
Abstract
Traumatic brain injury (TBI) initiates an excessive mediator release of e.g. neurotrophins, which promote neuronal survival, differentiation, and modulate synaptic plasticity. Paradoxically, mature forms of neurotrophins promote neuronal survival, whereas unprocessed forms of neurotrophins induce cell death through p75 neurotrophin receptor (p75NTR) signaling. p75NTR is widely expressed during synaptogenesis and is subsequently downregulated in adulthood. Repair mechanisms after acute cerebral insults can reactivate its expression. Therefore, the influence of p75NTR on secondary brain damage was addressed. mRNA levels of p75NTR and its ligands were quantified in brain tissue up to 7 days after experimental TBI (controlled cortical impact; CCI). Brain damage, motor function and inflammatory marker gene expression were determined in mice lacking the proneurotrophin-binding site of the p75NTR protein (NGFR-/-) and wild type littermates (NGFR+/+) 24 h and 5 days after CCI. In addition, the effect of TAT-Pep5 (pharmacological inhibitor of the intracellular p75NTR death domain) on lesion volume was evaluated 24 h after insult. p75NTR mRNA levels were induced nine-fold by TBI. In NGFR-/- mice, lesion volume was reduced by 29% at 24 h and by 21% 5 days after CCI. Motor coordination was significantly improved 24 h after trauma compared with the wild type. Pharmacological inhibition of the p75NTR signaling reduced lesion volume by 18%. The present study presents first time evidence that genetic mutation of the neurotrophin interaction site of p75NTR strongly limits post-traumatic cell death. In addition, we revealed pharmacological targeting of the intracellular p75NTR cell death domain as a promising approach to limit acute brain damage.
Introduction
T
The low regenerative properties of the central nervous system (CNS) after injury are attributed to a combination of events, including lack of support by trophic prosurvival pathways, 3 and increased initiation of cell death cascades. This imbalance of prosurvival and cell death signaling pathways is caused to some extent by a disturbed homeostasis of neurotrophic factors.
Neurotrophins are neurotrophic mediators, which are endogenous stimulators of neuronal survival, promote neuronal differentiation, and modulate synaptic plasticity. Nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), glial cell derived neurotrophic factor (GDNF), neurotrophin-3 (NT3), and neurotrophin4/5 (NT4/5) constitute the neurotrophin family, which act through high-affinity receptor tyrosine kinases (Trk A, Trk B, Trk C) and the low-affinity p75 neurotrophin receptor (p75NTR).
Paradoxically, the mature forms of neurotrophins promote neuronal survival via Trk activation, whereas unprocessed forms of the neurotrophins (proneurotrophins) display distinct and often opposing biological activities to their mature counterparts by inducing cell death through p75NTR signaling. 4 –7
Several studies have demonstrated alterations in the expression of the neurotrophin BDNF and its trophic support for neuronal survival in the acute and the chronic phase after different brain pathologies. 8 –11 Recent data suggest that elevated p75NTR levels seem to be required for BDNF mediated trophic support in injured neurons to promote neuronal survival. 12 The receptor p75NTR, a member of the tumor necrosis factor (TNF) receptor superfamily, is a key element in the proneurotrophin–mediated apoptotic pathway, which provides an ongoing cell death signal. 13,14 p75NTR has been implicated in playing a key role to establish an appropriate neuronal circuitry by regulating axon competition during development, but it also maintains the specificity of neuronal connectivity in the intact mature brain. 15,16
Adaptive properties of the CNS change during maturation and may coincide with alterations in expression levels of neurotrophins and p75NTR. Under the pathological conditions of brain injury, developmental-like programs of cell survival are initiated. 12 The p75NTR is highly expressed during synaptogenesis in the developing brain and is subsequently downregulated in the adult brain, and re-expressed in injured tissue. 17 –19
p75NTR contributes to the pathophysiology of a number of brain disorders. This has been shown for several animal models such as pilocarpine-induced seizures, 20 ex vivo corticospinal axotomy, 17 in vivo focal cerebral ischemia by middle cerebral artery occlusion, oxygen-glucose deprivation in hippocampal slices, 21 and mouse Alzheimer disease. The interaction between neurotrophins and p75NTR for secondary brain damage after mechanical brain injury such as TBI, however, has not been investigated in detail, yet.
Increasing evidence indicates that neurotrophins also modulate inflammatory response. The cross talk between neurotrophins and cytokines levels seems to be important for neuronal survival in several neurodegenerative disorders. The between neurotrophins and the inflammatory system has been described in detail for the cytokine interleukin (IL)-6, 22 as well as for the proinflammatory cytokines TNF-α and IL-1β 23 .
To get a deeper insight into the role of p75NTR, its regulation, and of its ligands, the neurotrophins BDNF and NGF were characterized after induction of mechanical brain injury. The consequences of a genetic p75NTR deficiency (NGFR-/-) with impaired neurotrophin binding on brain damage, mRNA expression of inflammation markers and neurotrophins, and functional outcome were studied after brain injury. In addition, the therapeutic effects on secondary brain damage after pharmacological inhibition of intracellular cell death signaling by p75NTR were investigated.
Methods
Experimental animals
All experiments were approved by the Animal Ethics Committee of the Landesuntersuchungsamt Rheinland-Pfalz, Germany (protocol number G 12-1-010). Before and during experiments, animal care was in accordance with the guidelines of the Johannes Gutenberg-University, Mainz, Germany.
A total of 127 C57BL/6 mice (weighing from 20.6 to 24.1 g) were investigated. Male C57BL/6 mice were analyzed in the experiments investigating the time dependent changes of mRNA expression after TBI and studies investigating the effect of TAT-Pep5 on lesion volume. C57BL/6 mice lacking exon III of the p75NTR extracellular domain (NGFR−/−) 24 and their wild-type littermates (NGFR+/+) were purchased from Jackson Laboratories (Bar Harbor, ME). These mice express a persisting isoform for a transmembrane protein that cannot bind neurotrophins, but interacts with Trk receptors. Mixed gender groups of the same age were used in experiments investigating NGFR-/- and NGFR+/+ mice (50:50 male:female).
P75NTR mutation in NGFR-/- was confirmed by quantitative real-time polymerase chain reaction (qPCR) at the mRNA and by genotyping at the DNA level. Mice were kept under 12:12 light and dark cycle with access to food and water ad libitum. All animals survived the observation period.
Experimental TBI
Mice were anesthetized with isoflurane (4 vol% induction; 2 vol% maintenance) via face mask and placed into a stereotactic frame. Rectal temperature was kept constant at 37°C using a feedback-controlled heating system (Hugo Sachs, March-Hugstetten, Germany). Trauma was induced by controlled cortical impact as described previously. 25,26 After craniotomy on the right rostrocaudal plane, a mechanical lesion was induced on the right parietotemporal cortex by a custom fabricated impactor (L. Kopacz, Germany). The following parameters were adjusted: tip diameter of 3 mm, 1.5 mm brain penetration, impact duration of 150 msec, and impact velocity of 8 M/sec.
After trauma, the craniotomy was rapidly sealed with histoacrylic glue (B Braun Melsungen AG, Melsungen, Germany). The wounds were sutured closed. Animals were returned to their own cages and placed in an incubator (33°C, 35% humidity; IC8000, Draeger, Germany) for 2 h. Animals woke up within 10 min after induction of trauma.
Treatment and drugs
TAT-Pep5 and TAT-Ctrl were obtained from PANATecs (Heilbronn, Germany). Treatment and controls were administered by intravenous injection at 6 h and by intraperitoneal injection 12 h after experimental TBI by an investigator blinded to the group allocation.
Histological evaluation of secondary brain damage
Animals were euthanized under isoflurane anesthesia (4 vol % for 1 min). Brains were quickly removed, frozen in powdered dried ice, and stored at −20°C. Brains were cut in the coronal plane using a cryostat (HM 560 Cryo Star, Thermo Fisher Scientific, Walldorf, Germany). Sections (10 μm) were collected at 500 μm intervals and stained with cresyl violet according to the manufacturer's instruction (Nissl-staining).
Areas of both hemispheres and the injured brain tissue were measured using a computerized image system (Delta Pix Insight, Delta Pix, Maalov, Denmark) by an investigator blinded to the randomization. Contusion volumes were calculated by multiplying contusion areas obtained from 16 consecutive sections with the distance-interval of 500 μm (0.5 * [A1+A2+A3 +…+ An]). 27,28
Rotarod testing
The rotarod test was used to measure motor coordination as described previously with minor modification by an investigator blinded to group allocation. 29,30 The rotarod test was performed on day 1 and day 5 after injury. The latency to balance on a rotating rod was measured using a five-lane rotarod device (Panlab Rota Rod, Harvard Apparatus, Holliston, MA). For acceleration, the speed was linearly increased from 4 rpm to 40 rpm over 5 min. Trials ended when the animal fell off the rod.
Four rotarod tests were performed before injury to score the baseline latencies for each animal. The average of these trials was taken as the baseline. After injury, animals were tested in two trials per investigated time point. Post-injury scores from these trials were averaged and evaluated relative to their pre-injury latencies to control for variability in pre-injury performance.
Gene expression (mRNA) analysis
For time series analysis, tissue preparation was performed as follows: brains were quickly removed and placed in a cooled brain matrix (Zivic Instruments, Pittsburgh, PA). For the 24 h study perilesional injured brain tissue was immediately dissected and rapidly frozen in liquid nitrogen. For the 5-day study, samples of perilesional injured brain tissue were collected during the cryosectioning procedure (−20°C). Brain tissue was stored at−80°C until processing.
Quantification of mRNA was performed by real-time PCR as described previously. 31 Absolute copy numbers of target genes were normalized against cyclophilin A (PPIA) as housekeeping gene.
Applied primers and probes are shown in Table 1. Same amounts of cDNA were amplified in duplicates using Kapa Probe Fast qPCR (peqlab biotechnology GmbH, Erlangen, Germany) for PPIA, Dynamo TM Color Flash (Biozym Scientific GmbH, Hessisch Oldendorf, Germany) for BDNF, ABsolute™ Fast qPCR Mix (Thermo Fisher Scientific, Dreieich, Germany) and Dynamo TN Color Flash (Biozym Scientific GmbH, Hessisch Oldendorf, Germany) for NGF, ABsolute™ Fast qPCR Mix (Thermo Fisher Scientific) for NGFR, ABsolute™ Fast qPCR Mix (Thermo Fisher Scientific) for iNOS, LightCycler® 480 Probes Master (Roche Deutschland Holding GmbH, Grenzach-Wyhlen, Germany) for IL-1β and IL-6, ABsolute™ Blue qPCR SYBR Green Mix (Thermo Fisher Scientific) for TNF-α and TrkA, and ABsolute™ Blue qPCR SybrgreenMix (Thermo Fisher Scientific) for TrkB according to the manufacturer's instructions.
PCR, polymerase chain reaction; Forw, sense primer; Rev, antisense primer; Cy5, Cyanine 5; Phos, Phosphate; FL, fluorescein; A
Statistical analysis
All experiments were performed by blinded investigators. Statistical analysis was performed using Sigma Plot 12.5 statistical software (Systat Software Inc., San Jose, CA). Exact Wilcoxon Mann-Whitney tests were used. Values were adjusted for multiple comparisons with the Holm-Bonferroni method. A value of p<0.05 was considered to be significant. Graph bars indicate mean and standard deviation throughout the figures.
Results
TBI increases p75NTR expression and changes neurotrophin and receptor Trk expression profiles
To investigate whether CCI influences neurotrophin and neurotrophin receptor expression, TBI was induced, and animals were randomized to 1, 3, 5, and 7 day survival after trauma and compared with nonoperated, naïve animals (n=9–10 mice/group). Gene expression analysis was performed by qPCR in perilesional cortical brain tissue (Fig. 1).
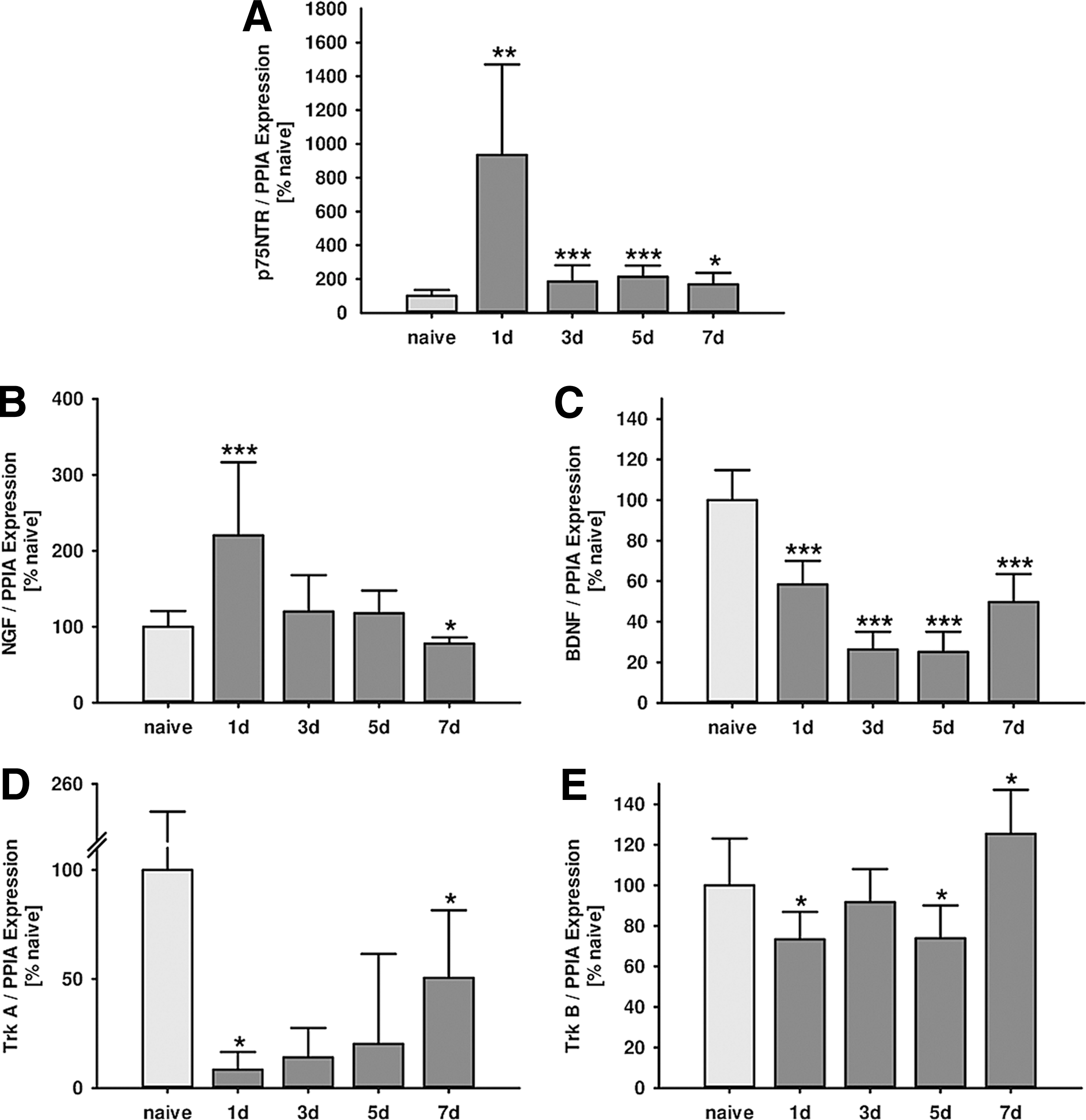
Influence of traumatic brain injury (TBI) on neurotrophin and p75 neurotrophin receptor (p75NTR) expression levels. Time course of perilesional cortical mRNA expression was determined by real-time polymerase chain reaction at (1, 3, 5, and 7 days) after experimentaI TBI. (
p75NTR expression increased in injured brain tissue within 7 days after trauma with a nine-fold peak at 24 h after CCI (naïve: 100±35 %naive; 1 day: 935±536 %naive, p=0.001; 3 days: 185±95 %naive, p=0.020; 5 days: 213±67 %naive, p<0.001; 7 days: 169±67 %naive, p=0.013; Fig. 1A).
Because p75NTR expression was strongly induced by CCI, mRNA expression of its ligands, NGF and BDNF, was analyzed. Real-time PCR revealed that NGF was upregulated by+110% in injured cortical tissue 24 h after TBI. Expression returned over time to naïve values at day 5 to finally become suppressed by −32% at day 7 post-insult (naïve: 100±21 %naive; 1 day: 220±96 %naive, p<0.001; 3 days: 120±48 %naive, p=0.567; 5 days: 118±30 %naive, p=0.391; 7 days: 78±8 %naive, p=0.022; Fig. 1B).
BDNF expression dropped (-42%) immediately after injury and expression level decreased even further with lowest values between day 3 and 5 after CCI (naïve: 100±15 %naive; 1 day: 58±12 %naive, p<0.001; 3 days: 26±9 %naive, p<0.001; 5 days: 25±10 %naive, p<0.001; 7 days: 50±5 %naive, p<0.001; Fig. 1C).
The expression of receptor Trk A and Trk B was also initially suppressed after CCI, with lowest values at day 1 post-insult (Trk A: −92%; Trk B: −27%). Trk B expression returned to baseline at day 3 and became induced at day 7 (+25%), while expression of Trk A stayed depressed until day 5 after injury (Trk A: naïve: 100±155 %naive; 1 day: 8±5 %naive, p=0.039; 3 days: 14±13 %naive, p=0.112; 5 days: 20±41 %naive, p=0.034, 7 days: 51±31 %naive, p=0.736; Fig. 1D; Trk B: naïve: 100±23 %naive; 1 day: 73±13 %naive, p=0.020; 3 days: 92±16 %naive, p=0.488; 5 days: 74±16 %naive, p=0.020, 7 days: 125±22 %naive, p=0.026; Fig. 1E).
The absence of p75NTR leads to reduced lesion volume after experimental TBI
Expression analysis showed a strong induction of p75NTR. In the next step, the role of p75NTR was investigated in animals with impaired functional interaction between neurotrophins and p75NTR protein. NGFR-/- mutant animals and NGFR+/+ littermates were randomly subjected to CCI, and brain lesion volume was quantified at 24 h (n=8 mice/group) and 5 days (n=10 mice/group) after insult in cresyl violet stained brain slices. In p75NTR, mutant mice lesion volume was significantly smaller at both 24 h (-21.2%; NGFR+/+: 46.3±7.3 mm3, NGFR-/-: 36.4±4.7 mm3, p=0.007; Fig. 2A) and 5 days (-29.2%; NGFR+/+: 24.1±6.7 mm3, p75NTR-/-: 17.0±5.2 mm3, p=0.031; Fig. 2B) after trauma.

Influence of p75 neurotrophin receptor (p75NTR) mutation on brain lesion volume. Mice lacking the neurotrophin binding site of P75NTR (NGFR-/-) and wild type (WT) littermates (NGFR+/+) were randomly subjected to experimental traumatic brain injury (TBI). Brain contusion volume was analyzed 24 h and 5 days after trauma by quantification of injured brain tissue in Nissl-stained cryosections. Lesion size was significantly lower in NGFR-/- animals compared with NGFR+/+ at 24 h (
p75NTR deficiency improves motor coordination 24 h after trauma
To test whether lack of functional p75NTR results in improved neurofunctional outcome, motor coordination was investigated by rotarod test (Fig. 3). Differences between baseline values and post-traumatic performance on the rotarod were analyzed at 24 h and 5 days after trauma in NGFR-/- and corresponding littermates (n=10 mice/group). The injury-induced impairment of motor coordination was attenuated at 24 h after CCI in NGFR-/- animals compared with NGFR+/+ littermates (NGFR+/+ time on rotarod: −61.2±25.5 sec, NGFR-/- time on rotarod: −32.1±20.3 sec, p=0.014). The performance improved over time in all animals, and differences persisted only by trend at 5 days after injury (NGFR+/+ time on rotarod: −37.9±9.9 sec, NGFR-/- time on rotarod: 20.4±16.3 sec, p=0.140).
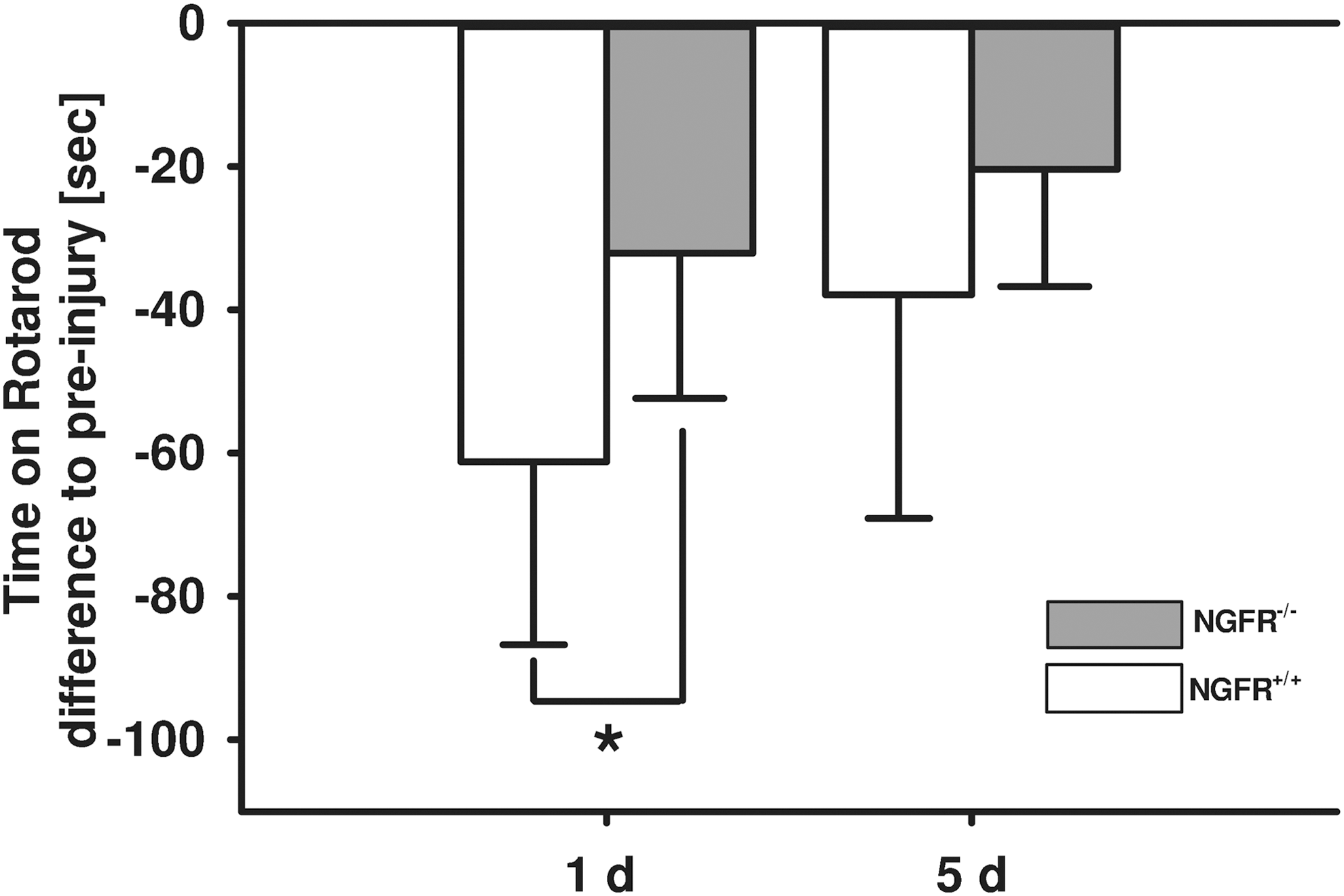
Effect of p75 neurotrophin receptor (p75NTR) mutation on motor coordination. Motor coordination was analyzed by rotarod test. The differences of time spent on the rod compared with baseline values were analyzed at 24 h and 5 days after trauma in p75NTR mutant (NGFR-/-) and corresponding littermates (NGFR+/+) (n=10 mice / group). NGFR-/- significantly improved motor function at 24 h after CCI (p=0.014) and by trend (p=0.140) at 5 days after injury (data are expressed as mean±standard deviation; (*) indicates p<0.05).
Marginal reduction of inflammatory marker gene expression of p75NTR mutant animals at 5 days after brain injury
Gene expression levels of the proinflammatory cytokines IL-1β, TNF-α, IL-6, and inducible nitric oxide synthase (iNOS) were investigated by real-time PCR at 24 h and 5 days after brain injury. Inflammatory marker gene expressions of TNF-α (NGFR-/-: 8415±4365 %naive; NGFR+/+: 6812±1678 %naive, p=0.421), IL-1β (NGFR-/-: 1589±913 %naive; NGFR+/+: 1216±322 %naive, p=0.690), IL-6 (NGFR-/-: 1589±913 %naive; NGFR+/+: 1216±322 %naive, p=1), and iNOS (NGFR-/-: 348±164 %naive; NGFR+/+: 271±85 %naive; p=0.690) were not affected by NGFR-/- in comparison with NGFR+/+ littermate gene expression levels at 24 h after trauma (Fig. 4A).
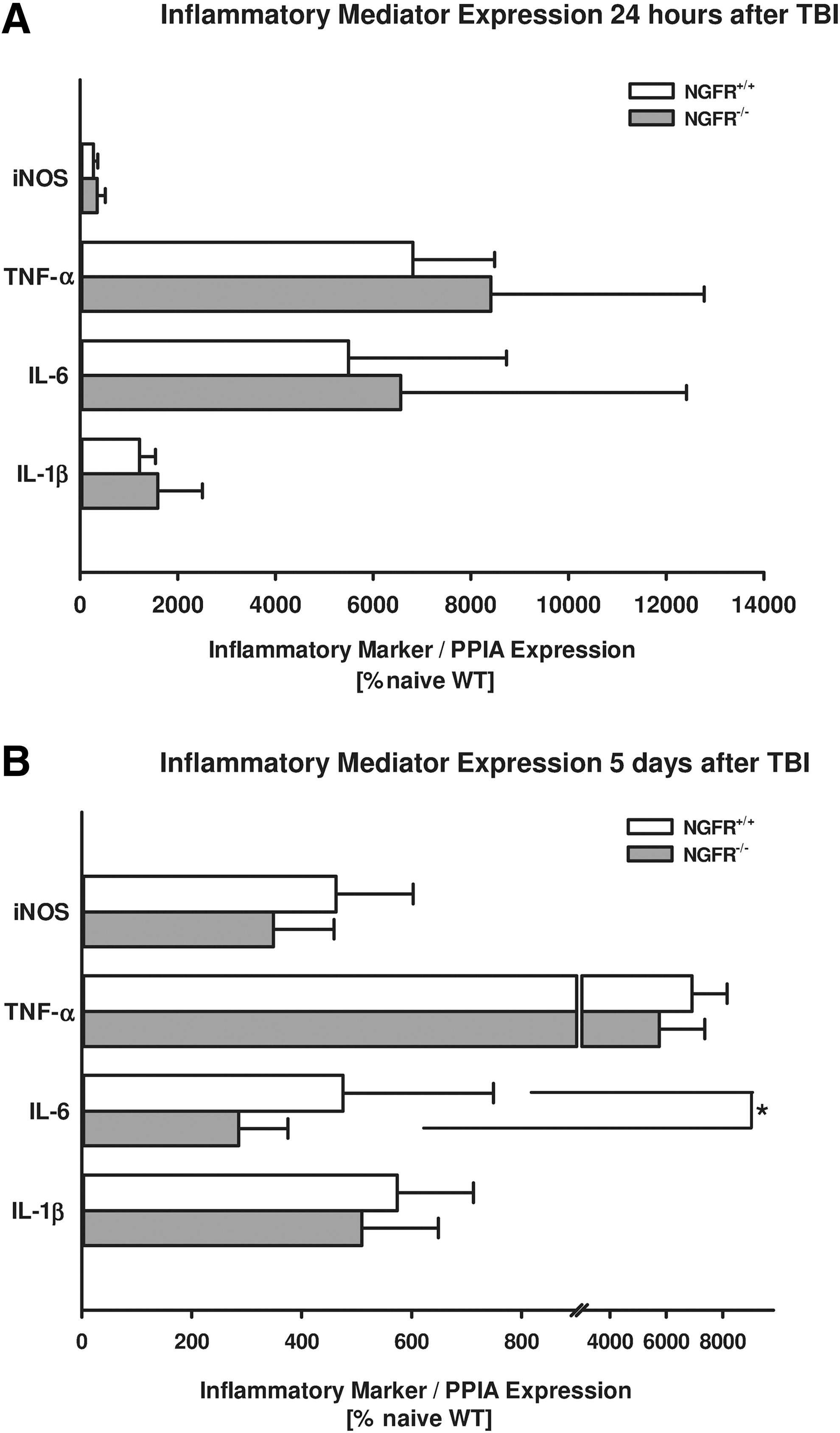
Effect of p75 neurotrophin receptor (p75NTR) mutation on inflammatory marker genes. Traumatic brain injury (TBI)-induced inflammatory marker gene expression levels were evaluated in perilesional brain tissue 5 days after brain injury. TBI increased proinflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and inducible nitric oxide synthase (iNOS). Inflammation markers were not changed in NGFR-/- compared with NGFR+/+ littermates 24 h after CCI. Five days after trauma, NGFR-/- showed reduced IL-6 expression compared with NGFR+/+ animals (n=10 mice / group). Other inflammation markers were downregulated by trend 5 days after trauma (data are normalized to the housekeeping gene cyclophilin A and are presented as mean±standard deviation in percentage of naïve animals; (*) indicates p<0.05). PPIA, cyclophilin A.
Expression of TNF-α (NGFR-/-: 5745±1620 %naive; NGFR+/+: 6910±1241 %naive, p=0.140), IL-1β (NGFR-/-: 509±139 %naive; NGFR+/+: 574±139 %naive, p=0.140), and iNOS (NGFR-/-: 349±110 %naive; NGFR+/+: 462±140 %naive; p=0.070), however, showed downward trends in NGFR-/- mice compared with NGFR+/+ littermates 5 days after brain injury (Fig. 4B). The proinflammatory marker IL-6, however, was significantly decreased at 5 days in NGFR-/- mice compared with NGFR+/+ animals (NGFR-/-: 285±89 %naive; NGFR+/+: 475±274 %naive; p=0.031).
p75NTR mutation of the neurotrophin interaction site slightly influences neurotrophin and neurotrophin receptor homeostasis after brain injury
p75NTR mutation with lack of the regular isoform was confirmed by real-time PCR in NGFR-/- and NGFR+/+ littermates (Fig. 5). Neurotrophin and neurotrophin receptor gene expression levels were not changed at 24 h post-insult in mice with p75NTR mutation (Trk A: NGFR-/-: 80±60 %naive; NGFR+/+: 70±25 %naive, p=0.1; Trk B: NGFR-/-: 84±9 %naive; NGFR+/+: 81±14 %naive, p=0.548; NGF: NGFR-/-: 112±60 %naive; NGFR+/+: 137±45 %naive, p=0.548; BDNF: NGFR-/-: 112±60 %naive; NGFR+/+: 137±45 %naive, p=0.548).
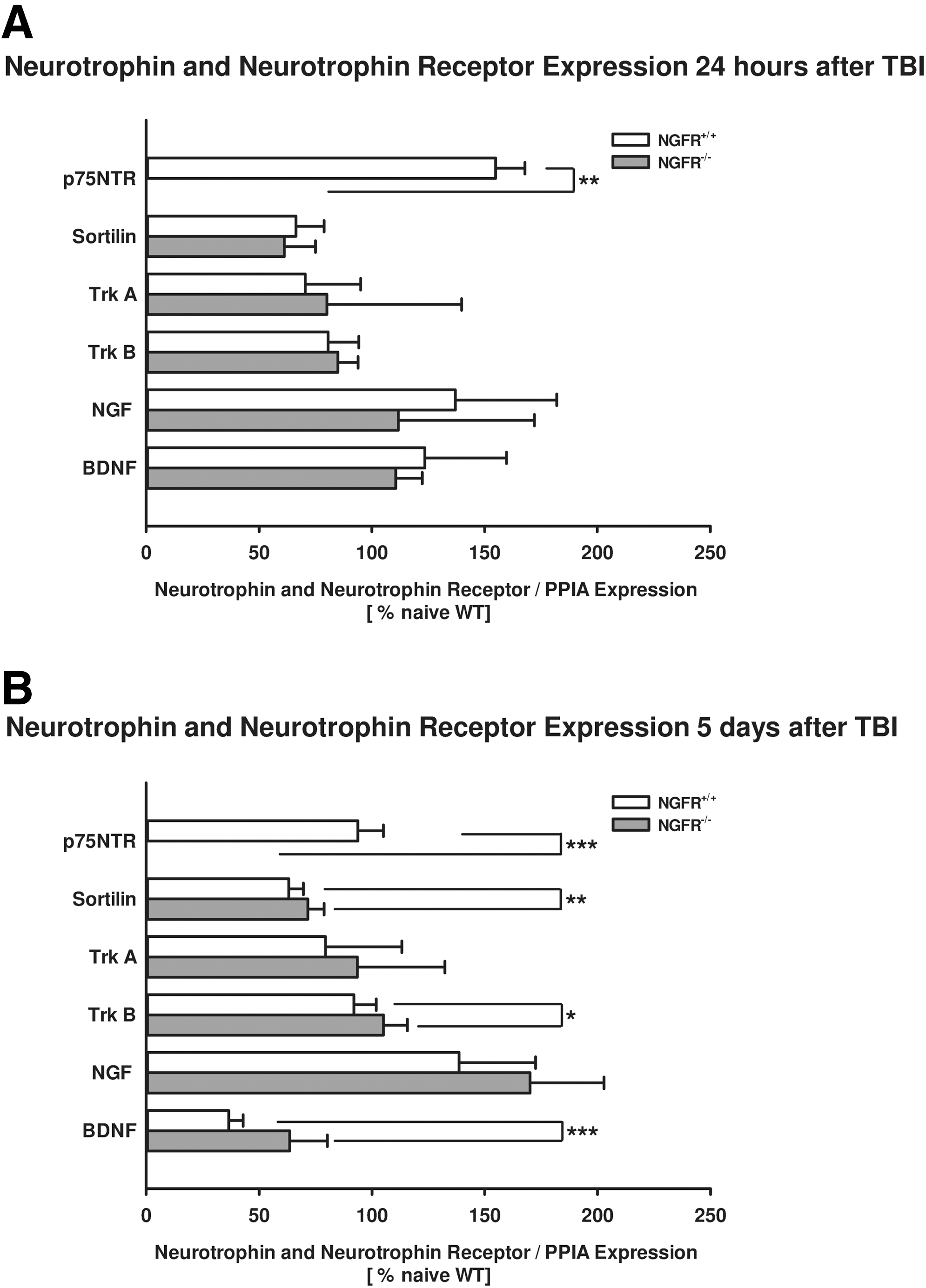
Effect of p75 neurotrophin receptor (p75NTR) mutation on neurotrophin and neurotrophin receptor expression. Lack of the wild type P75NTR isoform was confirmed by real-time polymerase chain reaction in NGFR-/- animals compared with NGFR+/+ littermates. Neurotrophin and neurotrophin receptor levels were not influenced in NGFR-/- in comparison with littermates 24 h after CCI (
Five days after brain trauma, Trk B (NGFR-/-: 105±11 %naive; NGFR+/+: 92±10 %naive, p=0.021) and BDNF (NGFR-/-: 64±17 %naive; NGFR+/+: 37±6 %naive, p<0.001) expression levels were slightly increased compared with littermates. Trk A and NGF levels were not significantly changed (Trk A: NGFR-/-: 94±39 %naive; NGFR+/+: 79±34 %naive, p=0.427; NGF: NGFR-/-: 170±33 %naive; NGFR+/+: 138±34 %naive, p=0.076).
Post-traumatic pharmacological inhibition of p75NTR signaling decreases lesion volume
Mice were subjected to CCI and randomized to low dose (1 μM) or high dose (10 μM) TAT-Pep5, or TAT peptide (TAT ctrl) treatment (n=12 mice/group) according to TAT-Pep5 and TAT ctrl dosages used in other studies in mice. 32,33 Lesion volume was determined 24 h after CCI.
To investigate effects of pharmacological p75NTR targeting, the p75NTR signaling inhibitor TAT-Pep5 was administered at two different doses after trauma. TAT-Pep5 low dose administration did not influence brain damage compared with control groups. High dosage of p75NTR signaling inhibitor TAT-Pep5 significantly decreased secondary brain damage (-18%) 24 h after CCI compared with the control treated group (TAT control: 46.1±7.1 mm3, 0.9% TAT-Pep5 low dosage: 47.2±11.7 mm3; TAT-Pep5 high dosage: 38.2±6.4 mm3, p=0.018; Fig. 6).

Influence of TAT-Pep5 on brain lesion volume after traumatic brain injury in Nissl-stained cryosections. Mice were randomly treated with low dose (LD, 1 μM) or high dose (HD, 10 μM) TAT-Pep5, or TAT ctrl (ctrl) 6 and 12 h after CCI (n=11/group). Data are presented as mean±standard deviation; p values are adjusted for multiple comparisons by Holm-Bonferroni adjustment; (*) indicates p<0.05.
Discussion
The present study demonstrates the important role of the p75NTR signaling pathway and the protective effect of its pharmacological inhibition in a well-characterized murine model of focal TBI. The present study reveals an increase in mRNA expression of p75NTR and changes in the expression pattern of its ligands, the neurotrophins NGF and BDNF, within 7 days after experimental TBI. Lesion volume was reduced and motor function was improved in mice with detect for neurotrophin-p75NTR interaction (NGFR-/-) after trauma—interaction between proneurotrophins and p75NTR may therefore be substantially required for lesion expansion after acute brain injury. Consistent with findings in NGFR-/- animals, TAT-Pep5, an intracellular p75NTR signaling inhibitor, significantly decreased brain damage after TBI.
p75NTR is an important neuronal signaling protein, which is widely expressed in the developing brain and becomes downregulated in adolescence. After brain injury, p75NTR levels become re-expressed, and neurotrophin levels are modulated under pathological conditions. 17,34 Its role as an apoptotic receptor has been established in developmental cell death 5,35 and has been implicated in promoting cell loss after various forms of CNS injuries. 36,37 p75NTR is a low affinity receptor to all members of neurotrophins. 38 Data from spinal cord injury suggest that proneurotrophins activate apoptosis via p75NTR in injured neuronal tissue to eliminate damaged cells. 34
The present study confirms increased mRNA expression levels of p75NTR and changes in expression levels of its ligands NGF and BDNF after acute brain injury—a pattern known to induce neuronal cell death. 4,39 Therefore, these data support a major role for p75NTR for brain damage formation. The question was addressed whether removal in exon III of the neurotrophin binding domain of p75NTR 37,40 results in reduced brain injury. NGFR-/- prevented impairments in motor coordination in the rotarod test and reduced lesion volume. Our findings are consistent with previous studies indicating that inhibition of expression, activation, or downstream signaling of p75NTR prevents cell loss and promotes improved functional recovery (reviewed in 18 ).
Among other mechanisms, increased expression of inflammatory mediators is believed to trigger the progressive lesion expansion after neuronal injury. 41 In the present study, however, inflammatory marker genes were not influenced in NGFR-/- 24 h after experimental TBI. Therefore, inflammatory processes induced by the proneurotrophin-p75NTR axis may play only a minor role for lesion expansion after experimental TBI.
Our results, however, demonstrate that NGFR-/- slightly blunted increases in the proinflammatory marker expression of IL-6 and TNF-α, IL-1β, and iNOS by trend at a delayed phase, 5 days after brain injury. The expression of proinflammatory mediators in the injured brain was shown to correlate with the extent of tissue damage. 42 Delayed reduction of the proinflammatory response may therefore be an indicator for less reparative processes as a consequence of decreased lesion volume in NGFR-/- animals.
Thus, reduced inflammation 5 days after TBI may be a secondary effect because of reduction in neuronal injury rather than a result of a direct influence on inflammation. This is consistent with findings of a study showing decreased microglia activation by pharmacological blockade of proNGF-p75NTR interaction 7 days after experimental TBI in rats, 39 but stays in contrast with the results of a study in diabetes and proNGF induced retinal inflammation and blood-retina barrier breakdown. Here, the authors concluded that the proNGF binding to p75NTR induced expression of inflammatory mediators. 43
In addition, the expression of BDNF and its receptor Trk B were slightly increased in the NGFR-/- animals. This suggests a shift toward proneurotrophic and prosurvival signaling in p75NTR mutant animals.
As in the first part of the present study, proneurotrophin-p75NTR interaction was confirmed to play a major role in the formation of secondary brain damage after acute brain trauma, the potential of the p75NTR signaling inhibitor TAT-Pep5 to ameliorate lesion expansion was investigated. Pep5 is a synthetic 15-amino acid residue peptide, which is directed to the intracellular death domain of p75NTR and a specific ligand to a binding site mapped by nuclear resonance spectroscopy onto a hydrophobic patch framed by helices 5 and 6. It is a peptide that specifically inhibits RhoA activation mediated by p75NTR. 44 –46 Pep5 is fused with the amino (N)-terminal protein transduction domain (11 amino acids) from the human immunodeficiency virus-type 1 trans-activating transcriptional activator TAT. 45 The biologically active TAT fusion proteins offer the possibility for direct delivery of proteins across the blood–brain barrier.
Beneficial effects of TAT fusion proteins as a therapeutic delivery system have been reported for stroke models. 47 –49 TAT proteins belong to the cell penetrating peptides, which are small peptides (5–25 amino acids) used to facilitate the delivery of normally nonpermeable cargos such as other peptides, proteins, nucleic acids, or drugs into the cells. Pharmacological modulation of p75NTR by a small-molecule p75NTR signaling modulator (LM11A-31) has recently been shown to decrease neuronal cell death and to improve neurogenesis and cognitive function after experimental TBI. 39
The present findings identify by TAT-Pep5 a different p75NTR inhibition strategy to reduce brain damage after experimental TBI. This may be of clinical interest, because fusion proteins like TAT-Pep5 can be intravenously administered and reach damaged brain regions to limit the cell toxic effect of p75NTR activation. 47,50 Further studies are needed to determine dose response and the treatment window of the beneficial effects of TAT-Pep5 in cerebral insults. In addition, the present study did not investigate apoptotic markers at the cellular level. We therefore cannot provide detailed information as to whether p75NTR deficiency or TAT Pep5 rescues all cell-types after acute brain injury.
Conclusion
The present findings demonstrate a robust and strong beneficial influence of the mutation of the neurotrophin-p75NTR binding on brain damage formation. Modulation of the p75NTR, as demonstrated by modulation of the p75NTR cell death domain, could therefore be a promising future target in neuroprotective strategies after acute brain injury.
Footnotes
Acknowledgments
This work was supported by the Federal Ministry of Education and Research (BMBF 01EO1003) to SCT, by the German Research Foundation (DFG) to SCT (TH1430/3-1) and KE (CRC1080/A9), by the Focus Program Translational Neurosciences (FTN) of the Johannes Gutenberg-University and Stage 1 funding of the Johannes Gutenberg-University to AS, MKES and SCT. Some data presented in this article are part of the doctoral thesis presented by CG to the Faculty of Veterinary Medicine of the Justus-Liebig-University Giessen, Germany.
Author Disclosure Statement
No competing financial interests exist.