
Abstract
Traumatic brain injury (TBI) caused by improvised explosive devices (IEDs) is a growing problem in military settings, but modeling this disease in rodents to pre-clinically evaluate potential therapeutics has been challenging because of inconsistency between models. Although the effects of primary blast wave injury have been extensively studied, little is known regarding the effects of noncontact rotational TBIs independent of the blast wave. To model this type of injury, we generated an air cannon system that does not produce a blast wave, but generates enough air pressure to cause rotational TBI. Mice exposed to this type of injury showed deficits in cognitive and motor task acquisition within 1–2 weeks post-injury, but mice tested 7–8 weeks post-injury did not retain any deficits. This suggests that the effects of a single, noncontact rotational TBI are not long lasting. Despite the transient nature of the behavioral deficits, increased levels of phosphorylated tau were observed at 2 and 8 weeks post-injury; however, this tau did not adopt typical pathological structures that have been observed in other TBI models that incorporate blast waves. This was possibly attributed to the fact that this injury was insufficient to induce changes in microglial activation, which was not affected at 2 or 8 weeks post-injury. Taken together, these data suggest that exposure to noncontact, rotational head injury only produces transient cognitive anomalies, but elicits some minor lasting neuropathological changes.
Introduction
T
Animal models of TBI have employed a variety of techniques to induce brain injury, including controlled cortical impact (CCI), fluid percussion injury (FPI), weight-drop closed TBI, rotational TBI, as well as blast wave models. 4 –13 The latter have relied on blast wave generators (BWGs) to model the physiological effects of blast in rodents. These BWGs produce BOP waves representative of close-proximity battlefield explosions, which can lead to behavioral deficits and neuropathological changes in mice reminiscent of the human condition. 3,14 –16 The neuropathology observed in animals after blast TBI varies depending on blast severity and duration of recovery, but consistent features include neuroinflammation, diffuse axonal injury (DAI), and alterations in tau phosphorylation and levels. 3,14,17 –19
Noncontact rotational TBIs across multiple species have been shown to produce DAI and other pathologies of TBI. 13,20 –22 Interestingly, it was shown that stabilizing the head prevented the effects of a blast-induced TBI, suggesting that head rotation is necessary for symptoms of TBI to manifest. 3 To test whether rotational injury caused by a blast of air is sufficient to induce cognitive and neuropathological changes, we built an air cannon system that does not produce a blast wave, but can cause rotational, noncontact TBI using pressurized air. We examined the behavioral and neuropathological consequences of this injury in mice at both 2 and 8 weeks post-injury and show that noncontact rotational injuries do not cause lasting physiological deficits or inflammatory changes, but do initiate some persistent tau dysfunction.
Methods
Animals
Three-month-old C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) were group housed under standard conditions, with food and water available ad libitum and a 12-h light-dark cycle (lights on at 6:00
Air cannon blast
Air cannon blast was performed on mice anesthetized with a cocktail of ketamine (100 mg/kg), xylazine (10 mg/kg), and acepromazine (2 mg/kg) with imipramine (2.3 mg/kg) administered as a prophylactic analgesic. A 70-liter Martin Hurricane industrial air cannon (Martin Engineering, Neponset, IL) was mounted to a sturdy steel base and charged with compressed air using a Craftsman air compressor to a pressure of 17.5 pounds per square inch (PSI), as measured using the cannon-mounted pressure gauge. A mechanical solenoid discharged 80% of the compressed air over 100 ms through the smaller aperture of the driven chamber, producing an effective surge measuring 50 PSI, which delivered a blast velocity of approximately 22 m/s (50 mph). Mice were secured to a holding device 1.0 m from the solenoid in a prone position with lead wrapped around the body to protect against thoracic injury (see Fig. 1A for cannon schematic). The holding device was a piece of 1-inch (in) flat steel bent into a U-shape so that it could be attached to the tube by four steel bolts. Extending from the center of the fixture were two 3⁄8;-in threaded steel rods 150 mm in length, spaced 25 mm apart, that were secured to the 1-in flat steel with steel nuts on both sides. A miniature digital gyroscope was mounted on the mouse head to measure head angular acceleration and displacement, triaxially, in response to the air blast. Gyroscopes measured 18 × 14 mm with a mass of 4 g.

Air cannon blast dynamics. (
Sham mice were anesthetized and placed outside the area of the blast while the air cannon was triggered to control for aural damage. After the blast, mice were behaviorally tested beginning either 1 (acute post-exposure [APE] mice) or 6.5 weeks (sub-acute post-exposure (SPE) mice) later in order to perform histology at 2 or 8 weeks post-injury, respectively. Behavioral testing for APE mice occurred in the following order: rotarod test; Y maze; and radial arm water maze (RAWM). SPE mice were left alone for 6.5 weeks before behavioral testing, which was run in the order of rotarod, Y maze, and Morris water maze (MWM).
Rotarod test
Motor function and motor learning were assessed using an accelerating rotarod test, as previously described. 23 The rotating bar gradually accelerated from 4 to 40 rpm over 5 min. Mice were placed on the bar, and latency to fall was recorded by a blind observer. Each specimen performed 4 trials per day over 2 consecutive days.
Y maze
Spontaneous alternation, a measure of working memory, was investigated in the Y maze as previously described. 24 Mice were placed at the center of a three-arm maze, which they were allowed to explore for 8 min. The sequence of arm entries was manually recorded by an observer blind to treatment. Alternation is defined as a triad of arm entries without re-entry into a previously visited arm. Percent spontaneous alternation was calculated as the number of correct triads divided by the maximum possible alternations (total arm entries minus 2) × 100.
Radial arm water maze
The RAWM was adapted from previous studies. 25,26 Briefly, a circular black tank with a six-arm metal insert was filled with water and a platform was submerged 1 cm below the surface of the water at the end of a designated goal arm. Animals were permitted 60 sec to locate the platform and given 15 sec on the platform before being dried and returned to the home cage to await the next trial. Mice were trained over 2 days with 15 trials per day, which were divided into 5 sessions of 3 trials each. An observer blind to treatment manually scored the number of errors, which were defined as an entry into an incorrect arm. A working memory error was defined as re-entry into an already visited incorrect arm within the same trial.
Morris water maze
The MWM was conducted in a black tank with an escape platform placed 1 cm below the surface of the water in the center of one of the four quadrants (target quadrant). 23 During acquisition testing, mice were permitted 60 sec to locate the platform, after which time they were gently guided to the platform by the experimenter. Mice were given 15 sec on the platform before removal into a heated holding cage. Four trials per day with a 60-sec intertrial interval were conducted until the sham control group reached a criterion mean latency of <15 sec. The probe trial was conducted 24 h later by removing the platform and allowing the animal to swim for 60 sec. The percent time spent in each quadrant was used as a measure of probe trial performance. All trials were recorded by a video camera and analyzed using ANY-maze software (Stoelting Co., Wood Dale, IL).
Immunohistochemistry
Free floating immunohistochemistry was performed as described previously. 27 Mice were euthanized either 2 or 8 weeks post-blast by overdose of Somnasol (Henry Schein, Dublin, OH) and transcardially perfused with physiological saline. Brains were removed and fixed in 4% paraformaldehyde for 24 h. After sucrose gradient cryopreservation, brains were sectioned coronally at 25 μM on a sliding microtome. Sections were then incubated in a 10% methanol/3% hydrogen peroxide solution to block endogenous peroxidases. After phosphate-buffered saline (PBS) washes, sections were permeabilized by 0.2% Triton X-100 with 1.83% lysine and 4% goat serum for 30 min before overnight incubation with one of the following antibodies: anti-pS199/202 tau (1:20,000; Anaspec, Fremont, CA); anti-pT231 tau (1:300; Anaspec; 1:300); or anti-Iba1 (ionized calcium-binding adaptor molecule 1; 1:3000; Wako Chemicals, Richmond, VA). Sections were then incubated in biotinylated goat antirabbit secondary antibodies before more washes and ABC amplification (Vector Labs, Burlingame, CA). Last, sections were incubated in 0.05% 3,3’-diaminobenzidine with 0.5% nickel sulfate and developed with 0.03% hydrogen peroxide. Sections were slide mounted, dehydrated in alcohol gradients, and cover-slipped before imaging.
A slide scanning microscope (Zeiss, Thornwood, NY) was used to image all tissue before analysis with Neuroquant IAE software (Zeiss). Six sections per mouse were used for each antibody stain. Regions of interest were outlined and a segmentation analysis was performed to evaluate the area ratio of positive cells within each region by an experimenter blinded to conditions. Area ratio was then averaged for all six sections per mouse.
Statistical analyses
Statistical significance for each analysis was determined with Student's t-tests or two-way analysis of variance with Bonferroni's post-tests to compare groups, where appropriate. All figures and statistics were generated using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA); each graph represents the mean ± the standard error of the mean (SEM).
Results
The air cannon was charged with compressed air to a pressure of 17.5 PSI, as measured using the cannon-mounted pressure gauge. When discharged through the smaller aperture of the driven chamber, this produced an effective surge measuring 50 PSI, which delivered a blast of air with a measured velocity of approximately 22 m/s (50 mph). Pressure measurements were recorded from sensors placed along the length of the driven chamber, as shown in Figure 1B. Angular head acceleration was measured using a miniature digital gyroscope mounted to the mouse head; head acceleration was 3843 ± 668.6 rad/s 2 (mean ± SEM; a representative graph of angular head movement is shown in Fig. 1C). Angular displacement of the head was observed to be 0.298 ± 0.06 radians, which is equivalent to ∼17 degrees. Unfortunately, it was determined in these initial pilot studies that the gyroscope was causing mortality in ∼30% of subjects for unknown reasons, perhaps putting increased strain on the head/neck, which necessitated removal of the gyroscope from subsequent experiments. No mortality was observed in any of the below experiments in the absence of the gyroscope.
After the blast, mice were behaviorally tested beginning either 1 (APE mice) or 6.5 weeks (SPE mice) later in order to perform histology at 2 or 8 weeks post-injury, respectively. Because APE mice had a short 1-week window in which to perform all behavioral testing, the MWM, the gold-standard spatial learning and memory test, could not be performed. Instead, the RAWM was used to examine learning and memory, as previously reported. 25
APE mice displayed cognitive deficits in the RAWM spatial learning task, compared to sham mice (Fig. 2A,B). Specifically, the blast disrupted the acquisition of the task across the first day of training, as measured by total number of errors (F 1,55 = 8.201; p < 0.01; Fig. 2A) and working memory errors (F 1,55 = 11.47; p < 0.01; Fig. 2B), which were classified as re-entry into a previously visited arm during the same trial. The blast did not affect recall of the task, because mice performed equivalently on the second day of the RAWM (p > 0.05 for total errors and working memory errors). Importantly, the total number of arm entries did not differ between sham and blast mice (p > 0.05), indicating that the deficit observed on day 1 was not attributed to differences in motoric function or motivation. SPE mice were tested in the MWM 7 weeks post-injury, but no differences were observed between groups in latency to find the platform during acquisition training (p > 0.05; Fig. 2C). Both sham and blast SPE mice spent significantly more time in the target quadrant, compared to all other quadrants, during the probe trial, indicating that they each learned the spatial location of the platform (p < 0.001; Fig. 2D).
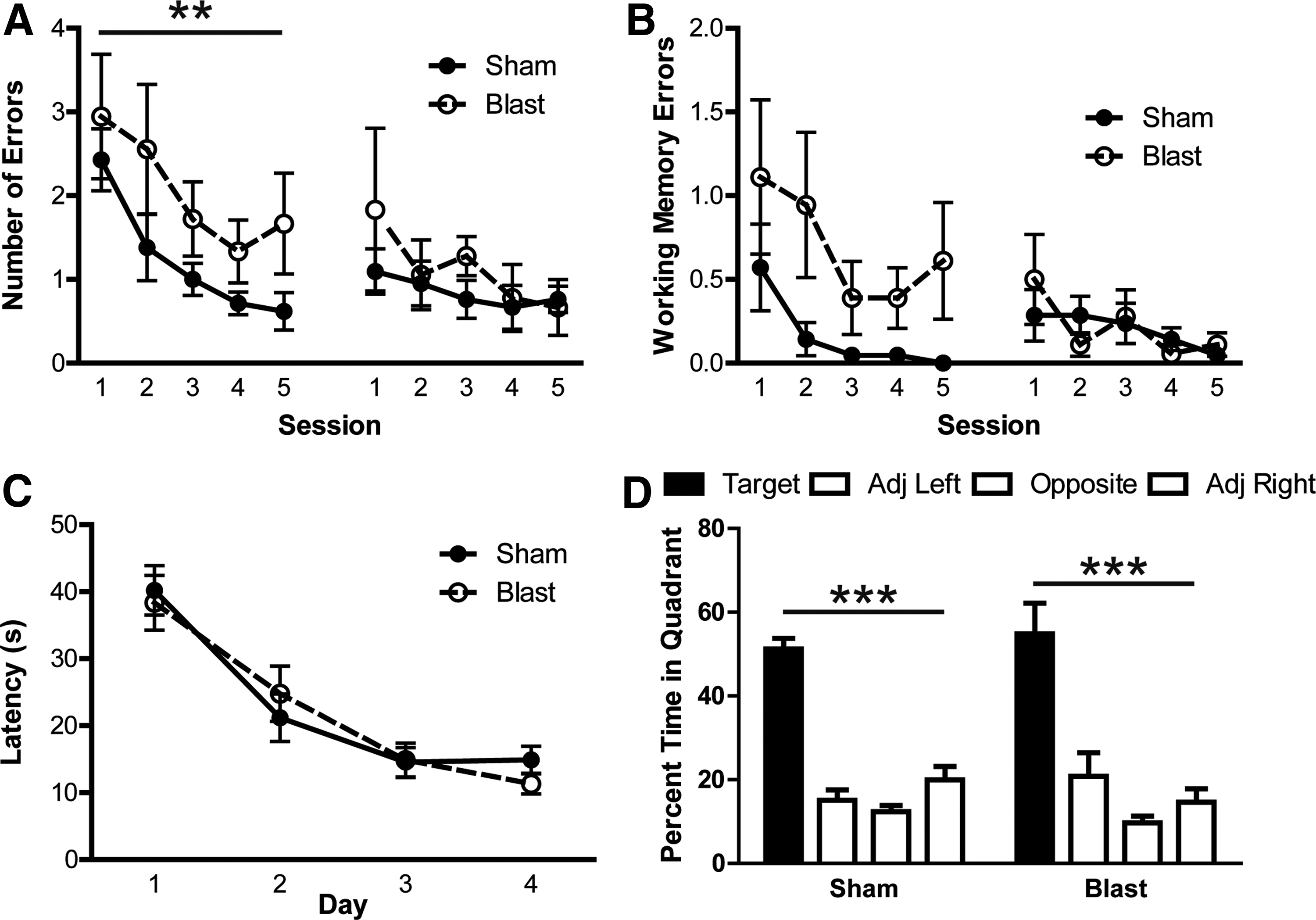
Mice displayed learning deficits 2 weeks, but not 8 weeks, post-injury. (
To investigate motor function after air cannon blast exposure, a 2-day rotarod test was conducted. In APE mice, there was a main effect of trial (F 7,88 = 3.334; p < 0.01) and treatment across the 2 days (F 1,88 = 11.97; p < 0.001; Fig. 3A), indicating that the blast impaired motor performance. APE blast mice had equivalent latencies with sham mice by the end of day 2, suggesting that the blast did not impair motor learning. In fact, linear regression analyses did not reveal any differences between groups (F 1,100 = 0.074; p = 0.786), confirming that APE blast mice were able to learn the task equivalently to controls. Figure 3B presents average latency for each day. The SPE mice did not display any deficits in motor performance, performing equivalently with sham mice on each day of training (p > 0.05; Fig. 3C,D).
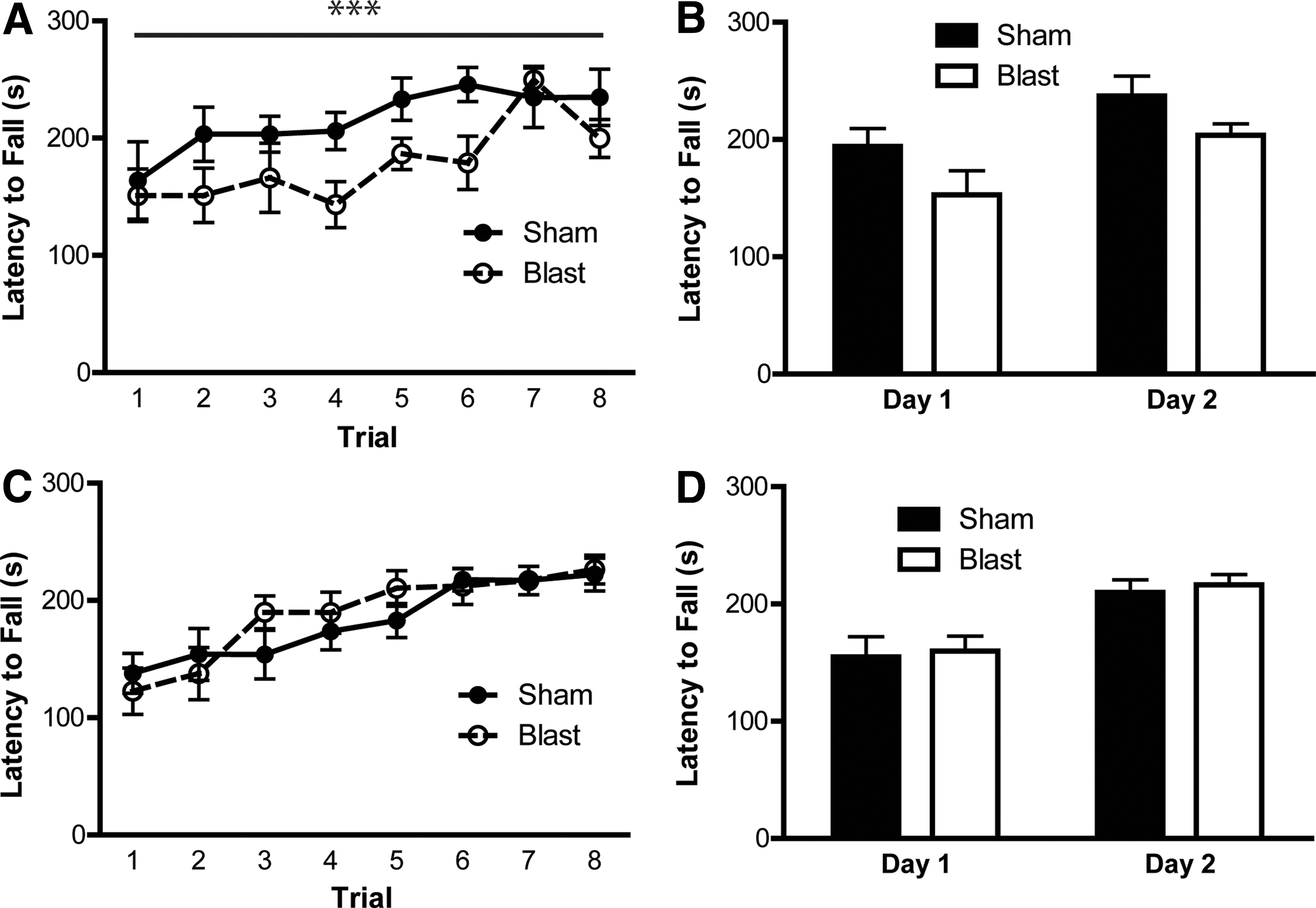
Air cannon blast disrupted motor function 1 week, but not 6.5 weeks, post-injury. (
Spontaneous alternation behavior, a measure of working memory, was also assessed in mice. The air cannon blast did not alter percent alternation in APE or SPE mice (p > 0.05; Fig. 4A,C) or total number of arm entries (p > 0.05; Fig. 4B,D), suggesting that working memory in a nonstressful environment was not impaired.
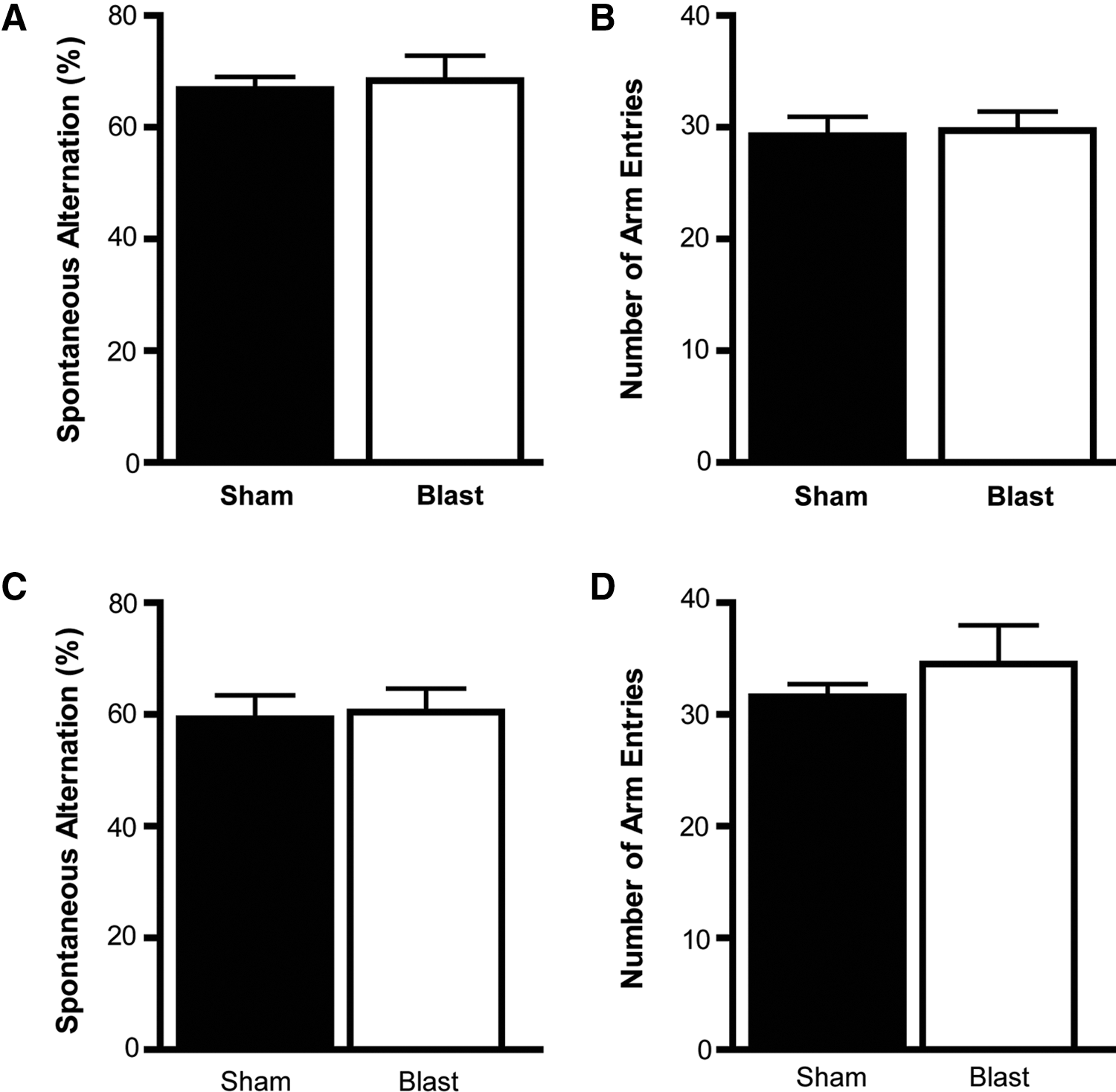
Spontaneous alternation behavior was not altered by the blast. The blast did not affect spontaneous alternation behavior in the Y maze 1.5 (
Tau phosphorylation and accumulation have been implicated as possible causative pathologies in TBI. 14,28 –30 Therefore, we evaluated levels of tau phosphorylated at Ser199/202 and Thr231 in the hippocampus and cortex of APE and SPE mice, because these phospho-epitopes have been implicated in TBI and tauopathies. 3,31 –33 Representative images from sham and blast mice for each cohort are presented for the hippocampus in Figure 5 and the cortex in Figure 6. Exposure to the air cannon blast significantly increased levels of tau phosphorylated at Ser199/202 in the hippocampus at 2 weeks post-injury (t 7 = 2.795; p < 0.05; Fig. 5A,C), but not 8 weeks post-injury (p > 0.05; Fig. 5B,C), as compared to sham mice. Conversely, levels of tau phosphorylated at Thr231 were elevated in the hippocampus 8 weeks post-injury (t 11 = 2.771; p < 0.05; Fig. 5E,F), but not 2 weeks post-injury (p > 0.05; Fig. 5D,F). In the cortex, levels of phospho-tau (pTau) at Ser199/202 were increased at 8 weeks post-injury (t 11 = 2.378; p < 0.05; Fig. 6B,C), but not at 2 weeks, despite a trend toward an increase (p = 0.082; Fig. 6A,C). Similarly, levels of pTau at Thr231 were significantly elevated in the cortex at 8 weeks (t 11 = 3.599; p < 0.01; Fig. 6E,F), but not 2 weeks, post-injury (p > 0.05; Fig. 6D,F). These findings suggest that tau phosphorylation is increased 2 weeks after the air cannon blast in the hippocampus, but not the cortex, whereas cortical pTau levels are elevated 8 weeks post-injury. When the whole brain was analyzed for tau phosphorylation, we did not observe statistically significant differences between sham and blast groups. However, a trend toward elevated pTau in both APE and SPE brains was still observed. Specifically, levels of tau phosphorylated at Ser199/202 were increased 1.635 ± 0.3392 (mean ± SEM) fold (p = 0.1474) at 2 weeks post-injury, whereas levels were increased 1.46 ± 0.2461 fold (p = 0.0813) at 8 weeks. Levels of tau phosphorylated at Thr231 were unchanged at 2 weeks post-exposure (p = 0.9725), but increased 1.489 ± 0.1926 fold (p = 0.0681) at 8 weeks. This suggests that the hippocampus and cortex were more affected by the injury than other brain regions.
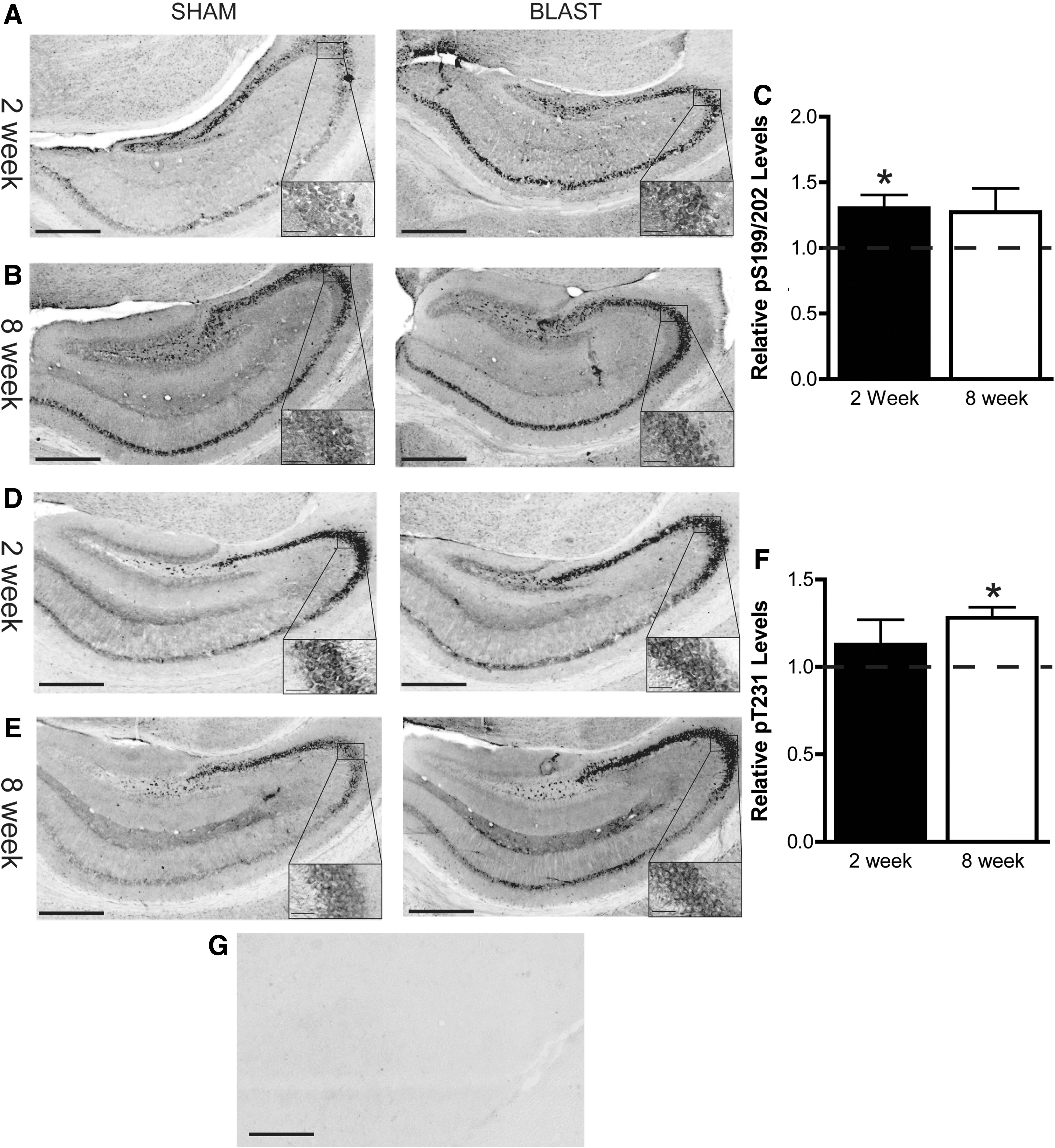
Tau phosphorylation in the hippocampus was increased by the blast. Levels of tau phosphorylated at Ser199/202 were increased 2 weeks (

Tau phosphorylation in the cortex was increased 8 weeks post-blast. Levels of tau phosphorylated at Ser199/202 were not changed 2 weeks (
Given that inflammation has also been linked to the physiological deficits caused by TBI, 18 tissue from these mice were immunostained for Iba1, a general marker of microglial activation. Quantitative analyses revealed no significant difference in the relative abundance of this microglial marker in the hippocampus (Fig. 7A–C) or cortex (Fig. 7D–F) at 2 or 8 weeks post-injury, compared to sham controls (p > 0.05).

The blast did not produce neuroinflammation in the hippocampus or cortex. Activated microglia were examined by Iba1 staining in the hippocampus (
Discussion
The current study demonstrated that a single blast of pressurized air transiently impairs motor function and spatial learning, while producing lasting, but subtle, changes in levels of phosphorylated tau. Mice tested 1 week after the rotational injury displayed impaired motor ability, but equivalent motor learning, indicating that despite a functional deficit, they were still able to improve. These mice were also impaired in the acquisition of the RAWM spatial learning task, but once again performed equivalently in the day 2 recall of the task. These data suggest that a noncontact rotational TBI impairs task acquisition, though the impairment is not severe enough to affect motor memory or hippocampal-dependent memory function. No deficits were observed when these same behaviors were examined 6.5–8.0 weeks post-blast, suggesting that the early deficits could be resolved. Subtle increases in phosphorylated tau levels were observed in the hippocampus and cortex at 2 weeks post-injury, with more-robust differences observed at 8 weeks, especially in the cortex. Despite the increased tau phosphorylation, no microglial activation was observed at either time point in the hippocampus or cortex.
Noncontact rotational TBIs have been shown to cause DAI and neuroinflammation, primarily in nonhuman primate and miniature swine models. 20,21,34,35 Smith and colleagues showed that this DAI was accompanied by tau accumulation in swine, suggesting that noncontact rotational injury was sufficient to induce this pathological change. 36 Although the mechanism of head rotation was quite different in the current studies, the effects on tau were similar. Behaviorally, Margulies and coworkers showed cognitive deficits in neonatal pigs after noncontact rotational injury, but no motor dysfunction. 37 Meanwhile, experiments investigating blast TBI utilizing BOP injury have revealed deficits in motoric and cognitive function that vary with the severity of the blast. Cernak and colleagues reported significant impairments in motor performance up to 2 weeks post-blast and memory performance up to 30 days post-blast, deficits that were amplified by a more severe blast. 16 A similar study by Koliatsos and colleagues discovered deficits in motor function and spatial learning at 1 week, but not 2 weeks, after exposure to a BOP wave. 38 Together with the data from the current study, these findings suggest that there is an initial period of motoric and cognitive dysfunction post-injury that resolves with time. In fact, our RAWM data in particular suggest that this type of injury can produce deficits consistent with findings from TBI patients showing that acquisition, but not retrieval, is impaired post-injury. 39
Tau accumulation in brains of individuals with TBI is a common pathological feature that is even being recommended for post-mortem diagnosis. 28,40,41 Tau from TBI brains typically presents at the depths of sulci, is found in astrocytes and neurons, and is perikaryal with ß-sheet structure. 3 Reports from animal models have demonstrated that several models of brain injury can induce or exacerbate a similar type of tau pathology, whereas tau knockout mice were protected from CCI-induced TBI injuries, highlighting the importance of tau pathology in pathogenic mechanisms of TBIs. 3 –5,14,32,36,42 The air cannon model used here is somewhat unique, showing that rotational injury without a pressure wave only increases levels of phospho-tau without promoting its aggregation. But this increased tau persisted and even worsened for at least ∼2 months in our model despite functional recovery, consistent with previous studies using BOP injury. 14 It is possible that repeated mild injuries could eventually exceed a threshold that precipitates the pathological condensation of tau into toxic aggregates.
Interestingly, the air cannon model did not induce any microglial activation, unlike other brain injury models. Microglial activation has been detected within 1 day of blast injury and shown to persist up to 3 weeks post-injury. 3,43 –46 The absence of any change in our study could be attributed to the timing of the experiments. It is possible that transient microglial activation occurred subsequent to our noncontact rotational TBI, but any differences were undetectable by 2 weeks post-exposure. However, Aungst and colleagues showed elevated microglial activation 4 weeks after an FPI, 9 suggesting that the air cannon may have produced a relatively mild injury, which accounts for the lack of sustained microgliosis. Future studies may be able to reveal whether our noncontact rotational injury produced any immediate microglial activation or even astrocytosis. 47,48
Because our study demonstrates that exposure to a noncontact rotational injury is sufficient to induce transient learning and motor deficits in mice shortly post-injury, there may be several practical applications as well. First, because there was no generation of a BOP wave used to induce these impairments, it indicates that the threshold of symptom presentation after blast exposure may be quite low. Moreover, the transience of these deficits suggests that there is a recovery period before optimal brain function returns that is at least 1–2 weeks long in rodents, even for mild exposures. Moreover, our findings that tau phosphorylation remains elevated, despite the recovery of cognitive function, could suggest that individuals exposed to even what would be considered “mild” events could be primed for future deficits that are accelerated by subsequent injuries.
Footnotes
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the James A. Haley Veterans' Hospital. The contents of this publication do not represent the views of the Department of Veterans Affairs or the U.S. government. This work was supported by NS073899 and BX001637 (to C.A.D).
Author Disclosure Statement
No competing financial interests exist.