
Abstract
Reactions of both astrocytes and microglia to central nervous system injury can be beneficial or detrimental to recovery. To gain insights into the functional importance of gliosis, we developed a new model of adolescent closed-head injury (CHI) and interrogated the behavioral, physiological, and cellular outcomes after a concussive CHI in leukemia inhibitory factor (LIF) haplodeficient mice. These mice were chosen because LIF is important for astrocyte and microglial activation. Behaviorally, the LIF haplodeficient animals were equally impaired 4 h after the injury, but in the subsequent 2 weeks, the LIF haplodeficient mice acquired more severe motor and sensory deficits, compared with wild type mice. The prolonged accumulation of neurological impairment was accompanied by desynchronization of the gliotic response, increased cell death, axonal degeneration, diminished callosal compound action potential, and hypomyelination. Our results clearly show that LIF is an essential injury-induced cytokine that is required to prevent the propagation of secondary neurodegeneration.
Introduction
P
Gliosis, or the glial responses to injury, are neither unitary nor stereotyped, and it is well accepted that the reactions of both astrocytes and microglia can be either beneficial or detrimental to recovery. A prevailing view is that once activated, astrocytes and microglial cells release an array of soluble mediators that may directly cause cell damage by increasing intracellular calcium or inducing oxidative stress, resulting in a wave of secondary cell death. 3,4 Further, signals produced by reactive glial cells recruit immune cells from the circulation that can further exacerbate the extent of injury. 3,5,6 However, reactive astrocytes also produce a variety of mediators that can reduce the extent of damage after injury. 7 Indeed, neuroprotective pre-conditioning paradigms, which have been shown to activate astrocytes, significantly preserve neurons, as well as higher order function. 8
Leukemia inhibitory factor (LIF) is rapidly induced in astrocytes in response to central nervous system injury and broadly affects neighboring cells. Its expression increases after cerebral ischemia, spinal cord injury, stroke, multiple sclerosis, Alzheimer's disease, Parkinson's disease, and seizure. 9 In studies of spinal cord injury, LIF gain of function has been shown to dramatically increase microglial cell proliferation and activation resulting in neuronal injury. 10 However, other studies have shown that LIF can reduce injury progression and promotes remyelination. 11,12 These studies establish LIF as an important initiator of both microglial and astroglial reactivity to injury, but they do not establish whether LIF might coordinate a neuroprotective or neurodegenerative program after mild TBI. Therefore, to glean new insights into mechanisms of both injury and recovery after closed-head injury (CHI), we developed a new model of pediatric brain injury and compared the extent of injury and functional recovery in wild type (WT) mice and in mice heterozygous (Het) for LIF. Our results clearly show that LIF is an essential injury-induced cytokine that elicits a neuroprotective program and resolution of secondary neurodegeneration.
Methods
Reagents
Common laboratory chemicals were purchased from either Sigma (St. Louis, MO) or VWR (Radnor, PA). The following antibodies were used: rat anti-GF2.2 (1/5, supernatant); rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1; Wako Chemicals, Richmond, VA;
CHI
All experiments were performed in accordance with protocols approved by the institutional animal care and use committee of Rutgers-New Jersey Medical School. The LIF null mice were provided by Dr. Douglas Fields with permission from Dr. Colin Stewart and were maintained on a CD1 background. CHI was performed on post-natal Day 18 (P18) on LIF heterozygous (LIF Het) and CD1 wild-type male and female mice based on a previously published protocol developed by Creed and colleagues. 13 Animals were initially anesthetized with a mixture of isoflurane and oxygen (5% induction, 3% maintenance). Once fully anesthetized, the scalp was cleansed and an incision along the midline was created to expose the skull. A sterile 5 mm rounded hemispheric metal impactor tip attached to a pneumatic piston was centered directly over the sagittal suture halfway between bregma and lambda and was brought down on the surface of the skull at a velocity of 5 m/sec, with a depth of 1.0 mm past zero point on the skull surface and a dwell time of 150 msec. The amount of time each mouse stopped breathing after impact (apnea) and the time taken by the mouse to right itself after being placed in a supine position (righting reflex) were evaluated to ensure that the mice were injured.
LIF real-time polymerase chain reaction
Tissue samples were snap frozen in Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA) and total RNA was extracted using the RNAeasy Mini protocol (Qiagen). Complementary DNA (cDNA) was generated using Superscript III reverse transcriptase (Invitrogen), according to manufacturer's instructions. A total of 100 ng of cDNA was used in each real-time polymerase chain reaction (qPCR) reaction well using an ABI Prism 7300 Sequence Detection System (Applied Biosystems, Grand Island, NY), QuantiTect LIF primers, and SYBR green for detection. As an internal standard, we used the Quantum RNA18S (Applied Biosystems) in a 4:6 primer:competimer reaction mix. Relative quantification was obtained using the ΔΔCT method.
Neurological severity score and elevated balance beams
Post-traumatic neurological impairments were assessed using a 12-point neurological severity score (NSS) paradigm adapted from tests previously used in experimental models of mouse TBI. 14 –16 Each task and criteria for passing are summarized in Table 1. Nonparametric data are presented as a composite score ranging from 1 to 12 representing performance on all tasks combined. High final NSS scores were indicative of task failures and interpreted as neurological impairment. Mice also were tested on 1 cm and 0.7 cm wide beams positioned at a 30° incline at 14 days post-injury (DPI). The time taken to traverse each beam and the number of foot slips off the beam were quantified over three trials and averaged.
Summary of the motor, balance, sensory, exploratory and reflex tests that go into the overall composite Modified Neurological Severity Score. Successful completion of each task results in a “0” score while failure results in a “1” score. Scores for each task are added to create a total composite score out of 12.
Glial fibrillary acidic protein (GFAP), Iba1, SMI-32, Rip, MBP, and Olig2 immunofluorescence
Immunofluorescence was performed on 40 μm frozen paraformaldehyde fixed sections as previously described 17 and sections were counterstained with 1 μg/mL DAPI for 5-10 min. Slides were cover-slipped with Fluoro-Gel. Sections were analyzed using an Olympus AX70 fluorescence microscope equipped with a Q-imaging CCD camera (Surrey, BC, Canada) and IP Lab imaging software (Biovision, Exton, PA) All secondary antibody combinations were carefully examined to ensure that there was no bleed through between fluorescent dyes or cross-reactivity between secondary antibodies. No signal above background was obtained when the primary antibodies were replaced with pre-immune sera.
In situ end labeling (ISEL)
DNA fragments in apoptotic cells were visualized on 40 μm frozen sections. Briefly, sections were dehydrated and re-hydrated, washed in distilled water and Tris-buffered saline, and incubated for 5 min in a buffer containing 2.5 M Tris, 5 mM MgCl2, 10 mM β-mercaptoethanol and 0.005% bovine serum albumin (Buffer 1). Slides were then rinsed two times for 5 minutes in Buffer 1 with 0.3% Triton X-100 added (Buffer 2) three times for 2 min. Sections were then incubated with 40 U Klenow fragments/mL and 0.01 mM Digoxigenin DNA in Buffer 1 for 1 h at room temperature. Slides were washed for 3 min each, then incubated in blocking solution for 1 h at room temperature. Slides were then incubated with anti-DIG-fluorescein (1:75) for 1 h at 37°C. Slides were washed four times and then cover-slipped with Fluoro-Gel.
Fluoro-Jade C
The Fluoro-Jade C staining protocol was adapted from Schmued and colleagues. 17 Briefly, 20 μm sections were rehydrated in graduated ethanol solutions before washing in distilled water, followed by transfer to 0.06% potassium permanganate solution for 15 min. After washing with distilled water, the sections were placed in a 0.0001% solution of Fluoro-Jade C (Millipore, Watford, U.K.) in 0.1% acetic acid. The sections were washed again in distilled water and dried in an oven at 60° C before clearing in xylenes and cover-slipped using DPX mounting medium.
Iba1, Fluoro-Jade C, ISEL, and Olig2 stereological cell counts
Sections were analyzed using the MicroBrightfield StereoInvestigator program (Williston, VT) interfaced with an Olympus BX51 microscope (Center Valley, PA). The span of the mouse brain directly beneath the area of impact in the coronal plane between bregma and lambda yielded approximately 80 sections. Every sixth section was systematically sampled, yielding 10-15 sections per brain. Under the Microbrightfield Optical Fractionator workflow, randomly-placed counting frames were used to sample co-localized markers within the region of interest at predetermined regular x and y intervals. Selection criteria for counting an object within the sampling frame were implemented using stereological counting rules. Inclusion and exclusion counting criteria were followed by an unbiased observer who recorded counts under 100× oil-immersion magnification only when cells positive for each marker fell within randomly placed 50 × 50 μm counting squares within preset grids in the retrosplenial area of the cortex, the corpus callosum between the lateral ventricles, and the hippocampal dentate gyrus. Completed cell count estimates were averaged and reported per section analyzed.
GFAP, SMI-32, Rip, and MBP fluorescence intensity quantification
Sections were analyzed using Image J software on an Olympus BX51 microscope. Every sixth section directly beneath the area of impact was systematically sampled, yielding 10-15 sections per brain. Integrated optical density (IOD) measurements for GFAP, SMI-32, Rip, and MBP stains were evaluated in the retrosplenial area of the cortex, the corpus callosum between the lateral ventricles and the hippocampal dentate gyrus.
BDA injections and quantification
WT and LIF Het injured and sham-operated control mice were stereotaxically injected with BDA (Sigma-Aldrich, St. Louis, MO) to label callosal axons 7 DPI. Animals were injected with 3 μL of BDA into three sites of the right somatosensory cortex relative to bregma: 1.2 mm lateral, 1.1 anterior; 1.3 mm lateral, 1.3 mm anterior; and 1.4 mm lateral, 1.5 mm anterior. Each injection was performed at 0.7 mm ventral to the brain surface. Mice were sacrificed and perfused with 3% paraformaldehyde 7 days after the injections (14 DPI). Pixel intensity measurements of BDA labeling were conducted in the corpus callosum between the lateral ventricles.
Compound action potentials
Naïve WT and LIF HET mice, as well as CHI WT and CHI Het mice, were assessed for changes in compound action potential (CAP) of the corpus callosum 14 days after CHI. Stimulation (125 micron, Teflon-coated stainless steel; AM Systems, Sequim, WA) and recording (75 micron, Teflon-coated stainless steel) electrodes were placed into the corpus callosum 1 mm rostral to bregma, 1.0 mm lateral to bregma, and 1.5 mm ventral to the brain surface. In vivo recordings were made from ketamine/xylazine anesthetized mice (100× amplification, 0.5 Hz-8 kHz bandpass, digitized at 20 kHz) and stored for offline analyses. Constant current square wave pulses (250 msec duration delivered at 0.1 Hz) were used to evoke CAPs. For analyses of CAP amplitude and latency, standardized input–output functions were generated by varying the intensity of stimulus pulses in steps from approximately threshold level to an asymptotic maximum (0.1–2.0 mA). Analyses were performed on waveforms that were averages of six successive sweeps. The difference from the first positive peak to the first trough was determined as the amplitude of the myelinated fibers (N1), while the difference between the second peak and the second trough was measured for the amplitude of the slower conducting unmyelinated fibers (N2). To examine refractoriness, pairs of pulses were presented for which the inter-pulse interval was increased in 0.5-msec steps from 3 msec to 12 msec using the maximum current level determined from the input–output curve.
Results
The CHI model implemented in these studies did not produce observable signs of brain injury to WT or LIF Het mice (Fig. 1A). However, LIF messenger RNA (mRNA) increased in the cortical brain tissue directly below the area impacted on the skull. LIF mRNA levels were elevated ∼10-fold in neocortical samples from WT mice at 24 h, peaked with a 15-fold increase at 48 h, and then returned to baseline at 72 h after CHI at P18 (Fig. 1B). In LIF Het mice, LIF mRNA was increased five-fold over sham controls at 24 h and eight-fold at 48 h, an ∼50% decrease versus WT LIF mRNA levels, as would be expected. Similar to WT injured animals, LIF mRNA levels in LIF Het mice returned to baseline levels by 72 h post-injury (Fig. 1B).

Wild type (WT) and leukemia inhibitory factor heterozygous (LIF Het) brains do not exhibit any gross signs of deformation or injury, yet LIF messenger RNA (mRNA) is significantly elevated.
LIF haplodeficient mice exhibit more severe and persistent behavioral deficits following pediatric CHI
To evaluate functional parameters of neurological injury we graded behavioral deficits following central nervous system (CNS) injury to produce a Neurological Severity Score (NSS; Table 1). An overall composite score was calculated, with higher scores reflecting higher overall impairment. Baseline evaluations conducted 1 day before injury showed no difference in performance between WT and LIF Het mice. Evaluation of NSS was conducted at 4 h after injury to establish whether each group of animals was affected differently by CHI immediately after injury. WT and LIF Het mice scored similarly on the NSS 4 h post-injury. Mice were then re-tested at 2 DPI, at which time the injured LIF Het mice performed significantly worse than WT injured mice (Fig. 2A). LIF Het mice showed a slight decline in NSS scores at 7 DPI, indicating a gradual recovery of functioning over time. NSS scores were not statistically different between injured LIF Het and WT mice at 7 DPI (Fig. 2A). Surprisingly, the average NSS for LIF Het mice at 14 DPI increased to an average of ∼6, indicating impairment in 50% of the behavioral tasks, while WT injured mice remained at baseline performance levels (Fig. 2A; Supplementary Videos 1 and 2; see online supplementary material at
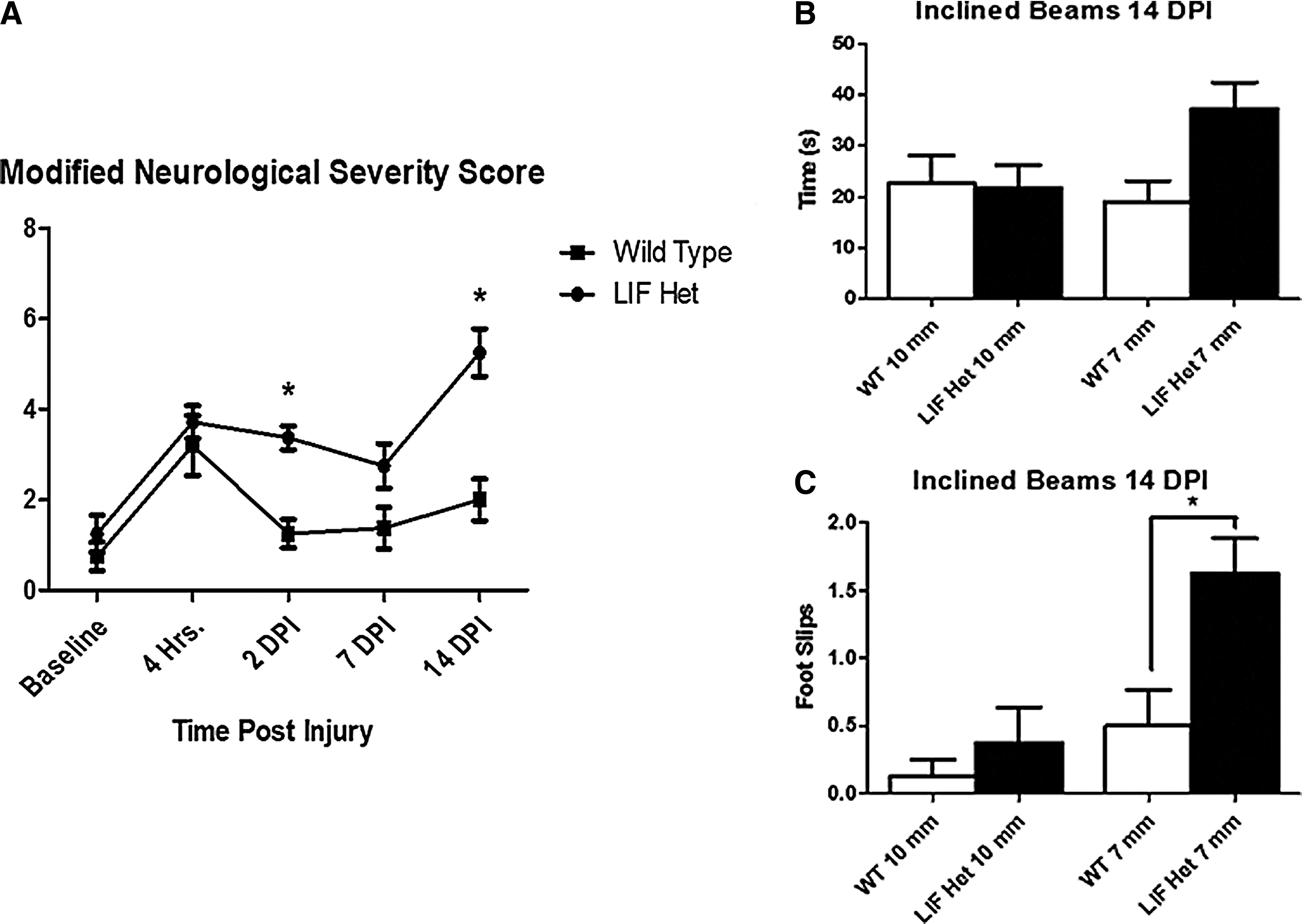
Leukemia inhibitory factor heterozygous (LIF Het) mice have increased behavioral deficits following closed-head injury (CHI). Wild type (WT) and LIF Het mice were injured at post-natal Day 18 (P18).
Decreased performance on the inclined beam has been linked with decreased subcortical white matter integrity after injury
18
; therefore, we next tasked the animals with traversing 1 cm and 0.7 cm wide beams at a 30° incline at 14 DPI. This time-point was chosen because uninjured animals were unable to successfully traverse the inclined beams prior to 21 days of age (unpublished observations). LIF Het mice tended to have longer latencies to traverse the smaller 0.7 cm beam, compared with injured WT mice, but this difference did not reach statistical significance (Fig. 2B). However, injured LIF Het mice displayed a three-fold increase in the number of foot slips off the smallest beam, compared with WT injured animals (Fig. 2C; p < 0.05; Supplementary Videos 3 and 4; see online supplementary material at
Astroglial responses are desynchronized in LIF haplodeficient mice after pediatric CHI
To establish whether the timing of the astroglial response to CHI was altered in the LIF Het mice, animals were sacrificed 2, 7, and 14 DPI, and brains were evaluated for GFAP immunofluorescence. Three brain regions were examined at each time-point from the rostral to caudal extent of the brain area directly ventral to the area of impact on the skull between bregma and lambda: the retrosplenial cortex, the corpus callosum between the lateral ventricles, and the hippocampal dentate gyrus. WT injured brains exhibited a 2.0- to 2.5-fold increase in GFAP immunofluorescence 2 days post-CHI in each region, compared with sham-operated controls (Fig. 3D-F). GFAP immunofluorescence was significantly lower in LIF Het CHI brains than in WT CHI brains at 2 DPI (p < 0.05), remaining at the same levels of WT and LIF Het sham-operated control levels (Fig. 3D-F). At 7 DPI, GFAP expression returned to control levels in all three regions in WT CHI brains, while GFAP staining intensity increased significantly in all brain regions of LIF Het CHI mice, compared with both sham controls and WT CHI 7 DPI brains (Fig. 3D-F). GFAP levels remained significantly increased in LIF Het CHI cortices, compared with WT injured cortex levels 14 DPI (p < 0.05; Fig. 3E). These data indicate that LIF deficiency delays the normal astrogliosis response following pediatric CHI that is followed by an exacerbated response.
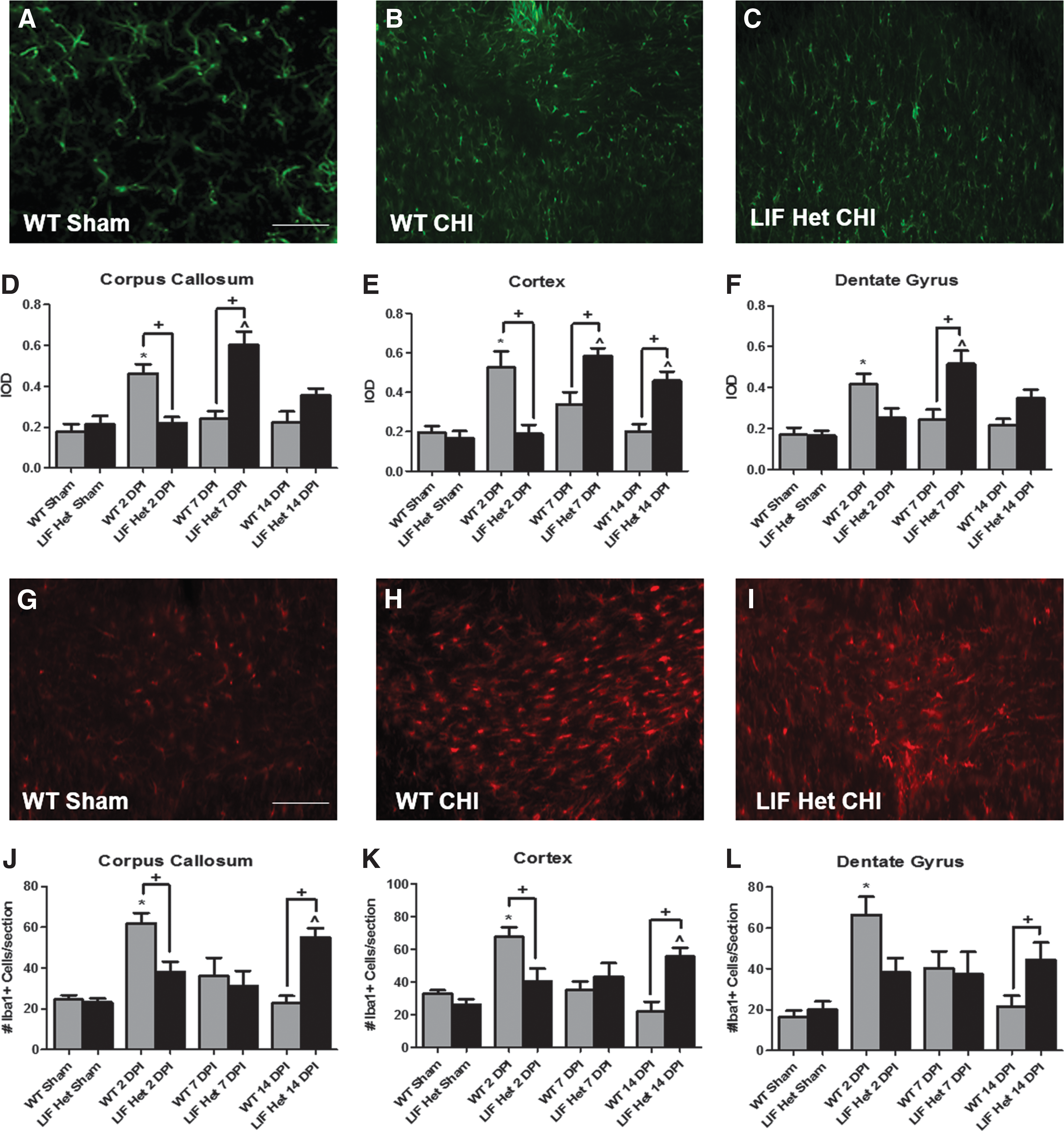
Leukemia inhibitory factor (LIF) deficient exhibit altered glial fibrillary acidic protein (GFAP) and microglial/macrophage expression after closed-head injury (CHI). Wild type (WT) and LIF heterozygous (LIF Het) mice were injured at post-natal Day 18 and sacrificed either 2, 7 or 14 days post-injury (DPI). Representative images (40× magnification) of the midline corpus callosum at the center of the impact zone on the skull halfway between bregma and lambda 2 DPI from
Macrophage and microglial responses are altered in LIF haplodeficient mice following pediatric CHI
To establish whether LIF haplodeficiency would alter the timing of the microglial and macrophage responses to CHI, animals injured at P18 were sacrificed 2, 7, and 14 DPI, and brains were immunolabeled for Iba1. Stereological cell counts of Iba1+ cells in WT injured brains revealed an approximately three-fold increase at 2 days post-CHI in each region, compared with sham-operated controls (p < 0.05; Fig. 3J-L). Iba1+ cell counts were significantly lower in LIF Het CHI brains than WT brains CHI at 2 DPI (p < 0.05), remaining at the same levels as sham-operated controls (Fig. 3J-L). At 7 DPI, Iba1+ cell numbers returned to sham-operated levels in all three regions in WT brains and remained at sham levels in the LIF Het mice (Fig. 3J-L). Interestingly, the number of Iba1+ cells increased 2.5- to 3.0-fold over WT mice at 14 DPI in all three brain regions (p < 0.05; Fig. 3J-L). Thus, similar to astroglial activation, these data indicate that LIF deficiency delays the normal microglial and macrophage responses after pediatric CHI that is followed by an exacerbated response.
LIF haplodefficient mice sustain greater cell death and axonal degeneration after pediatric CHI
To establish whether the delayed astrogliosis and microgliosis observed in the LIF Het mice after CHI would result in less cell death, apoptotic cells were labeled using ISEL and degenerating neurons were visualized using Fluoro-Jade C staining. The ISEL method was chosen as this method detects cells in the early stages of cell death and yields fewer false positives than terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Contrary to our predicted result, LIF Het mice had an ∼two-fold increase in ISEL+ apoptotic cells in all three brain regions examined, compared with injured WT mice at 2 DPI (p < 0.05; Fig. 4D-F). Similarly, Fluoro-Jade C+ neurons were significantly increased at this time-point in both LIF Het and WT cortices, compared with sham controls (p < 0.05; Fig. 4G-I); however, there were no differences between the genotypes. At 7 DPI, apoptotic cell numbers were still higher in both injured WT and LIF brain regions than in uninjured controls, but there were no significant differences between the injured genotypes (Fig. 4D-F). A small number of Fluoro-Jade C+ neurons were seen at this time-point in both injured groups, but unlike overall apoptotic numbers, LIF Het cortices had approximately twice as many degenerating neurons than WT injured brains (p < 0.05; Fig. 4I and 4J). At 14 DPI, injured LIF Het brains exhibited nearly two-fold more ISEL+ cells in the cortex and corpus callosum than injured WT animals, which had returned to uninjured control baseline levels (p < 0.05; Fig. 4D-F). Similarly, LIF Het CHI mice had two-fold more Fluoro-Jade C+ neurons in the cortex at 14 DPI, compared with injured WT mice (p < 0.05; Fig. 4I). These data demonstrate that the modified time-course of gliosis in LIF Het mice affects both early and delayed neurodegeneration following pediatric CHI.

Leukemia inhibitory factor (LIF) deficiency causes increased apoptotic cell death and neuronal degeneration after closed-head injury (CHI). Wild type (WT) and LIF heterozygous (LIF Het) mice were injured at post-natal Day 18 and sacrificed either 2, 7 or 14 days post-injury (DPI). Representative images of the corpus callosum 2 DPI from
LIF deficient mice exhibit altered axonal damage after CHI
We next evaluated the extent of axonal degeneration in the corpus callosum using SMI-32, an antibody that recognizes non-phosphorylated neurofilament H. Neurofilament H is phosphorylated under normal conditions. After mechanical injury, Neurofilament H becomes dephosphorylated and accumulates along the axon and specifically in the retraction bulbs of degenerating axons. 19,20 At 2 DPI, LIF Het corpus callosa had significantly higher SMI-32+ fluorescence levels than LIF Het sham controls (Fig. 5G; p < 0.05). Despite this increase, the levels were not significantly different between WT and LIF Het injured animals at 2 DPI. At 7 DPI, SMI-32 staining was significantly higher in both WT and LIF Het corpus callosa, compared with their respective sham controls, with the levels of SMI-32 fluorescence in LIF Het CHI mice approximately twice as high as WT injured levels (Fig. 5C, 5D, and 5G; p < 0.05). The levels of SMI-32 returned to sham-operated control levels at 14 DPI in WT corpus callosum but were ∼1.5 times greater in injured LIF Hets than WT injured brains and were significantly higher vs. LIF Het sham controls (Fig. 5E-G; p < 0.05). These data reinforce the conclusion that the altered gliotic response due to LIF haplodeficiency extended the duration of axonal damage within the corpus callosum after CHI.

Leukemia inhibitory factor (LIF) deficiency causes increased pathology in the corpus callosum after closed-head injury (CHI). Wild type (WT) and LIF heterozygous (LIF Het) mice were injured at post-natal Day 18 (P18) and sacrificed either 2, 7, or 14 days post-injury (DPI). Representative images of the corpus callosum 14 DPI
LIF deficient mice have altered callosal axon numbers following CHI
As changes in SMI-32 staining could have been due to reduced neurofilaments within existing axons rather than to reduced numbers of axons, we performed anterograde axonal tracing by injected BDA into the somatosensory cortex at 7 DPI (Fig. 5H). WT mice sustained an ∼50% decrease in callosal axons after CHI, whereas the LIF Het mice sustained an ∼75% loss of callosal axons (Fig. 5I-L, p < 0.05). These data support the conclusion that LIF normally exerts a protective role against white matter damage in response to mechanical brain injury in adolescent mice.
LIF haplodeficiency exacerbates hypomyelination in the corpus callosum after pediatric CHI
Recent studies have established that oligodendrocytes provide nutrients that support metabolism in axons. 21,22 Thus, the excessive axonal damage in the LIF Het mice could have been due to the demise of myelinating oligodendrocytes, myelin, and/or a change in the number of oligodendrocyte progenitor cells (OPC). Therefore, we examined oligodendrocyte and myelin integrity using Rip (which stains oligodendrocyte cytoplasm), MBP (which stains the myelin), and Olig2 (which stains oligodendrocyte lineage cell bodies). Injured WT and LIF Het mice exhibited significant decreases of Rip and MBP staining, compared with their respective sham-operated controls. (Fig. 6B-I; p < 0.05). By 7 DPI, LIF Het brains had ∼50% greater loss of RIP and MBP immunofluorescence in the corpus callosum than WT injured animals (Fig. 6C, 6H, 6N, and 6M; p < 0.05). At 14 DPI, LIF Het MBP loss persisted at ∼40%, compared with WT injured animals (Fig. 6M and 6N; p < 0.05). CHI was accompanied by approximately 50% and 60% increases in Olig2+ OPCs in both the neocortex and corpus callosum of WT injured animals, relative to sham-operated controls at 2 and 7 DPI, respectively (Fig. 6K, 6O, and 6P; p < 0.05). By 14 DPI, the number of Olig2+ cells was comparable to baseline sham levels. Compared with WT injured mice, injured LIF Het mice exhibited significantly smaller increases in Olig2+ cells of 25% and 40% over sham levels at 2 and 7 DPI, respectively. Olig2+ cell numbers were significantly increased at 7 DPI in both the cortex and corpus callosum and remained high at 14 DPI in the cortex in CHI LIF Het brains. Thus, contrary to our hypothesis, there was not a strong correlation between the degree of axonal loss and loss of oligodendrocytes in the LIF Het animals after CHI.

Leukemia inhibitory factor (LIF) deficiency causes increased myelin and oligodendrocyte loss and decreased oligodendrocyte progenitor cell (OPC) production after closed-head injury (CHI). Wild type (WT) and LIF heterozygous (LIF Het) mice were injured at post-natal Day 18 and sacrificed either 2, 7, or 14 days post-injury (DPI). Representative images of the corpus callosum of LIF Het Sham shows baseline levels of
LIF deficient mice have axonal conductance deficits after pediatric CHI
In vivo evoked CAPs were recorded from the corpus callosum of anesthetized mice at 2 weeks of recovery after the CHI to evaluate functional deficits in impulse conduction caused by the injury. Evoked CAPs were comprised of a biphasic waveform with an initial segment (N1) representing the fast-conducting myelinated axons, followed by a second component (N2) produced by the slower-conducting unmyelinated axons, as demonstrated previously (Fig. 7B1 ). 13 The CAP amplitude of the N1 myelinated fibers in LIF Het injured brains (Fig. 7B3 ) was reduced by 30–40% throughout a range of stimuli, compared with injured WT animals (Fig. 7B2 ; p < 0.001; Fig. 2C). The amplitude for the N2 component was not different between the sham-injured and brain-injured groups at any time-point post-injury (Fig. 7D). Additionally, the latency times from the stimulus artifact to the peak of N1 response were increased by ∼40% throughout a range of stimuli strength in LIF Het injured mice, compared with all other sham and WT injured mice (p < 0.01, Fig. 7E). No changes were observed in the N2 latency times (Fig. 7F). Refractoriness was measured over a range of time between equal strength paired pulse stimuli. The ratios of the amplitude size of the second paired pulse response (C2) over the first (C1) for the myelinated N1 fibers, were decreased by ∼25% from 2.5 to 5.5 msec interpulse interval in the LIF haplodeficient mice after CHI, compared with injured WT and sham-operated mice (Fig. 7G), whereas there was no change in refractoriness of the N2 component (Fig. 7H). These data provide evidence that LIF deficiency contributes to prolonged axonal conduction deficits in the myelinated fibers in the corpus callosum after CHI.
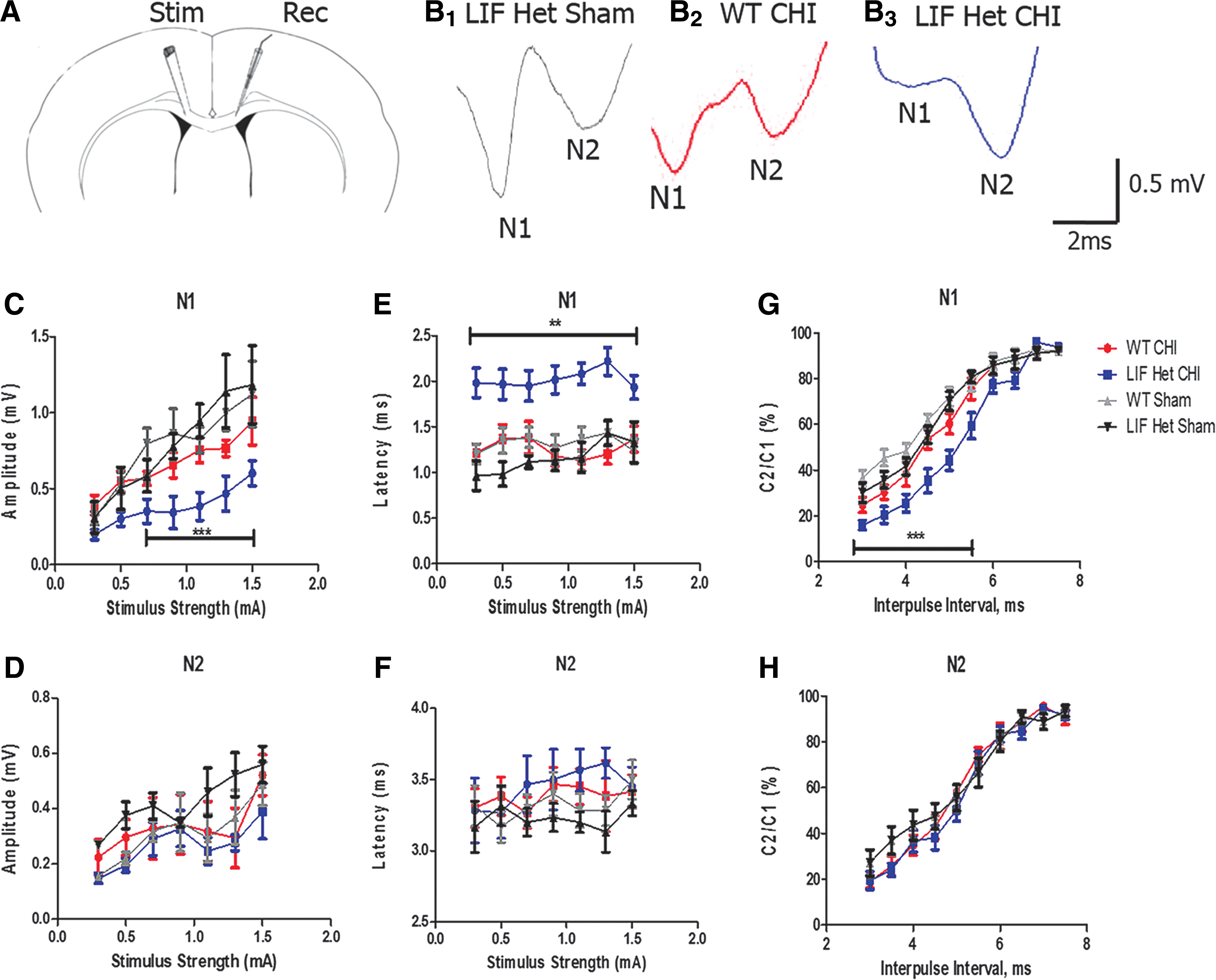
The myelinated (N1), but not the unmyelinated (N2), component of the corpus callosal compound action potential is altered in leukemia inhibitory factor (LIF) haplodeficient mice after closed-head injury (CHI).
Discussion
Taking into account both the time-course of LIF induction after injury, as well as the dramatic desynchronization of the injury response in the LIF haplodeficient mice, it seems evident that LIF is coordinating the acute stages of the injury response and that when a critical level of LIF is not reached, the evolution of injury is changed for the worse. Our studies demonstrate that reduced levels of LIF compromised both the initial astrocytosis and microgliosis. Whereas gliosis was once regarded as a stereotyped reaction to CNS injury, this view is no longer tenable, as it is clear that glial cells (both astrocytes and microglia) are capable of a spectrum of biochemical and physiological responses that are tailored to both the extent and nature of damage. 3 Their reactions are choreographed by extracellular signals that include small molecules, such as purines, neurotransmitters, steroids, and large polypeptides (e.g., cytokines, growth factors, and extracellular matrix molecules). 3 These signals in turn activate an array of second messengers and microRNAs. Increasingly, there is interest in deciphering the temporal sequence of signals that participate in coordinating gliosis and in identifying those that are beneficial versus detrimental so that new therapeutic interventions can be developed to promote better outcomes.
Astrocytes have been unambiguously shown to protect neurons from sustaining additional damage following brain injury. 23,24 For example, in a moderate traumatic brain injury model, ablating proliferating astrocytes exacerbates the extent of neuronal degeneration and inflammation, compared with control mice. 25 Other studies have demonstrated that activated astrocytes produce factors that prevent neurodegeneration and promote recovery. But which signals are truly important? One intracellular astroglial signaling pathway that is essential in limiting damage after CNS injury is the gp130/Stat3 signaling pathway. Deleting either gp130 or Stat-3 specifically in astrocytes reduces their response to injury, resulting in more widespread inflammation in models of traumatic injury, autoimmune disease, and infection. 26 –29 Since gp130 and Stat-3 are activated by the interleukin (IL)-6 family of cytokines that includes IL-6, LIF, ciliary neurotrophic factor (CNTF), oncostatin M, IL-11, cardiotrophin-1, and cardiotrophin-like cytokine factor 1, and since most of these cytokines increase subsequent to CNS injury, a priori it could not have been predicted that a 50% reduction in LIF expression would so dramatically affect the time course of gliosis and the severity of injury. On the other hand, LIF null mice have reduced astrocytic and microglial responses after seizure, sciatic nerve crush injury, and stab wound brain injury. 30,31
Astrocytes in vitro produce a wide range of trophic factors that include nerve growth factor, fibroblast growth 2, platelet-derived growth factor (PDGF), and CNTF. 32 –41 Thus, one might have predicted that these other trophic factors would compensate for the 50% reduction in LIF and that the LIF haplodeficient mice would fare as well as their WT littermates. Clearly, that prediction is not supported by our data. Interestingly, astrocytes, which normally express low levels of LIF receptor, markedly increase their expression of this receptor along with LIF itself following neurotrauma and in neurological diseases, 37,42 –46 indicating the establishment of an autocrine/paracrine loop. To date, the function of that loop is unclear, but our data suggest this loop may initiate a cascade of changes within the astrocytes that prevent damage. 47 –49
Deciphering the molecular mechanisms responsible for the neuroprotective actions of LIF will be both complex and time consuming—not simply because LIF receptors are expressed on virtually every cellular element of the CNS, but also because of the range of changes that LIF elicits. Our studies provide evidence that LIF is a critical neuroprotective signal for neurons after injury. The LIF haplodeficient animals clearly sustained greater traumatic axonal injury that was evident both electrophysiologically and histologically and also more severe secondary neuronal cell death. LIF gain of function studies have shown that LIF can limit the neurological deficits caused by ischemic brain injury. 50 Similarly, intravitreal LIF injections have been shown to prevent photoreceptor cell death caused by light-induced oxidative damage. 50,51 Studies in mice deficient for LIF, CNTF, or their common receptor subunit gp130 show that LIF receptor signaling is necessary for the neuroprotective and axon growth–promoting effects of conditioning lesions, 50,51 and these factors can rescue sensory and motor neurons from axotomy-induced cell death 52 ; some of these actions of LIF are likely direct, as LIF promotes neuronal survival in vitro, as well as to stimulate neurite outgrowth. 53 Some of the actions of LIF will be indirect. A recent study showed that one of the downstream signals that is induced by LIF in the spinal cord is insulin-like growth factor 1 (IGF-1). 45 Daily administration of LIF promoted oligodendrocyte survival after spinal cord injury. This effect was not mediated by a direct action of LIF on oligodendrocytes but rather via an ancillary cell type, with augmented IGF-1 expression. LIF also increased levels of neurotrophin-3 (NT3) in the spinal cord. 54 As IGF-1 is both a neuroprotective and glioprotective growth factor and NT3 is an important neurotrophic factor after both brain and spinal cord injury, decreasing or delaying the expression of these factors could easily contribute to the excess cell death seen in the LIF haplodeficient mice. 55,56
LIF acts directly on microglia and macrophages, but there are discrepancies in the literature. Hendriks and colleagues demonstrated that LIF inhibited free radical and tumor necrosis factor α production in macrophages and promoted an M2-like phenotype, which lead them to conclude that LIF elicits an anti-inflammatory and regenerative effect from macrophages. 57 However, Kerr and Patterson found that LIF over-expression in the spinal cord stimulated the proliferation of microglia/macrophages that resulted in severe hindlimb motor dysfunction. 10 Further, when microglial activation was inhibited by minocycline administration, the microgliosis and severity of these motor deficits was attenuated, suggesting that LIF elicits an M-1–like reaction from microglia. 55,57 Supporting this view, cell proliferation was significantly diminished and the microglial/macrophage response to spinal cord injury was decreased in LIF knockout (KO) mice, compared with wild type mice.
In our studies, white matter cell death and axon damage were accompanied by decreased axonal conductance within the corpus callosum, confirming the results of earlier studies using this CHI model. 13 However, a novel finding was the extensive reduction in the CAP of LIF Het CHI mice. This impairment was specific to the myelinated fibers 2 weeks after injury. It is likely that the sustained deficits in myelinated fiber conductance are due to myelin loss subsequent to axonal injury, as well as to the altered timeline of gliosis.
The increased numbers of Olig2+ cells in WT brains after CHI, are likely due to an increase in OPC proliferation in response to the injury. 12 Exogenous LIF has been shown to enhance the OPC response and promote remyelination after cuprizone-induced demyelination, and OPC production is reduced in CNTF KO mice during the late stages of experimental autoimmune encephalomyelitis. 58 It has been suggested that LIF directly stimulates OPC proliferation, especially in the presence of mitogens such as PDGF. 9,12,59 As LIF has been shown to promote OPC differentiation in vitro and in vivo, decreased levels of LIF may impair their differentiation resulting in an increase in the proliferation of the OPC and less myelin, as we observed. 60,61
In summary, we have provided compelling evidence for a vital role for LIF in orchestrating the glial response to a closed-head injury. Both astrogliosis and microgliosis were acutely diminished early with a delayed increase occurring at later time-points after injury. This desynchronized gliosis was accompanied by increased white and gray matter apoptosis, neuronal cell death and axonal degeneration. LIF haplodeficient mice also sustained greater callosal axonal loss, oligodendrocyte and myelin loss, blunted increases in OPCs, myelinated fiber conduction deficits, and they displayed more severe behavioral deficits, compared with wild-type injured mice. Altogether, these data demonstrate that LIF is an essential timing signal after brain injury, and that a 50% reduction in LIF expression is sufficient to elicit a cascade of events that result in a second wave of neurodegeneration and more severe neurological deficits. Clearly, additional studies will be required to fully understand and elucidate the mechanisms through which LIF affects these cellular responses to CNS injury.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.