
Abstract
Diffuse axonal injury is recognized as a progressive and long-term consequence of traumatic brain injury. Axonal injury can have sustained negative consequences on neuronal functions such as anterograde and retrograde transport and cellular processes such as autophagy that depend on cytoarchitecture and axon integrity. These changes can lead to somatic atrophy and an inability to repair and promote plasticity. Obstruction of the autophagic process has been noted after brain injury, and rapamycin, a drug used to stimulate autophagy, has demonstrated positive effects in brain injury models. The optimization of drugs to promote beneficial autophagy without negative side effects could be used to attenuate traumatic brain injury and promote improved outcome. Lanthionine ketimine ethyl ester, a bioavailable derivative of a natural sulfur amino acid metabolite, has demonstrated effects on autophagy both in vitro and in vivo. Thirty minutes after a moderate central fluid percussion injury and throughout the survival period, lanthionine ketimine ethyl ester was administered, and mice were subsequently evaluated for learning and memory impairments and biochemical and histological changes over a 5-week period. Lanthionine ketimine ethyl ester, which we have shown previously to modulate autophagy markers and alleviate pathology and slow cognitive decline in the 3 × TgAD mouse model, spared cognition and pathology after central fluid percussion injury through a mechanism involving autophagy modulation.
Introduction
T
Neuronal perturbations and axonal changes have long been described as a feature of human head injury. 1 –8 It has been demonstrated that a limited number of axons undergo primary axotomy from impact forces that create shear, tensile, and compressive strains and that a majority of injured axons degenerate via secondary axotomy. 9,10 Secondary axotomy may occur because of a combination of focal derangements in axolemmal structure and function, disruption of the microtubule/neurofilament network, ionic dysregulation, proteolytic activity, and impaired axonal transport. 8,11 –15
The demonstration of axonal swelling and disconnection occurring adjacent to intact, unaltered axons is consistent with the phenomenon of diffuse axonal injury (DAI) described in both animals and humans. 8,16,17 DAI is a common pathology noted in all severities of TBI 16,18 and can occur over long periods. 19 Evolving complications from TBI can be observed months to decades after the initial trauma, 20 contributing to psychiatric disorders, cognitive dysfunction, brain atrophy, chronic neuroinflammation, and the development of neurodegenerative disorders such as Alzheimer disease (AD) and chronic traumatic encephalopathy (CTE). 21
The family of collapsin response mediator proteins (CRMPs) plays a significant physiological role in neuron cell body and axon stability within the central nervous system (CNS). CRMPs are a family of neuronal phosphoproteins that regulate microtubule assembly. All CRMPs are expressed intracellularly at high levels during development in the CNS, but it is CRMP2 that remains at high levels in the adult mammalian CNS within neurons and specific oligodendrocytes. 22
CRMP2 is a microtubule-associated protein (MAP), phosphorylated by cyclin dependent kinase-5 (Cdk-5) and glycogen synthase kinase-β (GSK3β) enzymes. 23 Normally, CRMP2 stabilizes microtubules by binding tubulin 24 and facilitates protein trafficking by adapting kinesin-1 to protein cargo packages including neurotrophin receptors 25 and actin nucleation factors important for maintenance of synapse stability. 26 Studies have demonstrated that aberrant phosphorylation or cleavage of CRMP2 is detrimental to axon integrity and neuron survival. 27,28 Alterations in CRMP2 and CRMP4 protein patterns have been shown to be associated with the process of “protrusion-like” bead formation, a sign of axonal and neuronal degeneration. 29 Changes in CRMP2 expression, phosphorylation, or cleavage may have an adverse effect on its ability to bind cytoskeletal proteins and degrade the integrity of the axon.
Protein quality control and degradation play important roles in CNS homeostasis. The fidelity of the autophagic process is vital to CNS cells, and post-mitotic neurons in particular, that use autophagy to selectively dispose of misfolded or aggregated proteins and defective organelles. 30 Autophagy is essential to neurons because a reduction in autophagy has been implicated in neuronal cell death 31,32 and neurodegenerative diseases including AD, 33 Parkinson, 34 –37 Huntington, 38,39 and amyotrophic lateral sclerosis (ALS). 40,41
Spatial and mechanical processes ensure that autophagosomes move processively along the axon and multiple scaffolding proteins likely cooperate in this efficient transport. In the axon, autophagosomes undergo long-range microtubule-based transport that is coupled to compartment maturation. 33,42,43 The role of autophagy in TBI is clouded by conflicting reports of autophagy being both detrimental and beneficial. 44 -46 Although there is substantial evidence that markers of autophagy are increased in both human and animal brains after TBI, it is not clear whether those changes result in functional autophagy.
We have recently demonstrated that the axonal scaffolding protein, CRMP2, is involved in autophagy because engineered knockdown of CRMP2 reduces autophagy flux. 47 Further, lanthionine ketimine ethyl ester (LKE), previously demonstrated to dramatically reduce the accumulation of aggregated amyloid and tau in an AD mouse model, 48 binds to the microtubule-associated protein CRMP2 49 –51 and stimulates autophagy in mammalian CNS cells. 47
In this study, we used the mouse central fluid percussion model, a model of diffuse axonal injury. 52 The progressive nature of DAI suggests that there is a time in which a pharmacological treatment might be effective to stabilize neuronal architecture, stimulate productive autophagy, and allow repair mechanisms to function. To this effect, 30 min after a moderate TBI, and throughout the survival period, LKE was administered and mice were evaluated for learning and memory impairments at 3 and 5 weeks. Biochemical and histological analyses were also performed at 3 days and 5 weeks post-TBI.
Methods
Surgical preparation and injury induction
The procedures by which mice were subjected to central fluid percussion injury (cFPI) were modified, with appropriate scaling, from those described previously using rats. 53 Briefly, C57BL6/129SVJ mice, 4 months old and weighing 20–26 g, were surgically prepared for the induction of cFPI. Each animal was anesthetized in an anesthesia chamber with 3% isoflurane in O2. After induction, each animal's thigh was shaved for intraoperative physiological monitoring and placed in a stereotactic frame (David Kopf Instruments, Tujunga, CA) fitted with a nose cone to maintain anesthesia with 1–2% isoflurane in O2. A thermostatically controlled heating pad (Harvard Apparatus, Holliston, MA USA) was then placed under the animal and set to maintain body temperature at 37°C during the surgery.
A midline sagittal incision was made to expose the skull from bregma to lambda. The skull was cleaned and dried, and a 3.0 mm circular craniotomy (using a trephine drill bit inserted into a drill mounted to the stereotaxic frame; Harvard Apparatus) was then made along the sagittal suture midway between bregma and lambda, leaving the underlying dura intact. Care was taken not to generate heat during the craniotomy procedure.
A sterile Leur-lock syringe hub was then cut away from a 20-gauge needle and affixed to the craniotomy site using cyanoacrylate. On confirming the integrity of the seal between the hub and the skull, dental acrylic was then applied around the hub to provide stability during the induction of injury. After the dental acrylic hardened, the scalp was sutured around the hub, topical bacitracin and marcaine were applied to the incision site, and the animal was removed from anesthesia and monitored in a warmed cage until fully ambulatory (∼60–90 min).
For the induction of injury, 2 h after craniotomy surgery, each animal was reanesthetized with 3% isoflurane in 100% O2, and the male end of a spacing tube was inserted into the hub. The female end of the hub spacer assembly, filled with normal saline, was attached on to the male end of the fluid percussion apparatus (Custom Design and Fabrication; Virginia Commonwealth University; Richmond, VA). An injury of moderate severity (1.73 ± 0.03 atmospheres) was administered by releasing a pendulum onto a fluid-filled piston to induce a brief fluid pressure pulse on the intact dura. The pressure pulse measured by the transducer was displayed on a storage oscilloscope (Tektronix 5111), and the peak pressure was recorded.
After injury, the animals were visually monitored for recovery of spontaneous respiration. The hub and dental acrylic were removed en bloc, and the incision closed with VetBond veterinary adhesive before recovery from anesthesia/unconsciousness. The duration of transient unconsciousness was determined by measuring the time it took each animal to recover the following reflexes: toe pinch, tail pinch, and righting. After recovery of the righting reflex, animals were placed in a warmed holding cage to ensure the maintenance of normothermia and monitored during recovery before being returned to the vivarium.
For animals receiving a sham injury, all of the above steps were followed with the exception of the release of the pendulum to induce the injury. To verify a significant injury effect, righting reflex recovery times were analyzed by analysis of variance (ANOVA) and a Tukey post hoc analysis. All experiments were approved by the institutional animal care and use committee (protocol #2002-11) at the Veterans Administration-Greater Los Angeles Healthcare System (Animal welfare assurance #3002-01) in accordance with National Institutes of Health guidelines.
Treatment
LKE (R-LK-5-ethyl ester) was synthesized from 3-bromopyruvate (TCI America, Portland, OR) and L-cysteine-ethyl ester HCl (Alfa Aesar, Wood Hill, MA). 49,54 LKE was formulated at 1000 ppm (100 mg/kg/mouse/day) into AIN93M rodent diets (Dyets, Inc, Bethlehem, PA) blind-coded “A” or “B,” and stored at −80°C until the week of use. To ensure that mice received LKE in the first 3 days after surgery, 100 mg/kg/d LKE was delivered via intraperitoneal (ip) injection (in physiological saline) and then fed in the diet thereafter for longer survival time points.
Motor evaluation
To determine whether mice had motor impairments after injury, a battery of motor tasks was given to mice at three different time points. At 3 days after injury, a ledge test, hindlimb clasping, and gait analysis were performed. 55 The ledge test is a direct measure of coordination performed by observing the mouse as it walks along the cage ledge and lowers itself into its cage. A normal mouse will typically walk along the ledge without losing its balance and will lower itself back into the cage using its paws. This was assigned a score of 0. If the mouse lost its footing while walking along the ledge, but otherwise appeared coordinated, it received a score of 1. If it did not effectively use its hind legs, or lands on its head rather than its paws when descending into the cage, it received a score of 2. If it fell off the ledge while walking or attempting to lower itself, or was shaky and refused to move, it received a score of 3.
For hindlimb clasping analysis, the mouse was grasped by the tail near its base and lifted clear of all surrounding objects for 10 seconds. If the hindlimbs were consistently splayed outward, away from the abdomen, it was assigned a score of 0. If one hindlimb was retracted toward the abdomen for more than 50% of the time suspended, it received a score of 1, and if both hindlimbs were partially retracted toward the abdomen for more than 50% of the time, it received a score of 2. If its hindlimbs were entirely retracted and touching the abdomen for more than 50% of the time, it received a score of 3.
Gait is a measure of coordination and muscle function. Gait analysis was performed by removing the mouse from its cage and placing it on a flat surface with its head facing away from the investigator and observing the mouse from behind as it walks. If the mouse moved normally, with its body weight supported on all limbs, with its abdomen not touching the ground, and with both hindlimbs participating evenly, it received a score of 0. If it showed a tremor or appeared to limp while walking, it received a score of 1. If it showed a severe tremor, severe limp, lowered pelvis, or the feet pointed away from the body during locomotion (duck feet), it received a score of 2. If the mouse had difficulty moving forward and dragged its abdomen along the ground, it received a score of 3.
At day 15, a visible platform swim evaluation was performed to assess visual and swimming impairments. Ambulatory velocities were evaluated during the acquisition phase of the Barnes maze testing (days 30–33). These are described below in the respective sections. Treatment groups were blinded to the operator.
Morris water maze (MWM)
To test hippocampal-dependent spatial cognition, mice were trained in the standard MWM with a hidden platform as described previously 56,57 with minor modifications. The MWM consisted of a white, circular, water-filled pool (diameter, 1.20 m; height, 0.5 m at 24°C; maintained by a thermostat-driven heating system attached to the underside of the water tank) opacified by white powdered tempura paint. The tank was surrounded by solid white walls to eliminate distractions. Three solid black cues (different shapes) were affixed to the inside walls of the tank at three locations (N, E, and W). A white escape platform (15 cm diameter, height 24 cm) was located 1 cm below the water surface in a fixed position (NE quadrant, 22 cm away from the wall).
In each daily swim block (four swim trials per block, 30 sec intertrial interval), mice were placed at one of the starting locations, facing the wall, in random order (N, S, E, W, including permutations of the four starting points per session) and were allowed to swim until they located the platform. Mice failing to find the platform within 60 sec were placed on it for 15 sec (the same time as the successful animals). Mice floating/not actively searching for the platform or demonstrating >80% thigmotaxing behavior were eliminated from the analysis.
A probe trial was performed 24 h after the final training block. The platform was removed from the pool and the mice performed a 60 sec swim. At the end of every trial the mice were allowed to dry for 15 min in a heated enclosure and were returned to their home cage. The cued (visible platform) session was performed to test swimming speed and visual acuity. The visible platform was elevated 1 cm above the water and its position was clearly indicated by a visible cue (black tape around the elevated portion of the platform). Other than the black-taped platform, here were no other cues. The cued test (four trials) was performed 3 days before the hidden training protocol.
Anymaze software (Stoelting Co., Wood Dale, IL) was used to capture and analyze the MWM data. The person performing behavioral tasks was blinded to treatment condition. MWM was evaluated from 15–21 days. This time was chosen to allow for significant recovery post-injury and to allow LKE enough time to impact long-term outcome.
Barnes maze
The method used in our laboratory was adapted from a published protocol and review article. 58 A 20-hole Barnes maze apparatus was used (Any Maze). White curtains were used around the maze to reduce room cues. Three cues were placed in close proximity to the maze for use in spatial navigation. In the pre-training trial, the mouse was placed in the middle of the maze under a dark colored box allowing the mouse to be in random orientation before each trial. After 10 sec had elapsed, the chamber was lifted, and the mouse allowed to explore the maze for 3 min. Aversive stimuli (sounds, wind, harsh lighting) were not used.
Errors and latency were recorded during acquisition and testing. Errors were defined as nose pokes and head deflections over any hole that does not have the target box. Latency was defined as the time it took to locate the target box. Mice were trained for four trials per day for 4 days with an intertrial interval of at least 15 min. After each trial, the entire maze was cleaned with an unscented, mild, dilute soap solution. Mice not actively moving throughout the maze were eliminated from the analysis. At 48 h after the last training trial, a probe trial was conducted to evaluate short-term memory retention. Any Maze software was used to capture and analyze the data. Barnes maze was performed at 30–35 days (called the 5-week time point).
Tissue collection
At the end of Barnes maze testing, animals were anesthetized with 100 mg/kg pentobarbital and a blood sample taken via cardiac puncture followed by cardiac perfusion with HEPES buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 137 mM NaCl, 4.6 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4, 0.6 mM MgSO4 and 1.1 mM ethylenediamine tetraacetic acid [EDTA], 1 mM Na3VO4, and protease inhibitor cocktail [Roche complete ultra mini, Sigma Aldrich, St. Louis, MO]). Hippocampus and cortex were dissected from one hemisphere, snap frozen in N2(l), and stored at −80°C. The contralateral hemisphere was immersion fixed in 10% neutral buffered formalin (Thermo Fisher Scientific, Waltham, MA) for 24 h followed by step-wise (10-20-30%) sucrose cryopreservation and embedding in preparation for cryostat sectioning.
Immunohistochemistry
Formalin fixed, cryopreserved brains were sagitally sectioned on a Leica cryostat at 25 μm thickness and stored at −20°C until immunolabeling. Immunohistochemical labeling was performed on free-floating sections using a VectaStain Elite ABC kit (Vector Laboratories) with diaminobenzadine chromagen. Antibodies were: phospho-c-Jun Ser63 (1:1000; Cell Signaling Technologies) and phosphorylated neurofilament H (SMI-31,1:500; EMD Millipore, Billerica, MA). Sections were mounted on glass slides and coverslipped using Permount® mounting media (Thermo Fisher Scientific). Specificity of antibody immunoreactivity was confirmed by elimination of the primary antibody during the staining procedure. This negative control staining resulted in the loss of staining for these antibodies as predicted.
Western blot
Frozen tissue was homogenized in lysis buffer (10 mM Tris-HCl, 0.5 M NaCl, 0.3 M sucrose, pH 7.4 and containing 1% Triton-X100, 5 mM EDTA, 10 mM dithiothreitol, 1 mM Na3VO4, 1 mM NaF, and protease inhibitor cocktail (Roche, Complete Ultra Mini and PhosphoSTOP) at a ratio of 1:20 (1 mg tissue = 20 μL of lysis buffer). The resulting homogenates were assayed for protein concentration by the DC Protein Assay (Biorad, Hercules, CA). Samples were adjusted to equal protein concentration, mixed 1:1 with loading dye containing 2% B-mercaptoethanol, boiled and frozen at −20°C until use.
For LC3 blot
Total lysate (10 μg) was resolved on 4–20% TGX gel (Biorad) at 100 V for 100 min and transferred on a polyvinylidene difluoride membrane for 2 h at 60 V. Chloroquine-treated HeLa cells were used as the positive autophagy control (Cell Signaling Technology #11972S). All other proteins (10–30 μg) were resolved on 7.5% TGX gels (Biorad) and blotted onto a nitrocellulose membrane. Membranes were blocked in 5% dry milk, incubated in primary antibody overnight, and developed using ECL chemiluminescent reagents (SuperSignal West Pico, Thermo Fisher Scientific; or ECL Prime, GE Healthcare, Pittsburgh, PA). Blots were imaged using a Syngene G:Box and automated GENEsys control software (Syngene, Frederick, MD). Blots were normalized by reprobing with β-actin.
All primary antibodies were purchased from Cell Signaling Technologies unless otherwise noted and used at a 1:1000 dilution: Phospho-mTOR-Ser2448; mTOR (7C10); Phospho-ULK1 (S757); ULK1 (D8H5); Beclin1; Ubiquitin; LC3B/MAP1LC3B (Novus); CRMP2 (EMD Millipore); β-Actin (Pierce-Thermo Fisher Scientific).
Luxol fast blue (LFB)
Brain sections were removed from the −20°C freezer, thawed and rinsed 3 × 5 min in TBS followed by mounting on glass slides. Fat was removed from the sections by overnight incubation in a 1:1 ethanol/chloroform solution. Sections were rehydrated by successive 5 min incubations in 100/85/70% ethanol followed by H2O. Sections were then placed into a 1% Luxol fast blue solution (Sigma Aldrich, St. Louis, MO) for 2 h at 60°C. Staining was differentiated with three rounds of: 0.05% lithium carbonate for 30 sec followed by 70% ethanol for 30 sec and then H2O. Sections were dehydrated in 95% and 100% ethanol for 5 min each followed by 5 min in CitriSolve and then coverslipped with DPX (Thermo Fisher Scientific).
Cleaved tau enzyme-linked immunosorbent assay (ELISA)
Cleaved tau (C-tau) was measured using our previously characterized ELISA 59 and using three monoclonal antibodies (C-tau7, -8 and -12) that demonstrate noncompetitive binding to C-tau. Immulon 2 plates were coated with affinity-purified C-tau12 (1:200 in phosphate-buffered saline [PBS]; 100 μL/well) for 1 h at room temperature and then blocked overnight with 5% nonfat dry milk and 0.5% gelatin in Tris-buffered saline (TBS). Plates were washed 4× with 0.1% Tween in TBS (TBST), and samples added in triplicate, incubated with rocking for 1 h at room temperature and then washed 4× with TBST. Horseradish peroxidase (HRP)-conjugated C-tau7 and -8 (1:1000) were added for 1 h with rocking at room temperature. Plates were washed 4× with TBST and color developed using tetramethylbenzidine as substrate. The reaction was stopped after 25 minutes and the plate read at 450 nm.
A standard curve was generated using affinity-purified human C-tau. Negative controls included exclusion of C-tau12, deletion of sample, or deletion of HRP-conjugated C-tau7/C-tau8 from the assay. Experimental C-tau samples demonstrating optical density values less than the sensitivity of the assay were assigned a value of zero. The sensitivity of this ELISA is 30 pg per well and demonstrates an intra-assay variation of less than 7% and an interassay variation of less than 10%.
Plasma and brain LKE levels
Male C57BL/6 adult mice were purchased from Jackson Laboratories at 19–21 g body weight and randomized to three groups (n = 6 per group) fed AIN93M diet containing 0, 1, or 1000 ppm LKE. This treatment regimen provides daily oral doses of approximately 15 mg/kg LKE for the 1 ppm-modified diet and 150 mg/kg LKE for the 1000 ppm-modified diet, calculated from actual measured food consumption. Fresh food and water were provided ad libitum, and food intake and body weight were measured daily for 15 days. At the end of the study (10–12 AM on day 15), the mice were euthanized by CO2 inhalation followed by immediate exsanguination via cardiac puncture and harvesting of tissue specimens (brain cortex).
Blood samples (0.5–1 mL) were placed in heparinized eppendorf tubes on ice. Plasma was obtained by centrifugation and stored in sealed 1.5 mL tubes at −80°C until analysis. Plasma concentration of LKE was determined by LC-MS/MS analyses using tolbutamide as a standard for extraction efficiency.
Brain samples were carefully dissected to remove cortices, and meninges were discarded. Cortices were weighed by subtraction in pre-weighed eppendorf microcentrifuge tubes and frozen immediately, then stored at −80°C until analysis. Tissue was lysed by addition of two volumes of PBS and sonication. A 50 mL aliquot of brain lysate was mixed with 0.2 mL of extraction solvent and processed for LC-MS/MS.
Statistics
All data are graphically presented as mean ± standard error of the mean unless otherwise specified. In the case of single mean comparisons, data were analyzed by two-tailed unpaired t tests or Mann-Whitney tests appropriate to data distributions. In case of multiple comparisons, data were analyzed by one- or two-way ANOVA with post-hoc Bonferroni multiple comparisons using GraphPad Prism Software v 5.0a (GraphPad).
Results
Moderate cFPI in mice induced a transient behavioral suppression of the righting reflex in all injured animals of 6.3 ± 0.3 min that was significantly longer than that of 1.2 ± 0.03 min for sham-injured animals (p < 0.001). No significant difference in righting reflex suppression was noted between injury groups (p < 0.001), indicating that all groups received injuries of comparable severity. No hindlimb seizures were noted in any animals, although a transient apnea was observed in several animals after injury. This apneic episode was of short duration (<1 min) and resolved spontaneously. Of 54 injured mice used in the study, 49 survived the fluid percussion injury, resulting in a mortality rate of 9.9%, consistent with previous mortality rates. 52
Mice were assigned randomly to receive either LKE (100 mg/kg/d) or control diets. For the first 3 days after cFPI or sham surgery, mice received injections of LKE (LKE-cFPI) or saline vehicle (sham or cFPI mice) while being maintained on the control diet. From day 4 onward, injections were not necessary because food intake had returned to normal and LKE treatment could be delivered orally in the diet.
LKE is detected in plasma and cortex
During the course of the study, mice showed no aversion or preference to the LKE-formulated diets, confirming our previous observations in mouse models of AD (3 × Tg-AD mice) administered 1000 ppm LKE in diet during prolonged treatments (up to 11 months). The present data show no significant changes in daily food intake or body weight in C57BL/6 mice fed LKE modified or control diets ad libitum for 2 weeks (Table 1).
LKE, lanthionine ketimine ethyl ester; N.D., not detected.
Data presented as mean ± standard error of the mean for six animals per group. Changes in body weight were measured between days 1–15 of exposure to LKE in diet.
The plasma concentration of LKE was in the range of 2.9–3.8 mg/mL in mice fed 1 ppm LKE and 2.9–5.1 mg/mL in mice fed 1000 ppm LKE. These values correspond to mean plasma concentrations of 16.3 mM and 18.9 mM LKE, respectively. (Table 1).
Bulk brain concentrations of LKE correlate with plasma concentrations. Mice receiving 1000 ppm LKE in diet had approximately 11% greater brain LKE concentration than did mice receiving 1 ppm of the drug in diet. Mean brain/blood concentration ratios were 1.31 and 1.26 for 1 ppm and 1000 ppm dosage groups, respectively.
Cognitive dysfunction after cFPI is prevented by LKE
cFPI at this moderate intensity does not usually result in motor impairment. Mice were assessed at day 3 post-surgery for hindlimb flexion, gait, and motor coordination as described in Methods. No impairment in any of these measures was noted. At 15 days, all mice were capable of swimming and climbing up onto a visible platform protruding 1 cm above the water (Fig. 1A). At 28 days, all mice were ambulatory, walking normally, and exploring the Barnes maze with no differences in speed between the treatment groups (Fig. 1E).
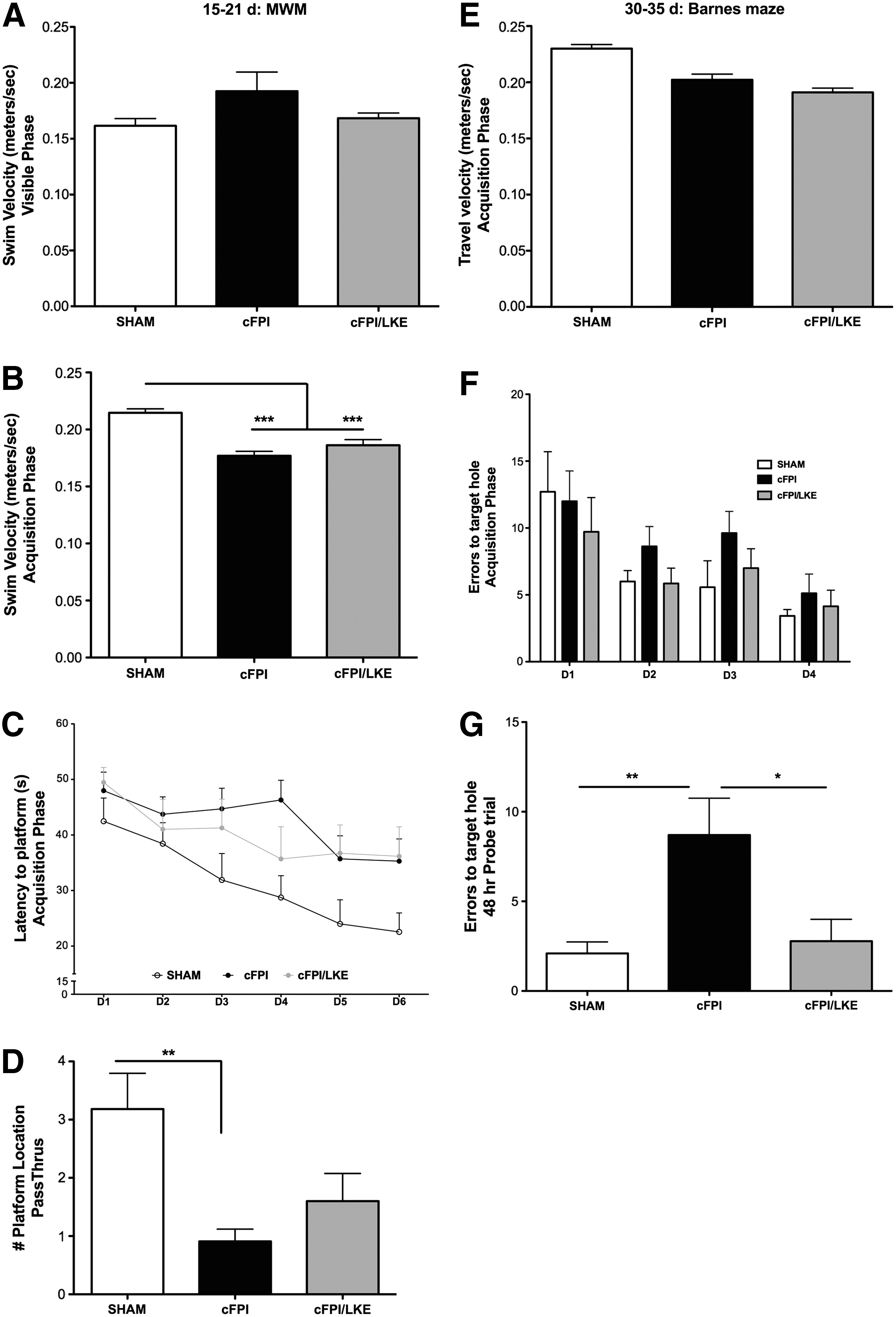
Spatial learning and memory retention. Morris water maze (MWM) and Barnes maze testing were used to longitudinally examine learning and memory at 15–21 days (
At 15–21 days after sham or cFPI surgery, a MWM analysis for learning and memory retention was conducted. Repeated measures ANOVA showed a statistical effect of treatment (F2, 160 = 4.88, P = 0.0141, Fig. 4A) and days of training (F5, 160 = 7.54, P < 0.0001, Fig. 4A) on latency to find the platform (Fig. 1C). On day 4, cFPI mice performed significantly worse than sham mice (*p < 0.05, Bonferroni post hoc analysis). By days 5–6, however, there was no significant difference in learning between any of the groups.
One-way ANOVA of the 48 h retention probe trial revealed a significant treatment effect on platform location crossings (F2, 28 = 6.4549, p = 0.0048). The cFPI group did not have good retention of the platform location after a 48 h probe trial and performed significantly worse than the sham-injured mice (**p < 0.01; Fig. 1D). The cFPI/LKE group performed better than the cFPI group on the probe trial but not as well as the sham-injured group.
At 30–35 days after sham or cFPI, a Barnes maze analysis for learning and memory retention was conducted
One-way ANOVA of the 48 h retention probe trial revealed a significant treatment effect on the number of errors to reach the target hole (F2, 28 = 6.455, p = 0.0053). LKE treatment significantly improved spatial memory retention compared with control cFPI mice. cFPI performance on the probe trial was significantly different from both sham mice (p < 0.01) and cFPI/LKE mice (p < 0.05).
cFPI results in long-term myelin changes
Myelin changes within the corpus callosum were investigated with LFB staining at 5 weeks post-surgery. In sham-injured mice, myelin was seen to be organized in a continuous and regular pattern with only minor alterations in intensity (Fig. 2A). Within the corpus callosum of cFPI mice, areas with reduced LFB staining could be observed. In addition, myelin stained with LFB commonly showed a disorganized structure (Fig. 2A, center panel). LKE treatment of cFPI mice prevented the long-term loss of LFB staining. We quantified the LFB staining with densitometry (Fig. 2B) and, compared with sham-injured mice, there was a decreased amount of LFB staining in the corpus callosum in the cFPI group, and the decrease was prevented by LKE (one-way ANOVA, p = 0.0012, **p < 0.001 vs. sham and cFPI/LKE).
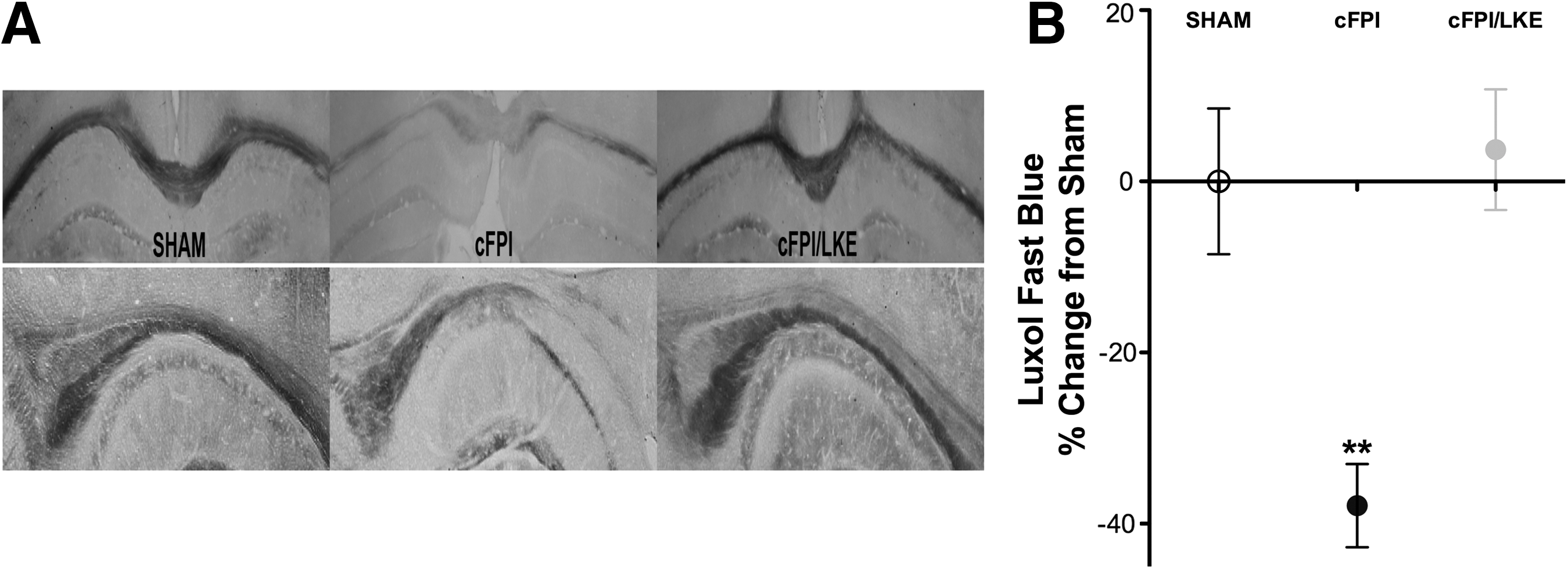
Luxol fast blue (LFB) stained sections 5 weeks after sham or central fluid percussion injury (cFPI). (
Persistent phospho-cJun expression after cFPI is reduced by LKE
cJun is a transcription factor and final messenger for the cJun N-terminal kinase (JNK) pathway whose activation correlates with degeneration. 60 Specifically, phosphorylation of cJun at Serine 63 (pcJun) has been shown to correlate with persistent atrophic changes in a mouse cFPI model. 52 The phosphorylation of cJun is mediated by JNK, and JNK has been shown to suppress autophagy. 61 After cFPI, pcJun immunoreactivity was observed throughout the hippocampal pyramidal cell layer, whereas there was no expression in sham-injured mice (Fig. 3). We found diffuse labeling of cells within the CA1 pyramidal cell layer in cFPI mice that was not seen in sham-injured mice and was reduced with LKE treatment (Fig. 3B) (one-way ANOVA, p = 0.0132, **p < 0.05 vs. sham and cFPI/LKE).

Representative sections and quantification of phosphorylated-cJun (pcJun) in the hippocampus 5 weeks after sham or central fluid percussion injury (cFPI). (
We also found abundant staining for pcJun in the corpus callosum of cFPI mice indicating expression of pcJun in cells other than neurons. Although pcJun is often studied in relation to neuronal degeneration, it is also found in other cell types such as glia. 62 This staining was not found in sham-injured mice and was reduced in cFPI mice treated with LKE.
cFPI results in a decrease in phosphorylated neurofilament H (NF-H) immunoreactivity
SMI-31 antibody targets select epitopes on the 170-200 kDa neurofilament subunits and reacts extensively with phosphorylated NF-H and to a lesser extent NF-M. A decrease in SMI-31 immunoreactivity indicates axonal damage. 63 cFPI resulted in a persistent loss of SMI-31 ir in the corpus callosum compared with sham-injured mice (Fig. 4) (one-way ANOVA, p = 0.037, **p < 0.05 vs. sham and cFPI/LKE). SMI-31 staining returned to sham levels in cFPI mice treated with LKE. Staining density was quantified and is shown in Figure 4B.

Representative sections and quantification of phosphorylated neurofilament (SMI-31) in the corpus callosum 5 weeks after sham or central fluid percussion injury (cFPI). (
C-tau and CRMP2 cleavage after cFPI
MAP-tau is a cytoskeletal protein demonstrated to be cleaved after both human and rodent TBI, 59,64,65 and c-tau is thought to be an indicator of the neuronal damage occurring after TBI. Consistent with previous reports, cFPI induced a significant increase in c-tau levels in the hippocampus 3 days after injury compared with sham-injured mice (Fig. 5). LKE reduced c-tau levels by approximately 40% from cFPI levels (one-way ANOVA, p = 0.009, ***p < 0.0001 vs. sham and *p < 0.05 vs. cFPI/LKE).
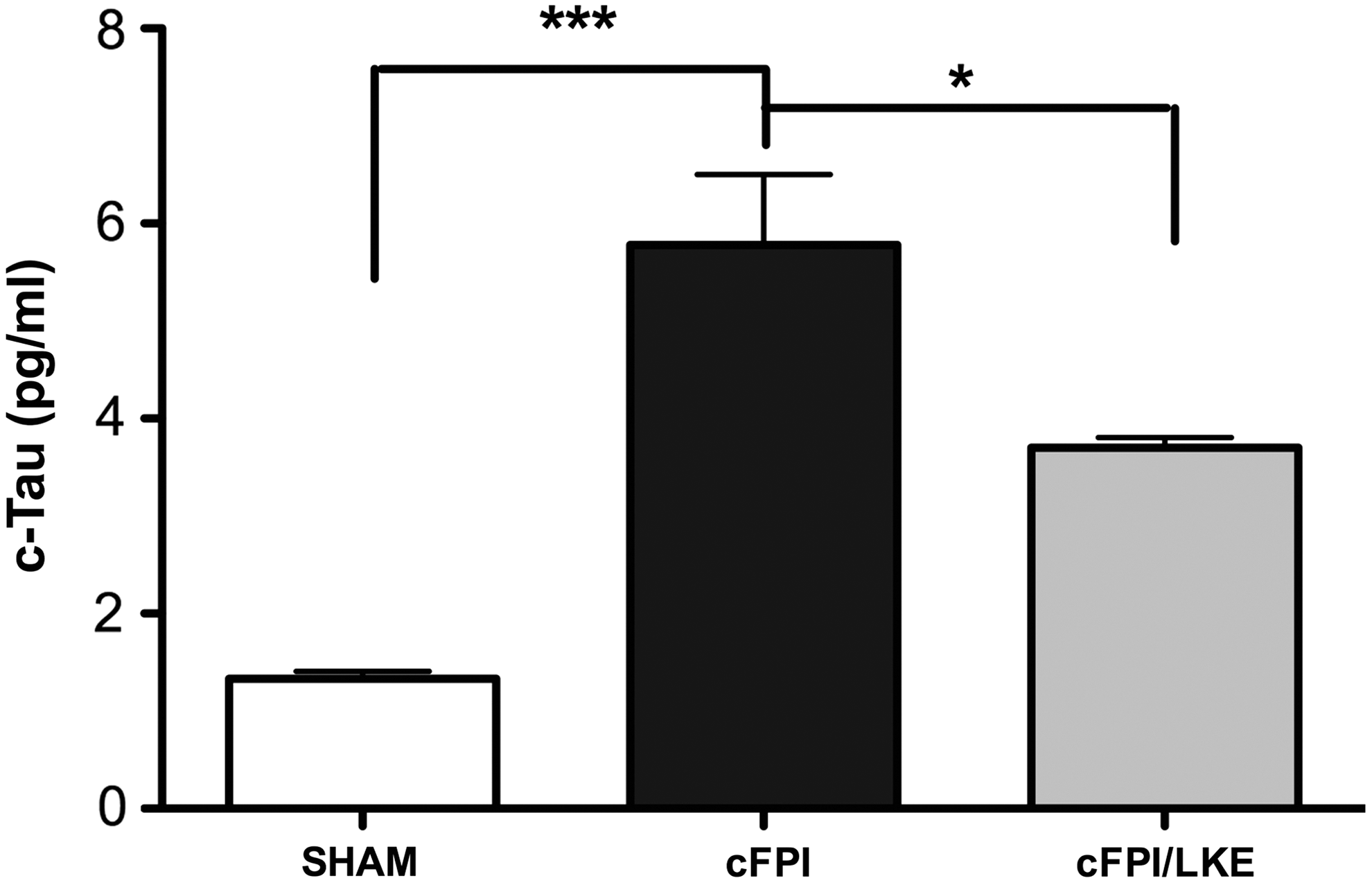
Cleaved tau (cTau) was determined in hippocampal extracts by enzyme-linked immunosorbent assay 3 days after sham or central fluid percussion injury (cFPI). Data show a four-fold increase in cTau after cFPI and a 35% reduction. N = 3/group. ***p < 0.001, *p < 0.05. LKE, lanthionine ketimine ethyl ester.
The MAP CRMP2 is a 62–66 kDa protein that is cleaved to a 55–58 kDa breakdown product (BDP). In the first 48 h after TBI in rodents, increased generation of the 55 kDa BDP has been reported.
66,67
We also find a decreased level of intact 62 kDa protein with an increased ratio of 55 kDa/62 kDa protein 1 day after cFPI (Supplementary Fig. 1; see online supplementary material at
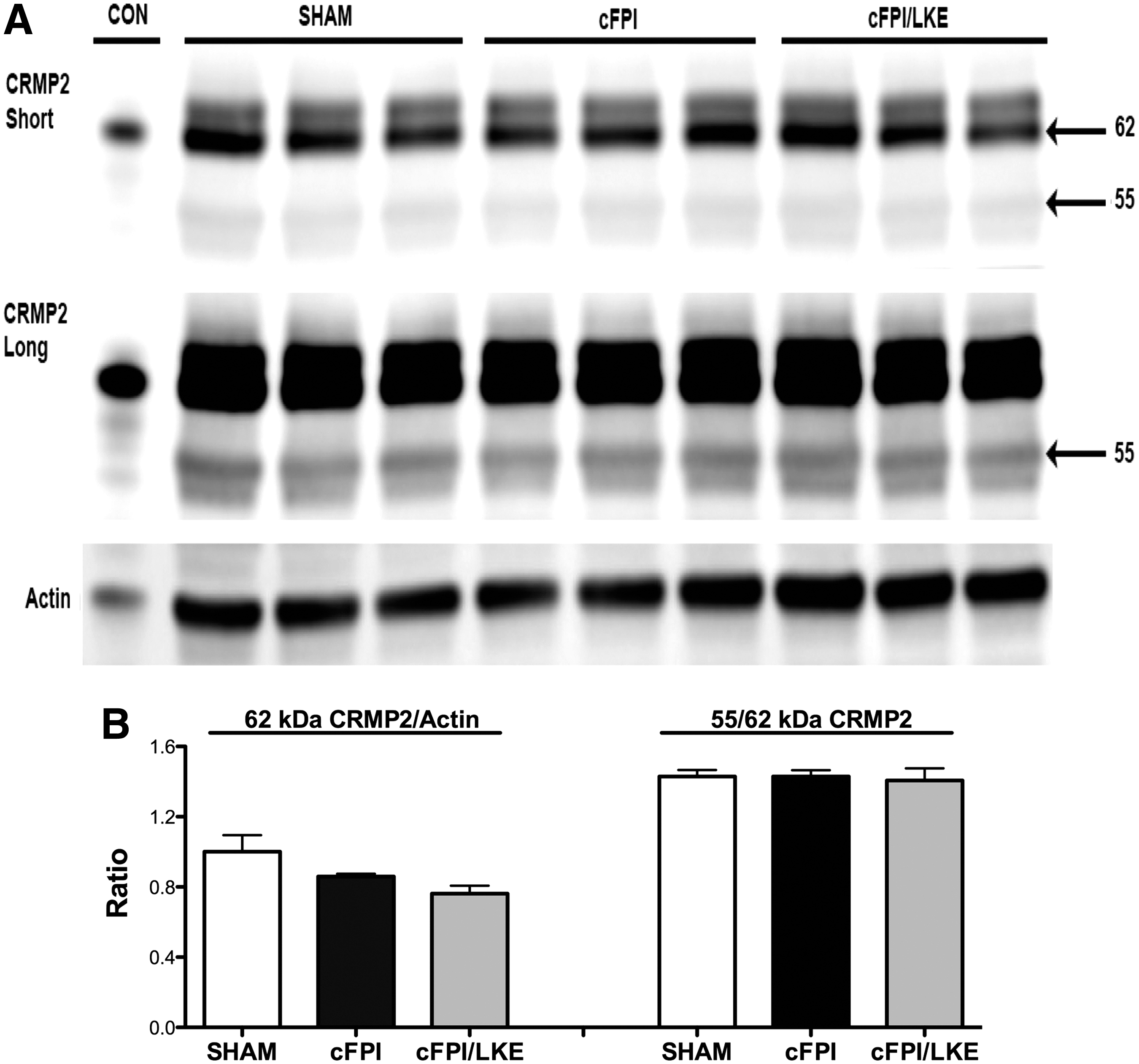
Western blot analysis of collapsin response mediator protein (CRMP2) from cortical extracts 3 days after sham of central fluid percussion injury (cFPI). Both the 62 kDa and 55 kDa bands were resolved by Western blot (
LKE modulates autophagy indicators after cFPI
Autophagy is a complex recycling process involving many different proteins, and primary among the regulatory proteins are the mammalian target of rapamycin complex-1 (mTORC1) and unc-51-like kinase-1 (ULK1). 68 –70 Previously published work has demonstrated impairment of autophagy after TBI, and we sought to determine if cFPI impairs upstream autophagy regulation. At 3 days after cFPI, phosphorylated mTor (p2448; p-mTORC1), total mTor (mTORC1), phosphorylated ULK1 (p-ULK1; p757), and total ULK1 (ULK1) as well as beclin1 protein levels were analyzed by Western blot. Compared with sham-injured mice, cFPI did not significantly alter individual p-mTor or mTor protein levels (Fig. 7). LKE treatment of cFPI mice decreased p-mTorC1 and increased mTORC1 protein resulting in a decrease in the p-mTORC1/mTORC1 ratio that would favor autophagy. 71

Western blot analysis of rapamycin complex-1 (mTORC1), unc-51-like kinase-1 (ULK1), and beclin1 from cortical extracts 3 days after sham or central fluid percussion injury (cFPI). Staining for actin was used as a loading control. N = 6/group. **p < 0.01, *p < 0.05. LKE, lanthionine ketimine ethyl ester.
The mammalian orthologue of yeast Atg1, the serine/threonine kinase ULK1, plays a key role in autophagy induction. Under normal conditions, the ULK1 complex is deactivated by mTorC1 kinase-dependent phosphorylation of ULK1, and inhibition of mTORC1 enhances ULK1 kinase activity by a reduction in phosphorylation of ULK1. 72 cFPI did not significantly change p-ULK1 or t-ULK1 proteins levels compared with sham-injury, but LKE treatment of cFPI mice reduced p-ULK1 while increasing t-ULK1 resulting in a >50% decrease in the p-ULK1/t-ULK1 ratio that would strongly favor autophagy stimulation. The ULK1 complex promotes downstream autophagy through the beclin-1/VPS34/AMBRA1 complex, which recruits downstream effectors to the site where nucleation occurs. Examination of beclin1 protein levels did not show a significant change after cFPI or with LKE treatment relative to sham-injured mice (Fig. 7C).
A common method to monitor procession of autophagy is to measure microtubule associated protein light chain 3 (LC3) by immunoblot—specifically, the conversion of LC3-I to LC3-II. 73 The basic principle is that the amount of LC3-II conversion is correlated with the number of autophagosomes formed. LC3-II itself, however, is degraded by autophagy, making interpretation of the results of LC3 immunoblotting a challenge. In addition, the amount of LC3 at any given time does not indicate autophagic flux, making it necessary to measure the amount of LC3-II delivered to lysosomes by comparing LC3-II levels in the presence and absence of a lysosomal protease inhibitor such as chloroquine.
Chloroquine is a lysosomotropic agent that prevents endosomal acidification, and it accumulates within endosomes and lysosomes. This accumulation leads to inhibition of lysosomal enzymes that require an acidic pH and prevents fusion of endosomes and lysosomes. Therefore, chloroquine inhibits autophagy because it raises the lysosomal pH, leading to inhibition of both fusion of autophagosome with lysosome and lysosomal protein degradation.
Using tissue from the 5-week post-injury time point, we evaluated LC3 by immunoblot and quantified it by densitometry (Fig. 8). There was a significant reduction in LC3-II (Fig. 8, right graph; one-way ANOVA, p = 0.0002, ***p < 0.0001, **p < 0.01, *p < 0.05) in both the cFPI (25%) and cFPI/LKE groups (50%). There was also a significant reduction in LC3-I in the cFPI/LKE mice versus sham-injured mice (Fig. 8, left graph; one-way ANOVA, p = 0.0107, *p < 0.05 sham vs. cFPI/LKE).
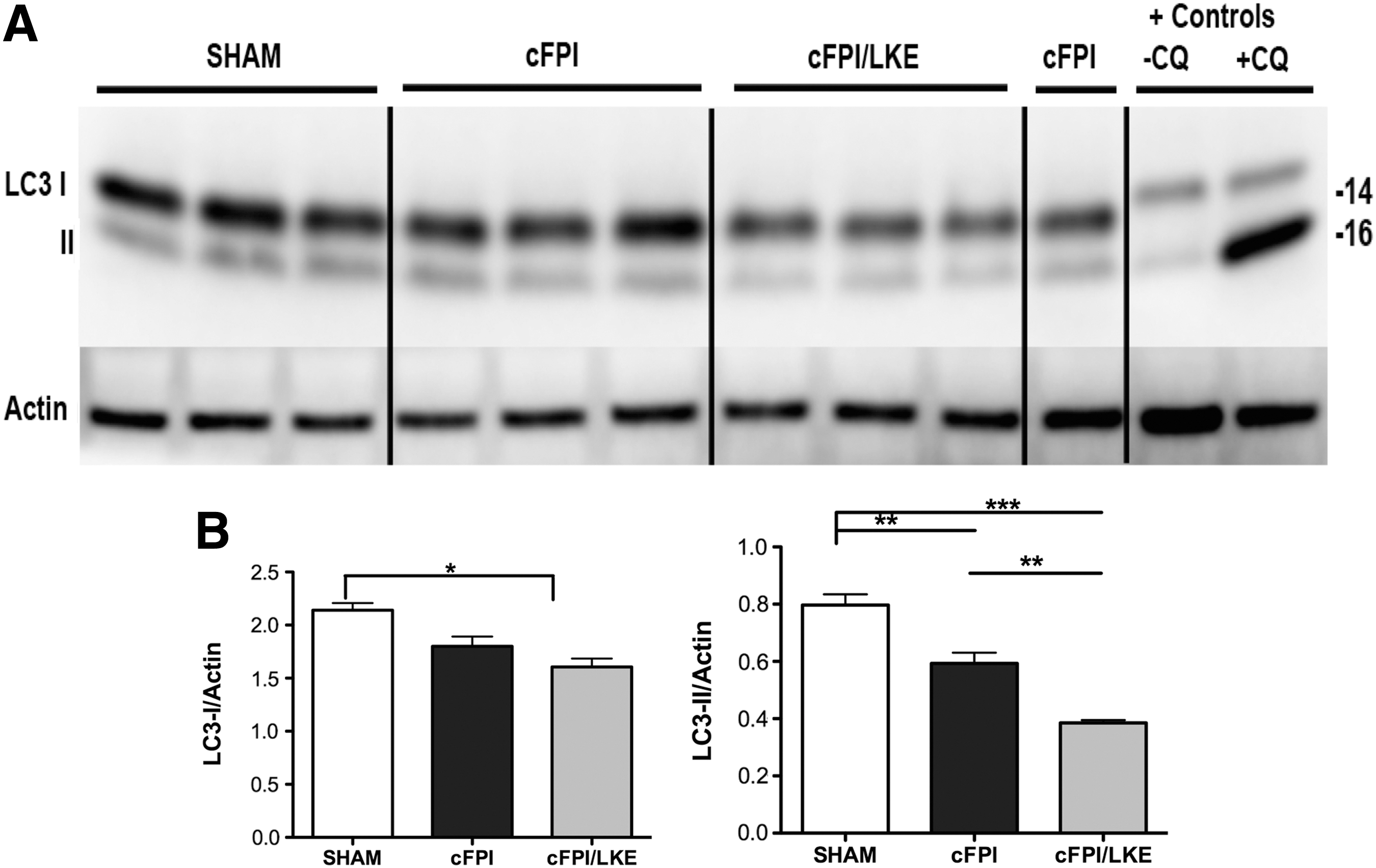
Western blot analysis of microtubule-associated protein light chain 3 (LC3) from cortical extracts 5 weeks after sham or central fluid percussion injury (cFPI). Both the 16 kDa (LC3-I) and 14 kDa (LC3-II) bands were resolved by Western blot (
Decreases in LC3-II can indicate either reduced autophagosome production or increased autophagosome clearance. To clarify, we performed a 3-day study using chloroquine in sham, cFPI, and cFPI/LKE mice. At 3 days after injury, mice were injected with chloroquine 4 h before tissue collection, and LC3 protein was determined by immunoblot and densitometry (Fig. 9). Similar to the 5-week time point, LC3-II was greatly reduced compared with sham- and cFPI-injured mice. Consistent with our previous in vitro studies demonstrating that LKE increases autophagy flux, 47 chloroquine treatment in cFPI/LKE mice resulted in a doubling of the LC3-II levels (*p = 0.05) whereas sham-injured and cFPI mice had only small increase with chloroquine treatment.
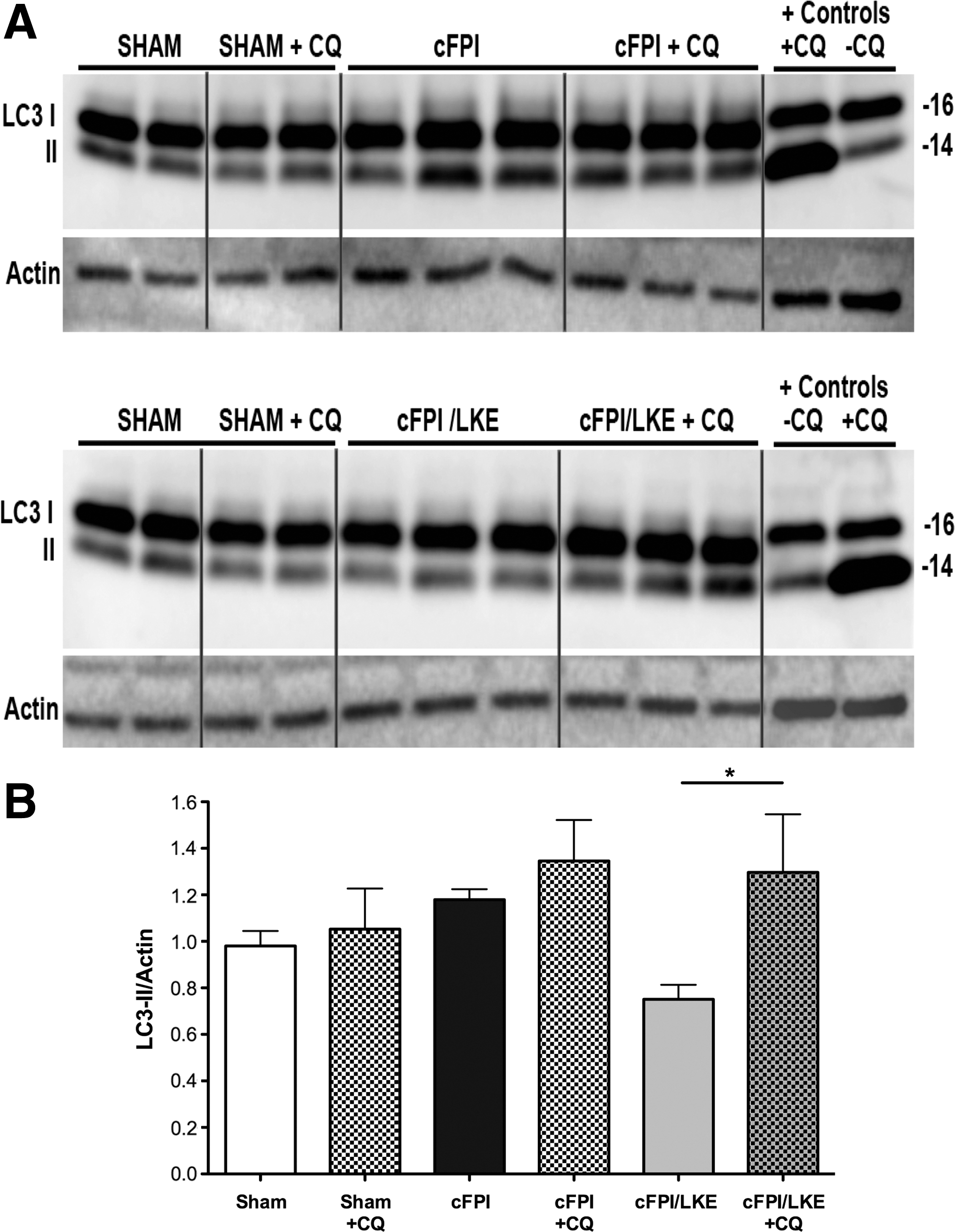
The use of chloroquine (CQ) and Western blot to determine changes in autophagy flux 3 days after sham or central fluid percussion injury (cFPI). On the third day after surgery, mice were given an inraperitoneal injection of chloroquine (CQ). Four hours after the chloroquine injection, tissue was collected for microtubule-associated protein light chain 3 (LC3) analysis. Both the 16 kDa (LC3-I) and 14 kDa (LC3-II) bands were resolved by Western blot (
Discussion
cFPI in the mouse is a diffuse axonal injury model characterized by axotomy with long-lasting neuronal atrophy 52 and behavioral derangements. 74 In the current study, we report persistent behavioral deficits, markers of neuronal atrophy, myelin loss, and autophagy alteration after cFPI. The work of Greer and associates 52 demonstrates that although neurons can display chronic signs of atrophy, there is also evidence for an enduring regenerative response after cFPI. This suggests the opportunity for pharmacological intervention to enhance recovery in DAI models. To this purpose, we have tested the novel therapeutic, lanthionine ketimine ethyl ester, LKE, delivered 30 min post-cFPI, and demonstrate significant improvement in both behavior and neuropathology.
Lanthionine ketimine (LK) is a natural sulfur amino acid metabolite formed through transulfuration pathway reactions 75 and originally discovered in 1983. 76 The ester derivative of LK, LKE, has superior cell permeability and possesses potent neurochemical functionality both in vitro and in vivo. 48,49,51,77 Protein binding studies indicate that lanthionine ketimine binds a number of proteins involved in key cellular functions, although precise mechanisms of action are still being determined. These binding proteins include beta-tubulin, beta-actin, myelin basic protein (MBP), myelin proteolipid protein (PLP), lanthionine synthetase-like protein-1 (LanCl1), and CRMP2. 49
The family of CRMPs plays a significant physiological role in neuron cell body and axon stability within the CNS. CRMPs are a family of neuronal phosphoproteins that regulate microtubule assembly. All CRMPs are expressed intracellularly at high levels during development in the CNS, but it is CRMP2 that remains at high levels in the adult mammalian CNS within neurons and specific oligodendrocytes. 22 CRMP2 is a MAP, phosphorylated by Cdk-5 and GSK3β enzymes. 23 Normally, CRMP2 stabilizes microtubules by binding tubulin 24 and facilitates protein trafficking by adapting kinesin-1 to protein cargo packages including neurotrophin receptors 25 and actin nucleation factors important for maintenance of synapse stability. 26
Studies have demonstrated that aberrant phosphorylation or cleavage of CRMP2 is detrimental to axon integrity and neuron survival. 27, 28 Alterations in CRMP2 and CRMP4 protein patterns have been shown to be associated with the process of “protrusion-like” bead formation, a sign of axonal and neuronal degeneration. 29 Changes in CRMP2 expression, phosphorylation, or cleavage may have an adverse effect on its ability to bind cytoskeletal proteins and degrade the integrity of the axon.
After TBI, CRMP2 is truncated to 55–58 KDa breakdown product by increased calpain activity. 67 A reduction in intact CRMP2 protein could impair critical cellular functions such as autophagy and axon transport. In the current study, we found a temporary increase in the proteolysis of CRMP2 at 1 day after cFPI with recovery by 3 days, consistent with previous studies. What is clear from the data presented is that LKE treatment does not increase the expression of intact 62 kDa CRMP2 or alter the ratio of 55/62 kDa CRMP2, making this unlikely to be the mechanism by which LKE improves cFPI outcome. Recent in vitro work from our group shows that LKE can restore autophagy flux in SH-SY5Y cell despite a challenge of partial CRMP2 knockdown and suggesting that LKE may still function after cFPI even while intact CRMP2 levels are temporarily decreased. 47
Given the central role of CRMP2 in axon stabilization and transport and growth cone dynamics, functional modulators to enhance CRMP2 activity are a logical choice to promote neuron repair and regrowth after TBI. LKE has a growing body of literature to support its activity as a CRMP2 enhancing compound. Recent work studying Unc-33, a homolog of mammalian CRMP2, demonstrates LKE's remarkable effects. Mutations of the unc-33 gene in nematodes reduce Unc-33 function and cause impaired locomotion and axogenesis resulting in failure and early termination of dorsoventral commissures before reaching the dorsal body wall muscle cells. 78 –80
Neuroimaging studies of unc-33 mutants demonstrate that LKE promoted neurite elongation, decreasing the number of dorsoventral commissures terminating before reaching the dorsal nerve cord. 51 Importantly, unc-33 null mutants, which do not express any Unc-33 isoforms, did not rescue the ventral nerve cord gaps, and the gap size was not significantly changed by the LKE treatment, highlighting the central role of Unc-33/CRMP2 in this process. Although we did not directly measure axonal injury, axon regrowth or growth cone dynamics in the current study, this is an important focus of ongoing TBI work in our laboratory.
Autophagy is a process for bulk cytoplasmic protein and organelle degradation that is characterized by double membrane vesicles that trap cargo and target it to lysosomes for degradation. Autophagy is critical for neuron survival, 81 and impairment of the autophagy system can be catastrophic for neurons attempting recovery. Evidence for dysregulation of autophagy after TBI has been highlighted in many studies beginning in 2005, 82 with some disagreement over the last 10 years on whether autophagy is harmful or beneficial. 44 –46
A variety of factors may contribute to this discrepancy including different TBI models and severity, methods used to analyze autophagy, time course of analysis, and the interpretation of these results. Recent work by Sarkar and colleagues, 83 however, shows that autophagy is stimulated but that flux is impaired as long as 7 days after controlled cortical impact and that this flux impairment is because of lysosomal dysfunction. Results of our upstream autophagy indices demonstrate a lackluster autophagic response to cFPI with no significant change in the ratios of phospho/total mTORC1 or phospho/total ULK1. Further, persistent pcJun expression after cFPI suggests increased JNK activity that may result in the suppression of autophagy. 61
Rapamycin, an immunosuppressive drug that induces autophagy via inhibition of mTOR, can improve outcome after TBI, 84,85 although clinical use of rapamycin is plagued by a long list of serious side effects that limit use, particularly for patients with brain injury. 86 We have recently demonstrated that LKE enhances autophagy flux in vitro suggesting that there may be endogenous regulators of autophagy of which LK may be one example.
LKE appears to operate in a manner that is functionally analogous to rapamycin but through a distinct mechanism that affects mTORC1 localization at the lysosome. 47 Engineered knockdown of CRMP2 using shRNA diminishes autophagy flux and LKE increased LC3-II in CRMP2-knockdown cells both with and without bafilomycin cotreatment, indicating increased autophagy flux induced by LKE even in the presence of diminished CRMP2. Our data indicate that intact 62 kDa CRMP2 is reduced at days 1 and 3 post-cFPI, yet LKE is able to stimulate autophagic flux despite this reduction. Interestingly, although we initially saw both upstream (ULK-1) and downstream (LC3-II) enhancement of autophagy by LKE, only the downstream effect remained long term.
The reduction in ULK-1 phosphorylation was transient and was not evident at 7 days (data not shown), but modulation of LC3-II levels was evident at both 3 days and 5 weeks. This suggests possible context-driven effects of LKE action or feedback control that prevents aggressive autophagy drive, an important point given that chronic overstimulation of autophagy can be deleterious. Further work is needed to determine the precise points of autophagy modulation by LKE.
TBI is established as a risk factor for development of neurodegenerative disease and dementia. 87 It is becoming evident that TBI is a chronic and progressive disease process with evolving pathological features. 88 Indeed, there are similarities between TBI and AD pathology that may partially explain the relationship. Autophagy dysfunction is a significant pathology common to both AD and TBI. More research is needed to determine the nature of long-term impairment of autophagy and proteostasis after TBI. Drugs like LKE, demonstrated to be safe for long-term delivery in a mouse model of AD 48 and to substantially reduce AD pathology and behavioral impairments, should also be considered for treatment of TBI to prevent neurodegenerative risk.
Conclusion
The LK derivative, LKE, is a potent neuroprotective agent in the mouse cFPI model and improves long-term behavioral outcome and neuropathology. We propose that LKE modulates late-stage autophagy to correct a cFPI-induced blockage consistent with previous work. 89 Future work will address the role of other LKE-binding proteins and how those interactions may benefit TBI recovery.
Footnotes
Author Disclosure Statement
P. Eslami, M.Johnson, A. Poteshkina, M. Harris-White, K.M. Venkova-Hristova, A.M. Hristov declare that no competing financial interests exist. S.P. Gabbita is an employee of P2D Bioscience, Inc., a company engaged in drug development but not involved in development of LKE. K. Hensley is the inventor on U.S. patent 7,683,055 covering composition and use of lanthionine ketimine derivatives including LKE and is cofounder of a company engaged in commercial development of the technology.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.