compared with WT sibling mice.
We next trained and tested uninjured Tsc+/KO and Tsc2+/+ mice in an abbreviated version of the Morris water maze task to determine if the loss of a Tsc2 allele affected their spatial learning ability. Mice were given 10 consecutive trials with a 4 min intertrial interval, and their performance averaged across every two trials. As shown in Figure 2D, there was no significant difference in the acquisition curves between the Tsc2+/KO and Tsc2+/+ groups. When tested for long-term memory 48 h after training, there was no significant difference in latency to the original platform location or the number of crossings through the platform area (Fig. 2D, E). Both groups performed equally well when tested using a visual platform and showed similar swimming velocity during the probe trial (Fig. 2F, G).
To further confirm that the loss of a Tsc2 allele does not impact HIPP function in uninjured animals, Tsc2+/KO and WT siblings were trained in a fear context discrimination paradigm. Previous studies have shown that performance in this task is dependent on the integrity of the HIPP, especially the dentate gyrus. 26,27 Animals were trained to differentiate between two similar, but distinct, contexts as described in the Methods section. In one cage, animals received a mild foot shock (the “shock” cage), while no foot shock was administered in the “safe” cage. Both Tsc2+/KO and WT siblings froze for a significantly longer period in the shock cage than in the safe cage indicating the heterozygous loss of a Tsc2 allele did not significantly impair performance in this task (data not shown).
mTORC1 activity is further elevated in TSC2+/KO mice after a mCHI
To test whether brain injury activates mTORC1, heterozygous Tsc2+/KO and WT littermate mice received a mCHI. HIPP and CTX tissues were collected 30 min after injury for western blot analysis. The levels of total S6 protein were not significantly different between the sham and mCHI animals in either the WT or Tsc2+/KO genotypes (Fig. 3A). When the phosphorylation of S6 was examined, no significant difference was observed between uninjured and injured Tsc2+/+ mice. Tsc2+/KO mice, however, exhibited significantly higher levels of phospho-S6 after a mCHI when compared with sham operated Tsc2+/KO mice (Fig. 3B). Note that the western blot results were normalized to sham operated controls. Therefore, this significant increase in phospho-S6 levels in mCHI Tsc2+/KO mice is in addition to the increased basal phospho-S6 levels observed in uninjured Tsc2+/KO mice (Fig. 2C; ∼60% in CTX, ∼25% in HIPP).
mCHI worsens cognitive functions of Tsc2+/KO mice
Additional cohorts of Tsc2+/KO and Tsc2+/+ siblings received a mCHI for cognitive function using the Morris water maze and context discrimination tasks. Immediately after the injury, animals were tested for apnea, tail pinch, and righting response times. For reference, the performance of representative uninjured animals is also shown. As shown in Figure 4A–C, there were no significant differences between the two injured groups in any of these acute neurological measurements.
Motor and vestibulomotor functions were tested on days 1–3 post-injury using the paw placement and beam balance tasks, respectively. Figure 4D shows that the Tsc2+/KO and Tsc2+/+ mice did not significantly differ in their ability to perform the foot fault task, nor was there an apparent difference in the number of foot faults in injured versus sham animals. It should be noted that these mice were not pre-trained in this task, likely giving rise to the initial deficit and subsequent improved performance over time that was observed in the sham controls. Vestibulomotor function was impaired in the mCHI mice compared with shams, but no significant difference in either the magnitude of dysfunction or the rate of recovery was observed between the Tsc2+/+ and Tsc2+/KO mCHI groups (Fig. 4E).
Cognitive assessments were performed as shown in the timeline depicted in Figure 5A. One week after mCHI, the animals were trained to find a hidden platform in an abbreviated version of the Morris water maze task. Figure 5B shows that the acquisition curves for both injured groups were not significantly different, and that they learned the platform location to a similar level. To test for memory of the platform location, animals were given a probe test 48 h after the last training trial. Figures 5B and C show that Tsc2+/KO injured mice took significantly longer to cross the previous platform location than Tsc2+/+ injured mice (Tsc2+/+ 29.1±8.1 sec; Tsc2+/KO 50.31±5.2 sec, p=0.046), and crossed the platform area fewer times (Tsc2+/+ 1.4±0.4 crossings; Tsc2+/KO 0.33±0.1, p=0.039). These differences in memory performance were not attributable to problems with either visual acuity or swimming speed (Fig. 5D, E).
We next examined the performance of these animals in a context discrimination task. Uninjured sham animals were able to discriminate between the safe and shock contexts after a single training trial, as indicated by the significant difference in their freezing behavior in the respective environments (Fig. 6A). Mild CHI-injured Tsc2+/+ mice acquired the task slightly slower, necessitating an additional exposure (Fig. 6B), while injured Tsc2+/KO mice needed two additional trials to be able to discriminate the different contexts (Fig. 6C). These results show that after a mCHI, Tsc2+/KO heterozygous mice underperformed compared with injured WT siblings and needed additional training exposures to learn to distinguish between the shock and safe contexts.
Histological examination
Animals were euthanized 6 weeks post-injury and brains extracted for immunohistochemical examination of neuronal loss, axonal damage, and inflammation. Immunohistochemical staining for the neuronal marker NeuN did not indicate any visible cell loss in the HIPP (or CTX, not shown) cell layers between sham, WT injured, and Tsc2+/KO injured mice (Fig. 7A–C). Glial fibrillary acidic protein (GFAP) immunoreactivity, however, appeared to be enhanced in both WT injured animals (Fig. 7E) and Tsc2+/KO injured mice (Fig. 7F) compared with sham controls (Fig. 7D). Further, activated microglia (as indicated by enhanced Iba-1 immunoreactivity) were observed in the corpus callosum of both injured groups (Fig. 7G–I). Axonal damage, examined using silver staining, was detected in the corpus callosum of both WT injured and Tsc2+/KO injured mice (Fig. 7J–L). These results suggest that heterozygous loss of a Tsc2 allele does not affect the inflammatory response initiated after mCHI. 20

Heterozygous loss of Tsc2 does not exacerbate neuronal loss, neuroinflammation, or axonal injury after mild closed head injury (mCHI).
Discussion
We investigated whether altering regulation of the mTORC1 pathway can influence the pathophysiology of a mCHI using a Tsc2+/KO heterozygous knockout mouse line. Our results show that uninjured Tsc2+/KO heterozygous mice have significantly increased phospho-S6 levels indicative of impaired mTORC1 regulation. When Tsc2+/KO mice received a mCHI, S6 phosphorylation in the brain was further enhanced compared with Tsc2+/+ injured mice. Tsc2+/KO injured mice performed significantly worse in the Morris water maze and contextual fear discrimination tasks when compared with injured WT littermates. Taken together, the results from this study suggest that enhanced mTORC1 activity after brain injury may be detrimental to neurobehavioral outcome.
A few previous studies have examined activation of mTORC1 signaling and its contribution to outcome after moderate-to-severe TBI. Using a moderate level of fluid-percussion injury (FPI), Chen and associates 16 observed significant increases in S6 phosphorylation in both CTX and HIPP tissue extracts at 30 min that persisted for up to 24 h post-FPI. Likewise, moderate CCI injury to mice resulted in significantly increased S6 phosphorylation in both CTX and HIPP tissue extracts that lasted for at least 24 h. 15 Park and colleagues, 15 however, found that intraventricular administration of the mTORC1 inhibitor rapamycin alone to moderately injured animals did not result in any improvement in spatial learning and memory, while coadministration of an Akt inhibitor with rapamycin was beneficial. In contrast, Erlich and coworkers 17 reported that intraperitoneal injection of rapamycin 4 h after injury using a weight drop model that caused a focal contusion improved motor function using a composite Neurological Severity Score. These results indicate that the role of enhanced mTORC1 activity on TBI-associated neurocognitive deficits is not yet fully understood.
To examine if activation of mTORC1 influences cognitive dysfunction after a TBI, we used a Tsc2+/KO heterozygous mouse line. 19 In response to energy availability and growth factor signaling, the GTPase-activating protein activity of Tsc1-Tsc2 is inhibited, which releases its repression on the G-protein Rheb (Ras homologue enriched in brain), leading to mTORC1 activation. 14 While loss of both copies of Tsc2 is embryonic lethal, loss of a single gene copy impairs the capacity of the Tsc1-Tsc2 complex to regulate mTORC1, leading to increased activity.
Consistent with this, we observed a small but significant increase in the phosphorylation level of ribosomal S6 protein, a direct substrate of mTORC1, in CTX and HIPP extracts prepared from uninjured Tsc2+/KO mice. Unlike the deficits reported by Ehninger and colleagues, 28 however, we did not detect significant learning and memory dysfunction in Tsc2+/KO as assessed by performance in abbreviated water maze and contextual fear tasks. While the reason for this discrepancy is not known, rats (e.g., Eker rats) and mice with dominant negative Tsc2 mutations have been reported to have either mild or no learning and memory deficits, suggesting that the degree of mTORC1 activation may be dictating factor in the expression of deficits. 29,30
We found that delivery of a mCHI to Tsc2+/KO mice further increased mTORC1 activity 30 min post-injury in the brain, but had no significant effect on mTORC1 activity in injured Tsc2+/+ siblings. The 30 min post-injury time point was based on previous studies that observed significant activation of mTORC1 signaling after moderate-to-severe TBI. It is possible that a transient change occurred in injured Tsc2+/+ mTORC1 activity at an earlier time, or that this mild CHI is insufficient to activate mTORC1 signaling when its regulatory pathway is fully intact. However, when mTORC1 activity is enhanced in the context of mild brain injury, it appears to be able to exacerbate mCHI-induced HIPP dysfunction, as indicated by worsened performance in both the abbreviated Morris water maze and context discrimination tasks.
This is consistent with recently published findings that demonstrate rapamycin treatment, which would inhibit mTORC1 activity, is beneficial in a variety of injury models. 31 –33 Treatment of injured animals with intracerebroventricular rapamycin alone, however, was insufficient to provide significant outcome benefits. 15 Although the reason for this lack of effect is unclear, Park and colleagues 15 observed that the combination of rapamycin with an Akt inhibitor was beneficial. These results suggest that when mTORC1 activity is enhanced as a result of brain injury, including mild TBI (mTBI), it likely contributes to poor cognitive outcome. Inhibiting pathologically activated mTORC1 by administration of drugs such as rapamycin may be able to improve learning and memory function after a TBI.
As loss of Tsc2 has been linked to an inability of neurons to control cellular stress and enhanced neuronal loss, 34,35 it is possible that the exacerbated learning and memory impairments we observed after mCHI could have arisen from increased neuronal loss in the Tsc2+/KO mice. Examination of NeuN immunostaining, however, did not reveal any obvious differences between sham, Tsc2+/+ injured, and Tsc2+/KO injured mice. This finding is consistent with previous reports that mCHI does not cause overt HIPP cell loss. 7,20,36
Recent reports have demonstrated acutely impaired axonal transport and delayed axonal degeneration in the corpus callosum after CHI. 37 Similarly, we observed silver-stained dystrophic processes in the corpus callosum of both the Tsc2+/+ injured and Tsc2+/KO , albeit to similar degrees in both genotypes. Consistent with previous reports showing an inflammatory response after an mTBI, we observed increased GFAP and Iba1 immunoreactivity in brain sections of injured mice 4–5 weeks after a mCHI. 38,39 Because the histopathological changes we observed occurred in both injured groups, they are unlikely to be the underlying mechanism for the worsened cognitive function observed in the Tsc2+/KO mice.
Previous studies have suggested that altered plasticity (e.g., long-term potentiation suppression) may contribute to learning and memory dysfunction in the absence of HIPP cell loss. 40,41 Because our evaluations were performed at the gross level, subtle alterations in morphology and/or impaired plasticity in the Tsc2+/KO mice cannot be ruled out. Because activation of mTORC1 stimulates energy consuming processes such as protein translation, our results suggest that activation of mTORC1 resulting from loss of Tsc2 regulation may exacerbate the cellular energy crisis that occurs after TBI. 6,9
Future studies to clarify the pathways involved in the observed worsening of cognitive function and directly measure changes in energy utilization in Tsc2+/KO mice after a mCHI will be needed to clarify the role of Tsc2 in TBI-related learning and memory dysfunction.
Footnotes
Acknowledgments
This work was supported in part by grants (NS087149 and NS060804) from the National Institutes of Health. The authors would like to thank Dr. Jing Zhao and Ms. Cristina Nelson for performing the animal surgeries, and Sara Orsi for her helpful discussions.
Author Disclosure Statement
No competing financial interests exist.