
Abstract
Traumatic spinal cord injury (SCI) causes major disruption to peripheral organ innervation and regulation. Relatively little work has investigated these post-SCI systemic changes, however, despite considerable evidence that multiple organ system dysfunction contributes to chronic impairments in health. Because metabolic dysfunction is common after SCI and the liver is a pivotal site for metabolic homeostasis, we sought to determine if liver pathology occurs as a result of SCI in a rat spinal contusion model. Histologic evidence showed excess lipid accumulation in the liver for at least 21 days post-injury after cervical or midthoracic SCI. Lipidomic analysis revealed an acute increase in hepatic ceramides as well as chronically elevated lactosylceramide. Post-SCI hepatic changes also included increased proinflammatory gene expression, including interleukin (IL)-1α, IL-1β, chemokine ligand-2, and tumor necrosis factor-α mRNA. These were coincident with increased CD68+ macrophages in the liver through 21 days post-injury. Serum alanine transaminase, used clinically to detect liver damage, was significantly increased at 21 days post-injury, suggesting that early metabolic and inflammatory damage preceded overt liver pathology. Surprisingly, liver inflammation was even detected after lumbar SCI. Collectively, these results suggest that SCI produces chronic liver injury with symptoms strikingly similar to those of nonalcoholic steatohepatitis (fatty liver disease). These clinically significant hepatic changes after SCI are known to contribute to systemic inflammation, cardiovascular disease, and metabolic syndrome, all of which are more prevalent in persons with SCI. Targeting acute and prolonged hepatic pathology may improve recovery and reduce long-term complications after SCI.
Introduction
M
A central role for the liver in initiating and prolonging systemic inflammation after SCI has been suggested by several studies showing that liver inflammation precedes and may exacerbate intraspinal inflammation and pathology after SCI. 1 –5 Experimental data evaluating hepatic and overall systemic inflammation in animal models of SCI are largely limited to the first 24 h post-injury, with a few studies extending into the first week. 6
A key question remaining unanswered is how long liver inflammation persists after SCI. This is important because liver pathology, including inflammation and fatty infiltration, is associated with metabolic syndrome, which occurs at increased incidence in the SCI population. 7 –9 Indeed, SCI patients commonly have many features of metabolic syndrome, including insulin resistance, increased abdominal adiposity, altered cholesterol levels, and abnormal metabolic hormone levels. 8,10,11 In fact, one study reported evidence of metabolic syndrome in 43% of persons with SCI examined. 8 The occurrence of metabolic syndrome and its hepatic manifestation termed nonalcoholic fatty liver disease (NAFLD) predict future cardiovascular disease, morbidity, and mortality, 12 –15 all of which are increased in the SCI population. 7 –9,16,17 Thus, a clear understanding of hepatic inflammation and pathology may provide new insights into intraspinal and systemic detrimental processes occurring after SCI.
Although few studies have examined liver pathology in patients with SCI, there are some anecdotal reports. For instance, a study using ultrasound in persons with chronic SCI revealed that ∼80% displayed some liver abnormality, including fatty infiltrates in the livers of ∼20% of subjects. 18 The current study was designed to determine if liver pathology occurs after SCI in rodents. Specifically, rats were subjected to controlled cervical, thoracic, or lumbar contusion injury after which clinical markers of liver pathology were assessed, including hepatic lipid accumulation, macrophage activation, cytokine expression, and serum levels of alanine transaminase (ALT).
The data indicate that hepatic lipid accumulation and inflammation occur acutely after cervical and thoracic SCI and are maintained for at least 3 weeks post-injury. Hepatic inflammation also occurred after lumbar SCI but was delayed until 14 days post-injury (dpi). The post-SCI hepatic pathology is consistent with the development of NASH (nonalcoholic steatohepatitis), which is the combination of fatty liver (NAFLD) and hepatic inflammation. NASH increases the risk of cirrhosis, fibrosis, and hepatic carcinoma. 12,19 NASH also predisposes persons to diabetes, cardiovascular disease, and metabolic syndrome, which, as stated above, are all comorbidities associated with SCI. 7 –9,17 Thus, the liver may represent a new therapeutic target for limiting morbidity and mortality in association with post-traumatic spinal cord pathology.
Methods
Spinal cord injury and drug treatment
Spinal cord contusions were performed using standardized protocols as described previously. 20 –22 All procedures conformed to National Institutes of Health and The Ohio State University Institutional Animal Care and Use Committee care guidelines. Briefly, adult female Sprague-Dawley rats (223g–280g; Harlan, Houston, TX) were anesthetized with intraperitoneal (i.p.) ketamine (80 mg/kg, i.p) and xylazine (10 mg/kg, i.p.), and a dorsal laminectomy was performed at the vertebral level C5 (n=3–5/group), T8 (n=3–4/group) or, T12 (spinal segment L1) (n=3–4/group). Rats then received a moderate midline spinal contusion injury using the Infinite Horizons device (Precision Systems and Instrumentation) with a preset force of 200 kD. The muscles overlying the spinal cord were sutured and the skin was closed with surgical clips. Animals were given 5 mL of saline and placed into warm recovery cages immediately after the injury. Postsurgical care included 5 days of antibiotic treatment (Gentamicin, 5 mg/kg) and saline to maintain hydration, and twice-a-day manual bladder expression until spontaneous voiding returned.
Hepatic macrophage analysis
At the appropriate time post-injury (n=3–6 per time point post-injury), animals were given a lethal dose of ketamine (120 mg/kg, i.p) and xylazine (15 mg/kg, i.p.) and transcardially perfused with 0.1M phosphate buffered saline (PBS) until the liver was cleared of blood. Next, animals were perfused with 400 mL of 4% paraformaldehyde (PFA). The livers were removed and post-fixed in 4% PFA for 2 h followed by phosphate buffer overnight. The next day, the tissue was transferred to 30% sucrose for 3 days before freezing and blocking for tissue sectioning. Tissue sections were cut at 10 μm using a cryostat and slide mounted (Superfrost Plus Slides, Fisher Scientific); slides were stored at −20°C until used. For tissue analysis, hepatic macrophages were visualized using immunohistochemistry for CD68 (Serotec, MCA341R, 1:2,000). Microscopic images of liver sections were digitized (MCID Image Analysis software, Imaging Research Inc), and three random fields were quantified per animal. The area of anti-CD68 labeling was divided by the total sample area to calculate the proportional area of liver CD68 immunoreactivity.
Hepatic lipid analysis
Oil red O staining was performed to visualize hepatic lipid accumulation. Liver sections were placed in 4% PFA for 20 min to further fix the tissue. Next, the tissue was placed in 70% ethanol for 10 min before being placed in a saturated solution of Oil red O dissolved in 70% ethanol at 60°C for 30 min. The tissue was differentiated in ethanol, washed in distilled water, and then coverslipped with Immu-mount (Thermo Scientific). Quantification of hepatic lipid content was performed using MCID Image Analysis software. Liver sections were digitized, and the proportional area of the liver positive for stained lipids was calculated as above. Each region of interest was centered on a central vein and the area of Oil Red O staining was normalized to the hepatocyte area to control for differences in the area of vasculature. To determine lipid droplet n size, Image J software (NIH, Bethesda, MD) was used to measure the size of lipid droplets within each section. The maximum droplet size at each time post-injury was compared with naïve using one-way analysis of variance (ANOVA).
Hepatic lipidomic analysis
A crude lipid extract from liver tissue samples was obtained using a modified Bligh and Dyer procedure as described previously. 23,24 Ceramide C12:0 (Avanti Polar Lipids, Alabaster, AL) was included in extraction solvent as an internal standard. Extracts were dried in a nitrogen evaporator (Organomation Associates Inc, Berlin, MA) and stored at −80°C. Dried extracts were resuspended in pure methanol just before analysis. Analyses of ceramides were performed on a high-performance liquid chromatography (HPLC) coupled electrospray ionization triple quadrupole mass spectrometer (API3000, AB Sciex Inc. Thornhill, Ontario, Canada) using instrument settings similar to those described in previous studies. 23,24
Samples were injected using a CTC PAL autosampler (LEAP technologies Inc., Carrboro, NC) into an HPLC (PerkinElmer, Wellesley, MA) equipped with a reverse phase C18 column (Phenomenex, Torrance, CA). The column was first pre-equilibrated for 0.5 min with the first mobile phase consisting of 85% methanol, 15% H20, and 5 mM ammonium formate, then eluted with the second mobile phase consisting of 99% methanol, 1% formic acid, and 5 mM ammonium formate at the flow rate of 400.0 μL/min. The eluted sample was injected into the ion source where the detection of each ceramide species was conducted by multiple reaction monitoring. Spectral analysis was conducted using MultiQuant software (AB Sciex Inc, Thornhill, Ontario, Canada), and each peak area was individually validated and normalized to the internal standard. Final data for each ceramide species were expressed as the ratio of analyte/internal standard.
Hepatic gene analysis
At the appropriate time post-injury (n=3–5 per time post-injury), animals were given a lethal dose of ketamine (120 mg/kg, i.p) and xylazine (15 mg/kg, i.p.) and transcardially perfused with 0.1M PBS until the tissue was cleared of blood. The liver was rapidly dissected and immediately homogenized with Trizol Reagent (15596018, Life Technologies). The homogenate was frozen in liquid nitrogen and stored at −80°C until processed for RNA isolation and cDNA generation as described previously. 21 Hepatic mRNA expression of CD68, interleukin (IL)-1α, IL-1β, chemokine ligand (CXCL)-1, tumor necrosis factor (TNF)-α, chemokine ligand (CCL)-2, and 18S was examined using a 7900HT Real-Time PCR System (Applied Biosystems) and the primers listed in Table 1. Ribosomal 18S expression was used to normalized values for each animal, and mRNA levels were calculated using the ΔΔCT method. 25
CCL, chemokine ligand; IL, interleukin; TNF, tumor necrosis factor; SPT, serine palmitoyltransferase.
Serum analysis of ALT
At the time of perfusion, cardiac blood was collected and allowed to clot at room temperature for 30 min. Serum was obtained by centrifuging the clotted blood at 3000×G for 10 min and then aliquoted and frozen at −80°C until analysis. Serum ALT levels were measured with a commercially available kit (Caymen Chemical, cat #700260). The assay was conducted according to manufacturers' instructions.
Statistical analysis
An investigator blinded to the groups conducted all analyses. Statistical analysis was performed using Graph Pad Prism 5.0 (San Diego, CA). For all end points, a one-way ANOVA was performed followed by Bonferroni post hoc analysis to determine between group differences. The minimum level to determine statistical significance was set at p<0.05.
Results
Thoracic SCI induces robust liver inflammation and lipid accumulation
Previous data show that leukocytes and cytokines are increased in the liver within 2 h of experimental SCI. 1 –5 Data in Figure 1 show that activated Kupffer cells, the resident hepatic macrophages, persist at and beyond 1 dpi in liver. Indeed, hepatic CD68 mRNA increased >100-fold by 1 dpi and remained at that level for at least 21 dpi (Fig. 1A). Immunohistochemistry was used to examine the expression and distribution of CD68+ Kupffer cells. CD68 immunoreactivity doubled at 1 day and 3 dpi and was significantly increased compared with laminectomy controls (Fig. 1B). CD68 immunoreactivity was again significantly elevated at 21 dpi, at which time enlarged macrophages were evident throughout the liver sections (Fig. 1B, C).
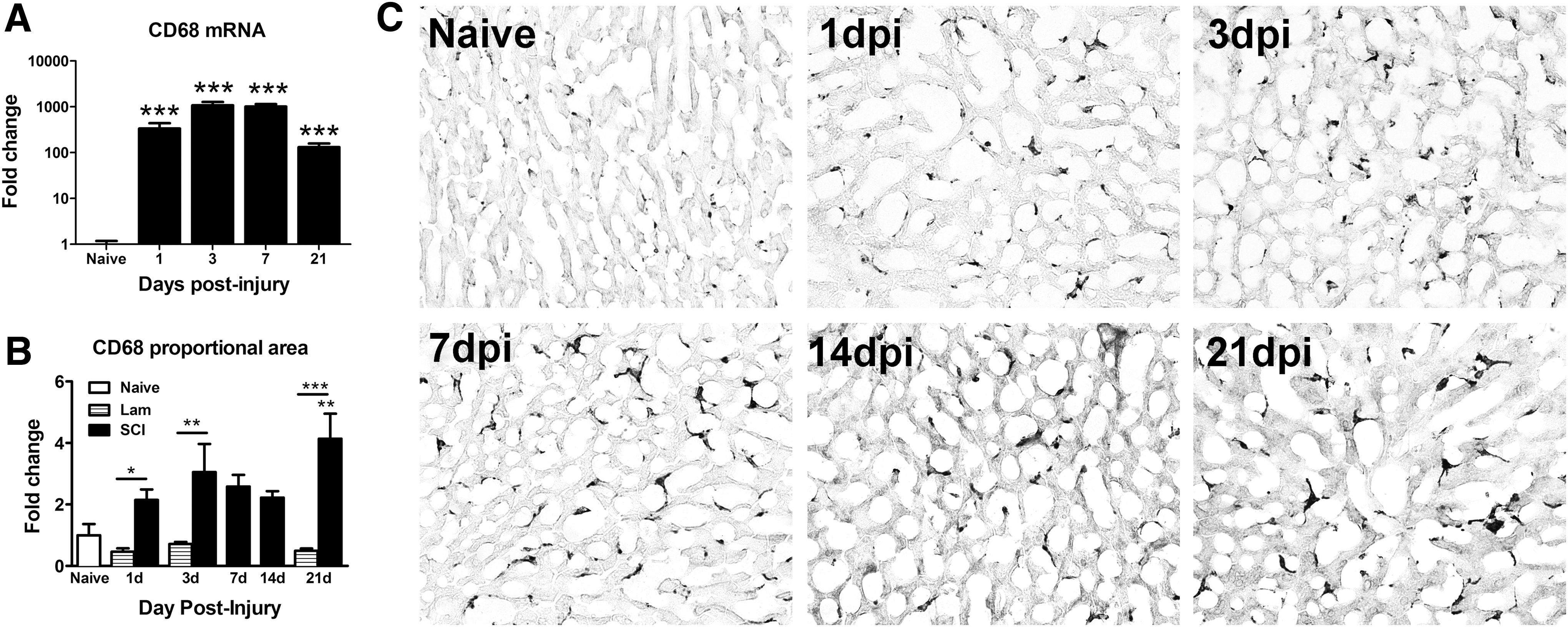
CD68 immunoreactivity increased in the liver after thoracic spinal cord injury (SCI). (
Sustained activation of hepatic macrophages was accompanied by enhanced and prolonged hepatic expression of inflammatory cytokine mRNA. IL-1α and IL-1β mRNA increased within 24 h, reaching levels 4–6-fold higher than in uninjured liver at 3–7 dpi (Fig. 2A,B). TNF-α mRNA exhibited a delayed increase, reaching ∼30-fold higher than controls by 14–21 dpi (Fig. 2C). mRNA for the monocyte chemoattractant protein CCL2 increased ∼3-fold at 14 dpi (Fig. 2D), and mRNA for the neutrophil chemoattractant chemokine CXCL1 increased ∼15-fold by 3 dpi but did not reach statistical significance (data not shown).

Increased hepatic inflammatory gene expression after thoracic spinal cord injury. (
Increased hepatic inflammation was paralleled by robust lipid accumulation in the liver (Fig. 3A). In contrast to CD68 expression that was not changed by surgical procedure alone, laminectomy did cause acute lipid accumulation in the liver at 1 dpi; this response, however, was rapidly resolved by 3 dpi when it was comparable to naïve levels (Fig. 3B). Lipids remained elevated in livers for at least 3 weeks after injury, with most lipids accumulating around the central veins (Fig. 3A). In addition to the elevated overall lipid, the maximum lipid droplet size increased significantly at 1, 3, 14, and 21 dpi, which is a feature of hepatic steatosis (Fig. 3C). 26 Lipid accumulation was positively correlated with Kupffer cell activation in the liver, suggesting a link between post-SCI liver inflammation and lipid accumulation (Fig. 3D). Collectively, the presence of hepatic inflammation and excess lipid is the defining characteristic of NASH.

Lipids accumulate in the liver acutely after thoracic spinal cord injury (SCI) and are maintained chronically. (
Thoracic SCI alters ceramide composition, vitamin E, and SPT mRNA in the liver
Ceramides are a subset of lipids that function as potent regulators of cellular functions, including inflammation and apoptosis. 27 –29 Notably, ceramide levels increase in the liver during NASH and can contribute to concomitant cognitive impairment and neurodegeneration. 30,31 Here, quantitative lipidomics was used to determine if liver ceramide levels change in a SCI-dependent manner. Of the 24 ceramides or ceramide precursors measured, 19 (79%) were significantly changed by SCI. Most ceramides and precursors (dihydroxyceramides) increased during the first 1–7 dpi and were two-fold or higher compared with naïve levels (Fig. 4). These early changes mostly comprised increased long-chain ceramide species (C16–C22) with relative sparing of the very long-chain species (C24–C26). One ceramide species in particular, galactosylceramide (18:1/16:0), increased two-fold by 1 dpi, then declined and was significantly reduced beyond 7 dpi (Fig. 4). Because this molecule can serve as a precursor to lactosylceramides (which increased after SCI), the reduction may reflect ongoing lactosylceramide synthesis. The direction and timing of changes in dihydroceramide and corresponding ceramide levels were similar, suggesting the involvement of de novo ceramide synthesis.
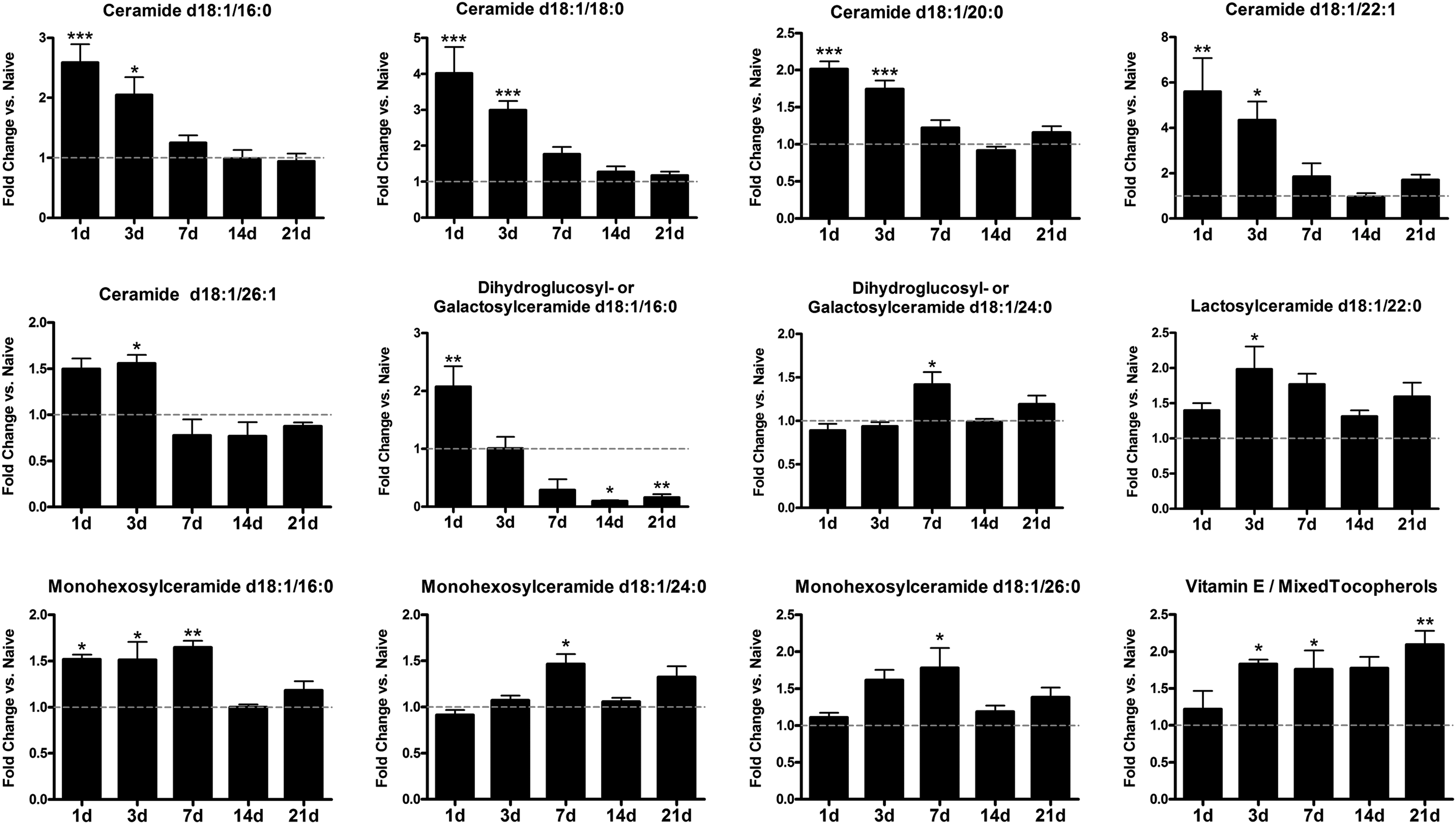
Spinal cord injury alters ceramide levels within the liver. Multiple ceramides were significantly elevated during the first week post-injury compared with naïve levels. In contrast, dihydroxy-glucose/galactose ceramide d18:1/16:0 increased at 1 day post-injury (dpi), then decreased significantly at 14–21 dpi. Mixed tocopherols including vitamin E increased at 3–21 dpi. All data are normalized to naïve levels, indicated by gray line in graphs. *p<0.05, **p<0.01, ***p<0.001 vs. naïve.
In addition to ceramides, hepatic tocopherols (including the lipid soluble antioxidant vitamin E) increased significantly after SCI (Fig. 4). Because vitamin E is fat soluble, its progressive accumulation likely reflects the ongoing rise in hepatic lipids after SCI. Indeed, analysis revealed a strong correlational trend for hepatic lipid and tocopherol levels (p=0.059).
To examine whether hepatic expression of enzymes involved in ceramide synthesis was altered by SCI, real-time polymerase chain reaction was used to examine mRNA level of serine palmitoyltransferase (SPT), the rate-limiting enzyme for de novo ceramide synthesis. SPT mRNA was two-fold higher than naïve by 1 dpi and was >4-fold higher at 3 dpi (p<0.01) (Fig. 5). Levels declined thereafter but then rose again at 21 dpi (p<0.01) (Fig. 5). Thus, hepatic SPT mRNA is dynamically regulated after SCI and may reflect acute and chronic phases of hepatic ceramide synthesis.
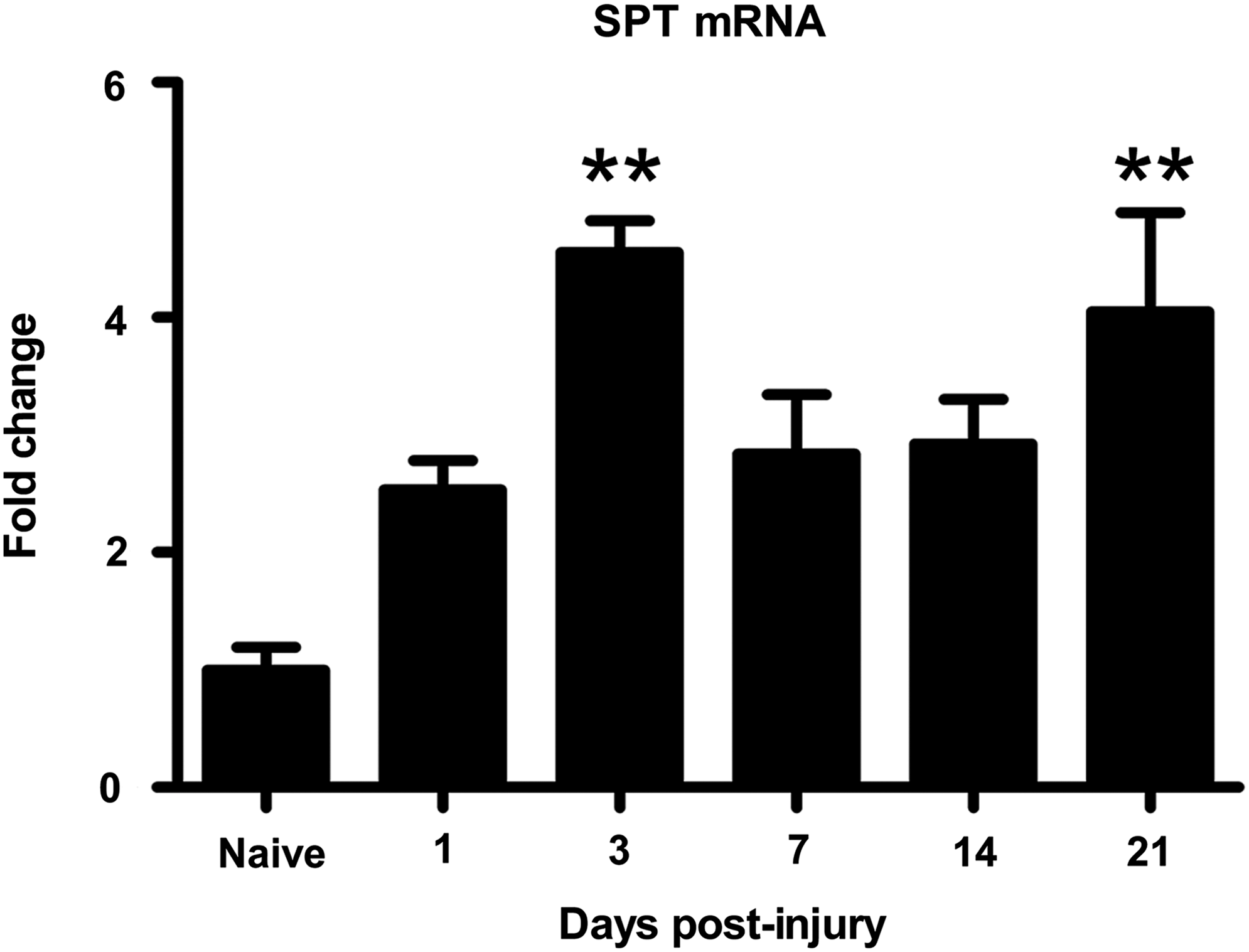
Increased mRNA expression of the ceramide synthesis enzyme serine palmitoyltransferase (SPT) after spinal cord injury. SPT is the rate limiting enzyme in the de novo synthesis of ceramide. SPT mRNA increased significantly compared with naïve at 3 days postinjury (dpi) and 21 dpi. **p<0.01.
Cervical SCI induces liver dysfunction
Previous studies have shown that post-SCI systemic alterations vary as a function of spinal injury level. 16,32 –35 To determine if post-injury changes in the liver were dependent on the level of SCI, we examined liver inflammation and lipid accumulation after midline cervical (C5 level) contusion injury in rats. Similar to thoracic SCI, hepatic CD68 mRNA significantly increased by 1 dpi, reaching levels ∼1000-fold higher than in naïve/uninjured livers (Fig. 6A). An additional 10-fold increase in expression of CD68 mRNA was evident by 3 weeks post-injury (702 at 1 dpi vs. 7840 at 21 dpi; Fig. 6A). Changes in mRNA expression were accompanied by a sustained increase in CD68 immunoreactivity (Fig. 6B), which was reflected by larger and more prominent macrophages, similar to that observed after thoracic SCI.
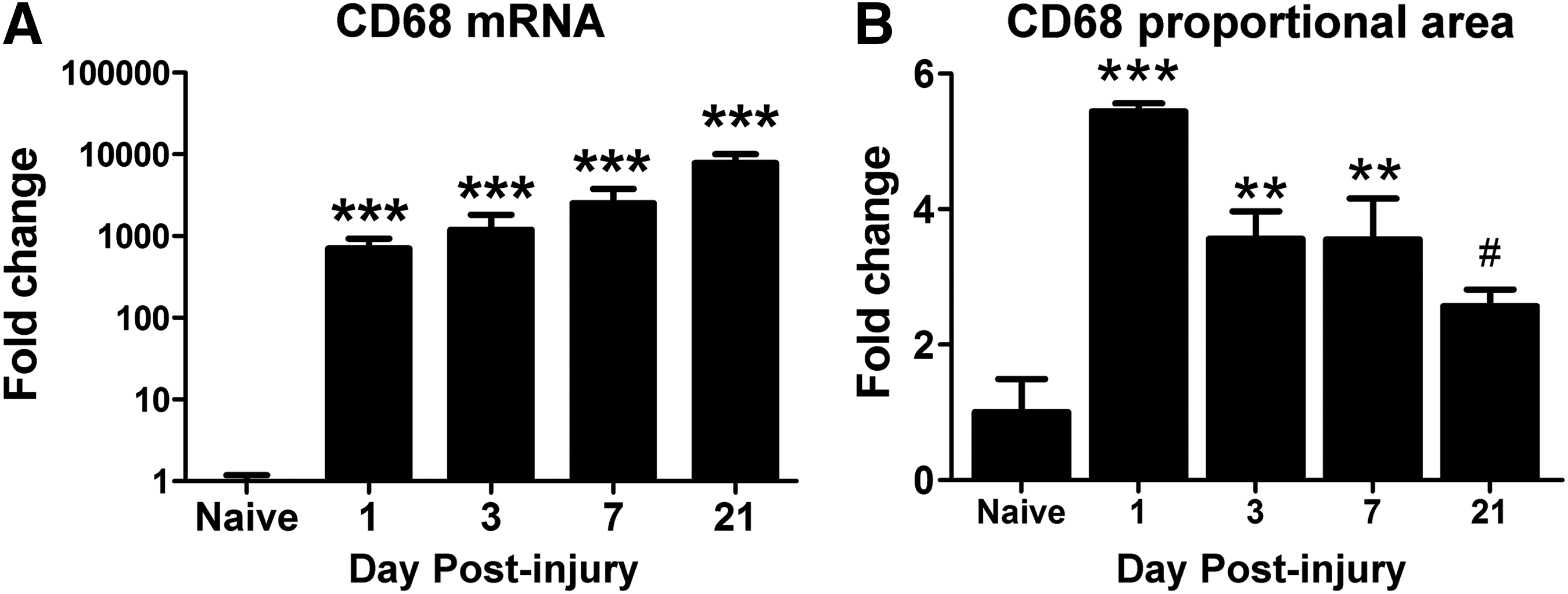
CD68 immunoreactivity increased in the liver after cervical spinal cord injury.
Hepatic cytokine and chemokine expression also were altered by cervical SCI. In contrast to thoracic SCI, IL-1α mRNA decreased 1–3 days after cervical SCI and returned to baseline thereafter (ANOVA, p<0.05) (Fig. 7A). After C5 contusion, IL-1β and TNF-α mRNA showed a delayed increased at 21 dpi (Fig. 7B,C). Similar to thoracic SCI, mRNA for the monocyte chemoattractant protein CCL2 increased ∼2-fold at 7 and 21 dpi (Fig. 7D).

Increased hepatic inflammatory gene expression after cervical spinal cord injury.
Hepatic lipid accumulation and droplet size also increased after cervical SCI. Lipid content was ∼8-fold higher at 1 dpi and 3 dpi (p<0.001), then declined but remained 2–3-fold elevated compared with naïve through 21 dpi (p<0.01; Fig. 8A). Lipic droplet size was increased at 1 dpi and 3 dpi and returned to baseline thereafter (Fig. 8B).
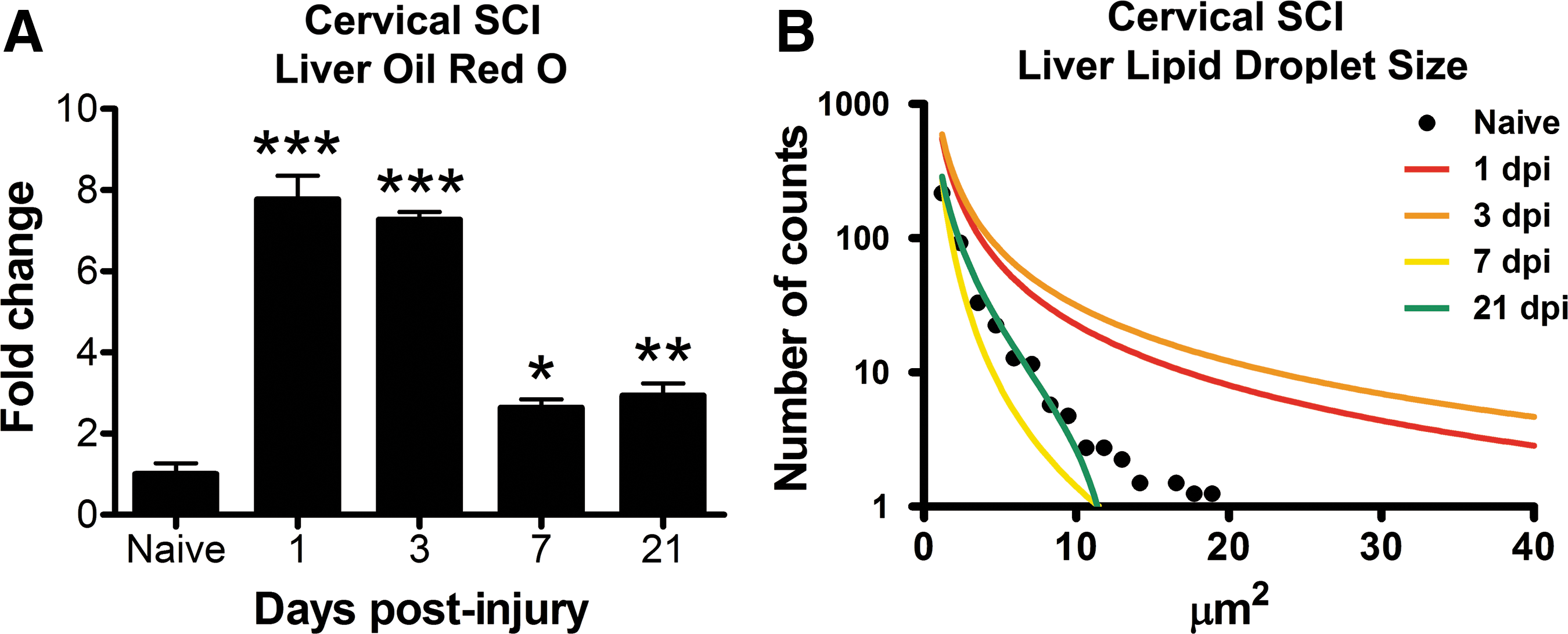
Lipid accumulates in the liver after cervical spinal cord injury (SCI).
SCI induces chronic elevation of serum ALT
Chronic inflammation and accumulation of lipids in the liver can cause prolonged liver pathology. Serum ALT is released from damaged/dying hepatocytes, and rising ALT levels are typically considered a marker of hepatic damage. After thoracic or cervical spinal contusion injury, serum ALT levels increased significantly 2–3 weeks post-injury compared with naïve and earlier post-injury time points (Fig. 9).
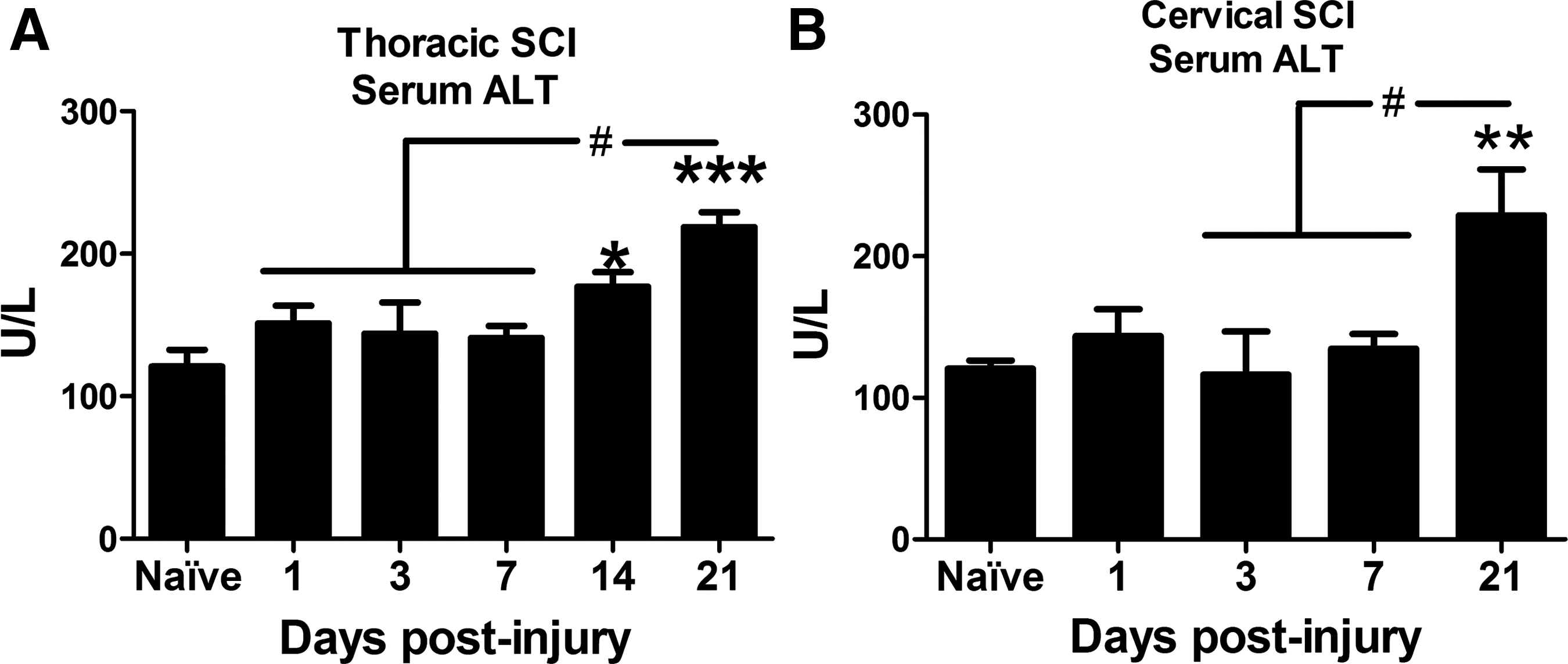
Serum alanine transaminase (ALT) increased after thoracic and cervical SCI. Elevated serum ALT is used clinically as a marker of liver pathology. (
Lumbar SCI induces liver inflammation but not lipid accumulation
One possible explanation for the above results is that post-SCI sympathetic outflow to the liver is disrupted by cervical and thoracic injuries, which in turn leads to hepatic pathology. To test if a lower level SCI would have similar effects, a cohort of rats received a spinal contusion at vertebral level T12, and hepatic macrophage activation and lipid accumulation was measured over the first 14 dpi. By 14 days after lumbar SCI, macrophage activation was significantly increased compared with controls (Fig. 10A). Lipid accumulation, however, did not increase over the first 2 weeks post-injury (Fig. 10B).
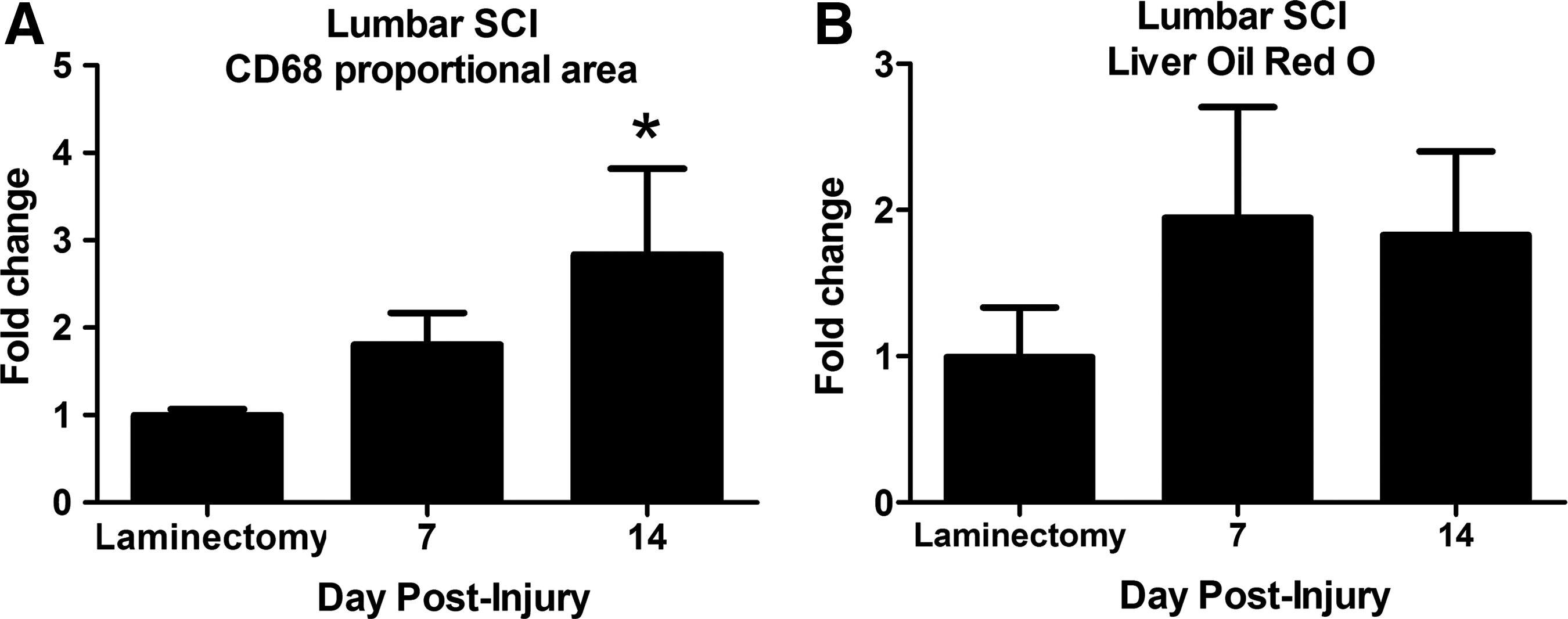
Lumbar spinal cord injury (SCI) caused hepatic inflammation but not lipid accumulation over the first 2 weeks post-injury. (
Discussion
The current results reveal that SCI induces acute liver inflammation and pathology that progresses for at least 3 weeks post-injury. SCI-induced changes include robust macrophage activation, prolonged expression of inflammatory cytokines, and marked lipid accumulation. It is notable that the areas exhibiting the most lipids were around the central veins because this region is particularly susceptible to metabolic perturbations and shows the earliest onset of lipid accumulation during steatosis. 36
The hepatic changes of excess lipid and inflammation are consistent with development of NASH, an advanced stage of NAFLD. 37 If left untreated, NASH can lead to irreversible fibrosis, cirrhosis, and, in some cases, hepatic carcinoma. 12,19 NASH itself results in suboptimal liver function and predisposes persons to diabetes, cardiovascular disease, metabolic syndrome, and liver cancer. 14,37,38 Notably, these conditions occur at increased rates after SCI and likely contribute to the decreased longevity in this population. 7 –9,17 Whether NASH occurs at a higher frequency in with SCI patients is unknown; however, liver pathology is a clinical feature of SCI. In one study, >70% of patients with SCI had abnormal liver changes, including fatty infiltration and/or parenchymal liver disease. 18 Other data show that hepatitis or cirrhosis is ∼7-fold higher in those with SCI compared with the general population. 39
Thus, SCI appears to place a person at increased risk for liver dysfunction and its associated metabolic problems. Notably, a study examining markers of liver pathology after SCI in patients with no abdominal injuries detected a delayed rise in serum markers of liver transaminases ∼18–22 dpi in >60% of patients, 40 which is in accordance with our current pre-clinical data. Importantly, the pathology associated with NASH is reversible if recognized and steps are taken to address it; otherwise, it can lead to irreversible fibrosis. Liver samples from our study did not show evidence of fibrosis based on the trichrome stain (not shown), but it will be important to follow up this work with more chronic time points to determine if NASH does convert to permanent fibrosis with time after SCI.
Aberrant liver function could contribute to the metabolic problems common after SCI. The role for the liver in metabolic syndrome is well documented and has been described as both “victim and culprit.” 41 The liver perpetuates metabolic syndrome (“culprit”) by releasing cytokines and low-density lipoprotein-bound fats, which induce systemic insulin resistance and inflammation. 42 This induction of cytokines by the liver activates leukocytes that traffic to the site of initial damage. 43 The liver in turn is damaged from the circulating inflammatory mediators, and lipid molecules present in metabolic syndrome (“victim”). These induce hepatic lipotoxicity including insulin resistance, oxidative damage, and hepatocyte death. 41 The delayed increase in serum ALT in our study suggests this mechanism may be at play after SCI.
Because certain lipids—in particular ceramides—are important signaling molecules and inflammatory mediators in NASH, 44 quantitative lipidomics was used to examine changes in hepatic ceramides after thoracic SCI. The data reveal that ceramide precursors (dihydroxyceramides) and mature ceramides increased significantly in the liver over the first week post-injury. Because SPT, the rate-limiting enzyme for ceramide synthesis, was also elevated, de novo synthesis likely contributes to the acute increase in ceramides. There was a secondary peak in SPT mRNA at 21 dpi; however, ceramide levels were not significantly elevated at that time.
It is possible that the increased hepatic vitamin E contributes to this discrepancy. Treatment with vitamin E can reduce ceramide accumulation in the brain and liver. 45,46 In the liver, this reduction in ceramide levels by vitamin E occurs, at least in part, through inhibition of both SPT and sphingomyelinase. 46 In this way, vitamin E inhibits two pathways of ceramide synthesis and thereby may prevent ceramide elevation in the liver at 21 dpi despite increased SPT mRNA. Ceramide production is also induced by inflammatory cytokines, such as TNF-α and IL-1β, 44 both of which are increased after SCI and may contribute to ceramide formation through the induction of hydrolytic or recycling pathways, although this remains to be determined.
Because ceramides are associated with liver pathology, 47 the increased hepatic ceramides may contribute to liver damage after SCI. Given that Oil red O increased in the liver 1 day after laminectomy surgery, it is possible that part of the ceramide increases at 1 dpi were from a surgery effect and not to the SCI, per se. Of the ceramides measured (12 species), however, 50% were significantly changed at 3 dpi or beyond, suggesting that injury to the spinal cord indeed alters the ceramide composition within the liver.
Ceramides play important regulatory roles, and when overproduced for prolonged periods can induce apoptosis through effects that ultimately perturb mitochondrial function. 44 Ceramides also can elicit inflammation, 28,29 in part by stimulating Toll-like receptor-4 (TLR4), which is expressed by most cells in the liver. 48 In hepatocytes and Kupffer cells (hepatic macrophages), TLR4 activation in turn drives the production of ceramides, 49,50 which can then lead to more pathology as a feed-forward cascade is established. Ceramides also form lipid rafts that cluster TNF family receptors and enhance death signaling events. 51,52 Delayed increases in TNF-α in the liver may contribute to delayed hepatotoxicity and the rise in serum ALT.
The ceramide species lactosylceramide, which increased early after SCI, is an active inflammatory mediator that induces superoxide formation and upregulates adhesion molecules that recruit monocytes. 52,53 Therefore, lactosylceramide in particular may play a role in acute post-SCI liver damage, which is thought to drive intraspinal inflammatory pathology. 54 Ceramides also contribute to metabolic syndrome because they attenuate insulin signaling in hepatocytes, macrophages, and muscle. 44,55 In addition to these local effects, ceramides are thought to circulate as lipoprotein complexes that may have CNS access, and could exacerbate intraspinal pathology after SCI, because ceramides impair energy metabolism, induce oxidative stress, and reduce viability in neurons. 31 Indeed, hepatic ceramides are thought to aggravate cognitive impairment and neurodegeneration in Alzheimer's disease and alcohol-mediated neurodegeneration. 56,57
Previous work suggests that acute liver inflammation precedes and exacerbates intraspinal pathology after SCI, 4 and these systemic changes have led others to test therapeutics targeting this post-SCI systemic inflammatory response. 1,2 Our data extend these results by showing that liver inflammation is nonresolving (or resolves very slowly), and persists into chronic stages of recovery. Thus, chronic hepatic inflammation may contribute to nonresolving intraspinal inflammation and chronic lesion pathology 58 and/or limit the amount of spontaneous recovery.
Indeed, it is known that levels of circulating C-reactive protein (CRP), an acute phase protein, remain elevated chronically in people with SCI despite the absence of overt infection. 59 Accordingly, the typical use of measuring systemic CRP to detect infections is of limited use in those with SCI. Although the current results show increased macrophage activation in the liver after SCI, the source of these cells remains undetermined. Resident hepatic macrophages become activated during NASH and contribute to disease progression 60 ; however, infiltration of new macrophages to the liver also occurs and contributes to NASH-induced pathology. 61,62 Future studies will be needed to evaluate the role of resident versus recruited macrophages in post-SCI hepatic pathology after SCI.
The hepatic inflammatory response may induce the post-SCI hepatic lipid changes. For instance, TNF-α upregulates expression of lipogenesis genes, such as fatty acid synthase and sterol regulatory element binding protein-1c, and can stimulate the formation of ceramide. 63 Therefore, inflammation can lead to excess lipids, which in turn can damage cells and exacerbate inflammatory cascades. Given that infections are a common problem after SCI, this pathological cycle may be initiated every time a spinal cord injured person gets a bacterial infection. Specifically, bacteria stimulate TLR4, which in turn stimulates cytokine production and synthesis of ceramides. Because persons with SCI are often thought to have chronic systemic inflammation, this hepatic component may be an important piece of the post-SCI syndrome leading to enhanced metabolic syndrome, diabetes, and cardiovascular disease.
In the current study, serum ALT, a metabolic enzyme abundant in the liver, was used as an indicator of liver damage. Damage to hepatocytes causes ALT to accumulate in the circulation. 64 As such, ALT levels are used clinically to detect liver abnormalities 65 and a rise in serum ALT typically signifies liver damage. In our study, serum ALT was elevated at 2 and 3 weeks post-injury, which may suggest the liver loses the ability to mitigate damage and enact repair as the local pathological effects become more chronic. This signifies the chronic nature of liver damage after SCI and reveals a potentially large window during which therapeutic intervention is feasible.
In the present study, both cervical and thoracic SCI caused comparable acute changes in the liver. This is in contrast to previous work showing more severe effects after higher-level injuries on autonomic dysreflexia, inflammation, and cardiovascular problems. 16,66,67 Both injury levels used here, however, completely (cervical) or partially (thoracic) disrupt descending control of sympathetic neurons innervating the liver and, thus, differences in hepatic changes between cervical and thoracic injuries were subtle. To further test this, a cohort of animals received a lumbar SCI to spare sympathetic innervation to the liver. In these animals, delayed inflammation but not lipid accumulation increased in the liver. This suggests that post-SCI hepatic inflammation is not completely dependent on sympathetic pathways innervating the liver. These results suggest that the majority of people with SCI may have underlying hepatic alterations, with those sustaining higher level injuries likely exhibiting more acute pathology.
How might altered liver function be important to the SCI population? Drug metabolism is a key consideration. People with SCI take many different drugs, and aberrant liver function will affect the metabolism and bioavailability of these drugs. It is also possible that these drugs will exacerbate pathology in an already fragile or dysfunctional liver. Notably, a recent clinical trial in patients with SCI using riluzole (100 mg/d) showed moderate to severe elevations in ALT in ∼30% of patients. 68 In contrast, when the identical riluzole dose was used to treat >7800 patients with amyotrophic lateral sclerosis, elevated ALT was detected in only 4.3% of patients. 69 These data might indicate that enhanced liver dysfunction is a unique feature of SCI compared with other diseases of the spinal cord. It is important to know whether increased liver enzymes are caused by SCI or are an indirect indication of drug-induced hepatotoxicity in those with SCI. Knowing this will inform the design and data analyses/interpretation for ongoing or future clinical trials.
Collectively, the present study builds on other work showing that SCI does more than injure the spinal cord. Indeed, robust and prolonged pathology occurs in the liver. The liver would be expected to react rapidly to SCI, as it would to any trauma, by initiating an acute phase response (APR). Normally, the APR would resolve; however, this does not seem to happen after SCI. These and other published data indicate that the systemic pathology associated with SCI does not develop “passively” as a result of disuse atrophy or denervation. Instead, chronic liver dysfunction may be an active and sustained mechanism of chronic neuroinflammation and systemic metabolic syndrome after SCI.
Indeed, it is known that traumatic brain injury induces neurogenic-derived pathology in peripheral organs that cause these organs to have a poor outcome on transplantation. 70 –72 It is quite possible that the SCI initiates a similar “neurogenic disease” state in peripheral organs and contributes to the overall poor health and reduced longevity in the SCI population. The peripheral liver dysfunction in turn could potentially contribute to spinal cord dysfunction because hepatic myelopathy has been described in patients presenting with progressive spasticity and paresis. 73 While metabolic and cardiovascular problems after SCI are often thought to be because of reduced physical activity, there is perhaps more than meets the eye in the SCI “syndrome.” Finding therapies that minimize post-SCI hepatic (and other systemic) changes may provide a novel approach to improve the overall health, well-being, and longevity of persons with SCI.
Footnotes
Acknowledgments
The authors gratefully acknowledge Drs. Jan Schwab and Richard Bruno for helpful discussions on this study and critical review of the manuscript. The authors also acknowledge Ping Wei, Zhen Guan, A. Todd Lash, Jackie Lovett, and Joelle Dorskind for excellent technical assistance. This work was funded by DOD AMRAA W81XWH-10-1-0946 (DMM), NINDS R01NS082095 (DMM), NINDS P30-NS045758 (DMM), The Craig H Neilsen Foundation Grant 260853 (ADS), R01MH077542 (NJH), and P30MH075673 (NJH).
Author Disclosure Statement
No competing financial interests exist.