
Abstract
Chemokines and their receptors are of great interest within the milieu of immune responses elicited in the central nervous system in response to trauma. Chemokine (C-C motif)) ligand 2 (CCL2), which is also known as monocyte chemotactic protein-1, has been implicated in the pathogenesis of traumatic brain injury (TBI), brain ischemia, Alzheimer's disease, and other neurodegenerative diseases. In this study, we investigated the time course of CCL2 accumulation in cerebrospinal fluid (CSF) after exposures to single and repeated blast overpressures of varied intensities along with the neuropathological changes and motor deficits resulting from these blast conditions. Significantly increased concentrations of CCL2 in CSF were evident by 1 h of blast exposure and persisted over 24 h with peak levels measured at 6 h post-injury. The increased levels of CCL2 in CSF corresponded with both the number and intensities of blast overpressure and were also commensurate with the extent of neuromotor impairment and neuropathological abnormalities resulting from these exposures. CCL2 levels in CSF and plasma were tightly correlated with levels of CCL2 messenger RNA in cerebellum, the brain region most consistently neuropathologically disrupted by blast. In view of the roles of CCL2 that have been implicated in multiple neurodegenerative disorders, it is likely that the sustained high levels of CCL2 and the increased expression of its main receptor, CCR2, in the brain after blast may similarly contribute to neurodegenerative processes after blast exposure. In addition, the markedly elevated concentration of CCL2 in CSF might be a candidate early-response biomarker for diagnosis and prognosis of blast-induced TBI.
Introduction
B
Induction of inflammatory processes has long been recognized to be among the first responses of the body to a localized injury. Neuroinflammation is prominently involved in acute and chronic neuropathological conditions, 12 and TBI-induced neuroinflammatory responses are now recognized as critical steps in the development of a variety of brain diseases. 13,14 Inflammation is mediated by cytokines and eicosanoids, which are released predominantly from injured tissue and from accumulated macrophages and contribute to both secondary tissue damage and repair. 15 Chemokines are a family of small cytokines that are chemoattractants for leukocytes and guide the directional migration of monocytes, neutrophils, and other effector cells to the site of tissue damage. In particular, chemokine (C-C motif) ligand 2 (CCL2), also known as monocyte chemotactic protein-1, is secreted by leukocytes in the blood and is also produced by astrocytes, microglia, and neurons in the brain. It is involved in cellular migration and intercellular communication through interactions with glycol-protein receptors coupled to a G-protein-signaling pathway. Its pattern of neuroanatomical localization and codistribution with neurotransmitters and neuroregulatory peptides has prompted speculation that CCL2 might act as a modulator of neuronal activity and neuroendocrine function. 16 CCL2 has also been shown to act as a key factor in both initiation and maintenance of neuroinflammation in the spinal cord after peripheral nerve injury 17 and has been strongly implicated in the pathogenesis of brain ischemia, 18 autoimmune encephalomyelitis, 19 epilepsy, 20 TBI, 21 –29 and neurodegenerative diseases. 30 –32 Notably, Semple and colleagues 22 detected >20-fold immediate elevations in CCL2 in the cerebrospinal fluid (CSF) of severe TBI (sTBI) patients, which, although diminished, persisted for 10 days post-trauma. 22 They additionally found transiently increased levels of CCL2 in the cortices of mice subjected to closed head inury (with peak levels by 4–12 h) and established that the absence of CCL2 in CCL2–/– mice was associated with improved neurological recovery and a delayed reduction in lesion volume, macrophage accumulation, and astrogliosis, pointing to a potentially important role of CCL2 as both a biomarker and mediator of acute secondary brain degeneration post-trauma.
In addition to its role as a mediator of acute neurotrauma, CCL2 has also been implicated in chronic neurodegenerative disorders 31 –35 and may potentially serve as a link between the former and the latter, with cerebrospinal fluid (CSF) CCL2 levels providing diagnostic insights. For example, Westin and colleagues found that in patients with prodromal Alzheimer's disease (AD), CCL2 levels in CSF correlate with a faster cognitive decline observed during follow-up, 35 suggesting that CCL2 in CSF may have predictive diagnostic utility of AD-related pathology, and that CCL2-related signaling pathways might provide therapeutic targets to counter progression of this and perhaps other neurodegenerative disorders. In the present study, we used a well-defined bTBI model in rats to determine the acute changes in CCL2 levels in the brain, CSF, and plasma after blast exposure(s) and assessed its association with the acute functional disruptions and pathological changes resulting from these traumatic insults.
Methods
Animal and blast injury
All animal experiments were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals (NRC publication, 2011 edition). Male Sprague-Dawley rats, weighing 325–350 g at 8–10 weeks of age (Charles River Laboratories Inc, Frederick, MD), were housed at 20–22°C (12-h light/dark cycle) with free access to food and water ad libitum. After 4% isoflurane gas anesthesia in an induction chamber for 8 min (O2 flow rate, 1.5 L/min), rats were immediately tautly secured in a transverse prone position 2.5 feet (ft) within the mouth of a 1-ft-diameter compressed air-driven shock tube with the right side of the animal facing the oncoming shockwave. Animals were exposed to a single blast at a peak total pressure of 12 pounds per square inch (psi), single low-level blast (SLB) or 19 psi (single high-level blast (SHB) or were exposed to closely coupled repeated 19-psi blasts separated by a 1-min interval (double high-level blast; DHB). All blast overpressures (BOPs) had an 8- to 9-msec positive phase duration. Sham control animals were included in all individual experiments and were treated in the same fashion without exposure to blast waves.
Biosamples preparation
Blood, CSF, and brain tissue samples were collected at different times (1, 6, and 24 h and 7 days) post-blast. Blood was collected under isoflurane anesthesia by cardiac puncture, and plasma was separated immediately by centrifugation at 1000 rpm for 10 min. For CSF collection, isoflurane-anesthetized rats were secured in a stereotaxic frame (Stoelting Inc, Wood Dale, IL), and after a midline incision, the cervicospinal muscle was separated to expose the atlanto-ocipital membrane. CSF was removed using a needle puncture of the atlanto-occipital membrane. Tissue protein extraction was carried out by homogenizing brains in tissue protein extraction reagent (T-PER; Thermo Fisher Scientific, Rockford, IL) containing protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO) using an ultrasonic homogenizer. The homogenate was centrifuged at 17,700g for 15 min at 4°C, and the supernatant was collected and stored at −80°C.
Enzyme-linked immunosorbent assay
CCL2 concentrations in plasma, CSF and homogenate supernatant of brain tissue were determined using enzyme-linked immunosorbent assay (ELISA) kits (ThermoFisher Scientific, Carlsbad, CA) in accord with the manufacturer's instructions. All samples were assayed in duplicates. Standards and control samples were run simultaneously for validation.
RNA extraction and polymerase chain reaction analyses
Total RNA from brain tissue was isolated using QUAzol reagent and purified using an RNeasy kit after being treated with DNase, according to the manufacturer's instructions (QIAGEN, Valencia, CA). An equal amount (1 μg) of RNA from each sample was reverse transcribed using the RT2 first-strand kit (QIAGEN). The custom RT2 profiler polymerase chain reaction (PCR) array was used and the complementary DNA was amplified with the RT2 SYBR Green ROX quantitative PCR (qPCR) Mastermix on an ABI 7500 Fast real-time PCR system (Thermo Fisher Scientific, Carlsbad, CA). The RT2 qPCR primer assay was used for verification of relative gene expression.
Histopathology
Rats were anesthetized with isoflurane and perfused transcardially with saline, followed by 4% paraformaldehyde (PFA). After 6 h of further immersion fixation in PFA, the brain was cryoprotected with 20% sucrose for 72 h and rapidly frozen in isopentane pre-cooled to −70°C with dry ice. Serial coronal sections (50 μm) were cut from the cerebrum and brainstem/cerebellum (bregma 2.68 to −4.84 mm and bregma −5.68 to −6.40 mm, respectively), immersed in 4% PFA for 5 days, and then processed using the FD Neurosilver™ kit II (FD NeuroTechologies, Ellicott City, MD). Brain sectioning and silver staining were performed by FD NeuroTechnologies. Silver stained sections were scored based on relative visible intensity of silver precipitates by an experienced, blinded neuropathologist using a bright-field microscope. Scores were assigned from 1 to 4 on a rank scale to indicate an increasing severity of axonal degeneration.
Rotarod testing
The Rotamex-5 rotarod apparatus (Columbus Instruments, Columbus, OH) was used to assess motor coordination and balance. A fixed-speed training regimen was conducted at 10 and 20 rpm with 120 sec/trial and three trials per day for each fixed speed. Rats were trained for 4 consecutive days, followed by a 2-day break, and then received 1 additional day of training. The baseline testing was recorded on the day of blast exposure, and the testing paradigm was the same as that used for training. Rats failing to establish a ≥60-sec baseline runtime were excluded.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (version 6; GraphPad Software Inc., San Diego, CA). Data were analyzed using the Student's t-test or repeated-measures analysis of variance (ANOVA). For multiple comparisons, following ANOVA, significant differences among treatment groups were identified using Tukey's multiple comparison tests. Differences were considered to be significant at the level p < 0.05, and values are expressed as the mean ± standard error.
Results
Blast exposure impaired motor coordination
Rats with baseline performances over 60 sec with both 10- and 20-rpm fixed speeds on the rotarod were randomly assigned to the DHB, SHB, and sham treatment groups. Run times (latencies to fall) and % change in run times relative to baseline performance for each rat were analyzed by one-way ANOVA with Tukey's multiple comparison. At 10 rpm (Fig. 1A and B), DHB rats' runtimes and % changes in run times were significantly different from those of sham controls at 1 and 2 days post-injury. Compared to SHB, DHB rats presented a significant decrease in % changes in run times at 1 day post-injury. At 20 rpm (Fig. 1C and D), DHB rats displayed significantly reduced runtimes (e.g., 31.7 ± 11.3 sec at 24 h post-injury) and greater % changes in run time from baseline (e.g., −71.1 ± 9.1% at 24 h post-injury) compared to the values recorded for both SHB (71.3 ± 10.9 sec, −27.9 ± 8.3%) and sham controls (87.2 ± 5.4 sec, −8.7 ± 4.6%). The changes persisted through 7 days post-injury. The % changes in runtimes in SHB rats were also significantly different from shams on each test date through 7 days post-injury.

Rat's performance on the rotarod. (
Blast-exposure–induced axonal degeneration
The extent of axonal degeneration after blast exposure was determined by silver staining and scored by a blinded neuropathologist, with higher scores reflecting increasing severities of axonal degeneration. Our previous studies consistently showed that blast-induced axonal degeneration was particularly pronounced in cerebellum compared to other brain regions. 36,37 In the present study, the greatest axonal degeneration was again observed in the cerebellar white matter at 7 days post-BOP (Fig. 2A). Double blast exposures resulted in a significant increase in axonal degeneration relative to that observed after a single blast exposure (p < 0.01; Fig. 2B).
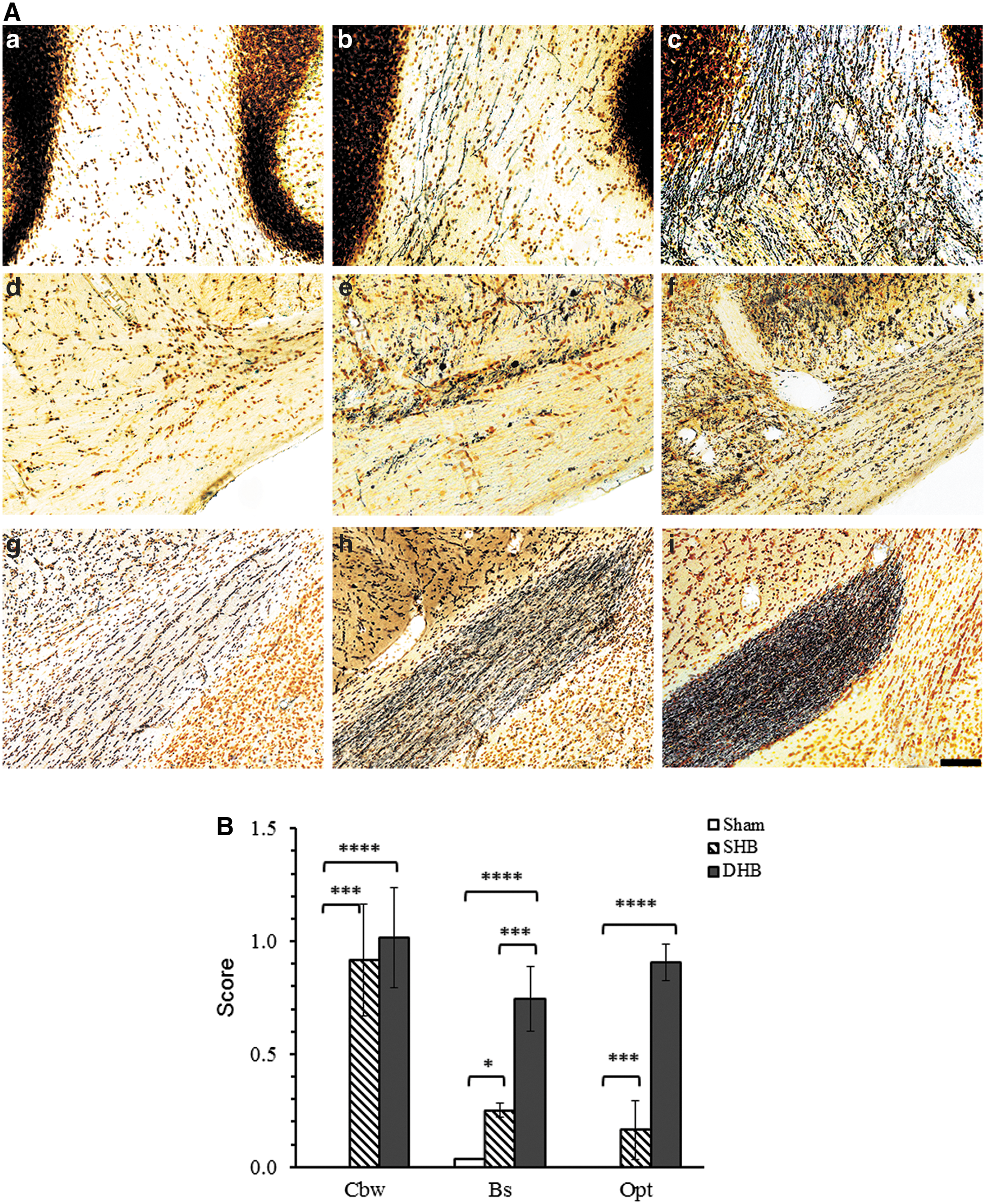
Silver staining showing axonal degeneration induced by blast exposure. At 7–9 days after single blast (
Chemokine (C-C motif) ligand 2 levels in cerebrospinal fluid increased after blast injury
Figure 3A illustrates the CSF concentrations of CCL2 at 1, 6, and 24 h and 7 days after blast exposure(s). These data show that CCL2 levels in CSF were significantly elevated by 1 h and remained increased up to 24 h after double blast exposures, with the greatest CCL2 concentrations measured at 6 h post-injury. At 6 h after DHB exposure, levels of CCL2 were significantly higher than at 1 h post-exposure. In order to address whether the magnitude of the CSF CCL2 changes is injury-severity dependent, we exposed rats to a single blast with 12- (SLB) or 19-psi (SHB) peak total pressure or a double blast (DHB) with 19-psi peak total pressures and measured CCL2 levels at 6 h post-insult. Compared to sham controls, CCL2 levels in CSF were significantly increased after SLB, SHB, and DHB (Fig. 3B). CCL2 concentrations in CSF post-SHB were significantly greater than post-SLB, and elevated CCL2 concentrations elicited by DHB were significantly greater than measured in all other treatment groups.

Effect of blast exposure on CCL2 levels in the CSF. (
Chemokine (C-C motif) ligand 2 levels in plasma increased after blast exposure
Plasma CCL2 were unchanged after exposure to a single 12-psi blast overpressure (SLB). In contrast, plasma CCL2 levels increased dramatically at 6 h post-insult in the 19-psi single blast (SHB) and double (DHB) exposed groups and were significantly different from CCL2 levels measured in both sham and SLB rats (Fig. 4A). The time course of changes in plasma CCL2 after SHB and DHB reveals a peak elevation around 6 h post-exposure (Fig. 4B). There was no difference between the plasma CCL2 levels measured in the DHB and SHB treatment groups at any time point studied.
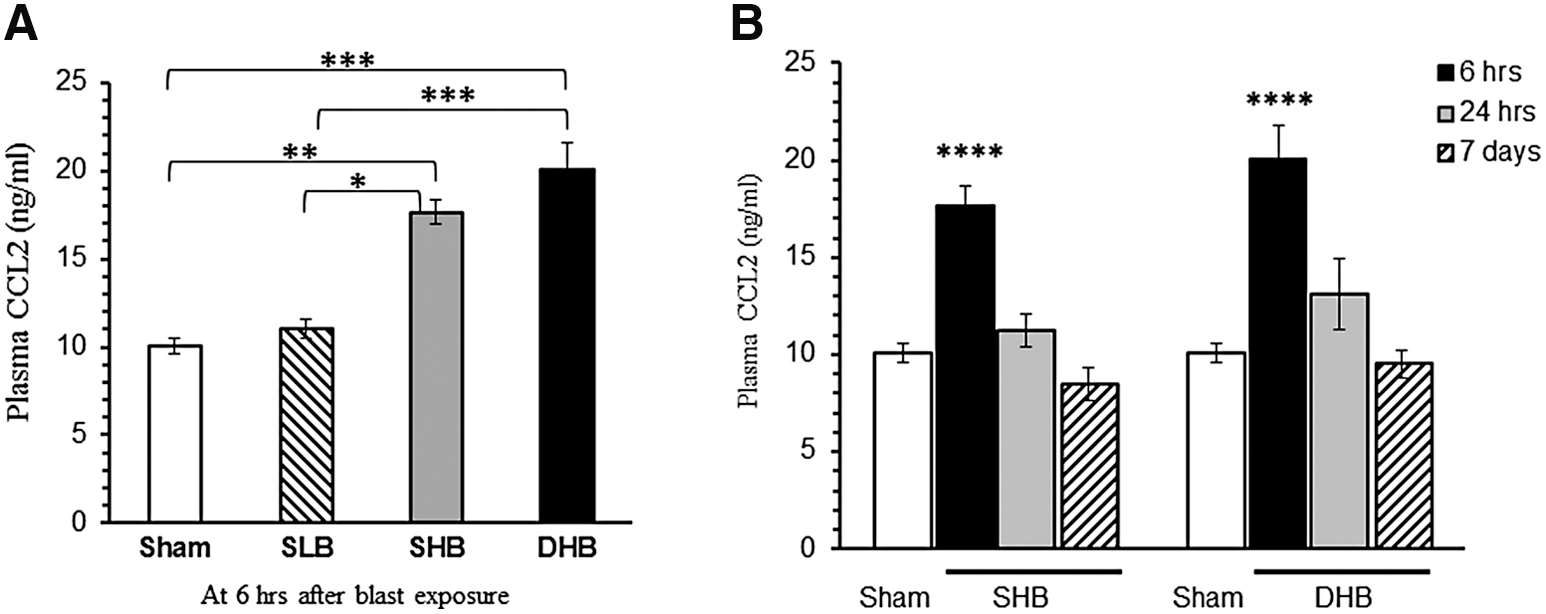
Effect of blast exposure on CCL2 plasma levels. (
The increase in chemokine (C-C motif) ligand 2 in the cerebrospinal fluid after blast exposure was greater than that in plasma
Comparison of the relative increases in CCL2 in CSF and plasma post-DHB reveal that the changes in CCL2 after repeated blasts persisted longer and were considerably greater in CSF than in plasma (Fig. 5A). Specifically, the fold changes of CCL2 in the CSF at 1, 6, and 24 h post–double exposures were 8.70 ± 1.32, 16.49 ± 2.27, and 11.35 ± 2.89, respectively, whereas in the plasma at these times the fold changes were 1.73 ± 0.23, 2.00 ± 0.17, and 1.24 ± 0.23, respectively. The data also showed a positive correlation between CCL2 levels in CSF and plasma (Fig. 5B; Pearson's r = 0.76; p < 0.0002). When compared to sham, the ratio of CSF/plasma CCL2 showed no significant change in rats exposed to a 12-psi single blast. In contrast, after exposure to a 19-psi single blast (Fig. 5C) the ratio increased significantly at 6 and 24 h. Further, rats exposed to double 19-psi BOP had significantly greater CSF/plasma CCL2 ratios at both 6 and 24 h post-injury compared to the other experimental groups.

Comparison of CCL2 concentration in CSF and plasma. (
Upregulation of chemokine (C-C motif) ligand 2 in the cerebellum after blast exposure
Compared to the sham controls (Fig. 6), CCL2 protein levels increased significantly at 6 and 24 h post-SHB. DHB elicited significantly higher levels of CCL2 in the cerebellum than SHB at 6 h post-injury, and these increases persisted at 24 h and 3 days post-DHB.

Blast-induced upregulation of CCL2 protein in the cerebellum. Values are expressed as mean ± standard error of the mean for each group. The Student's t-test and one-way analysis of variance with Tukey's multiple comparisons test were carried out for statistical analysis (n = 8). *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001. CCL2, chemokine (C-C motif) ligand 2; DHB, double high-level blast; SHB, single high-level blast.
Expressions of chemokine (C-C motif) ligand 2 and C-C chemokine receptor type 2 genes increased after blast exposure
We further examined the changes in messenger RNA (mRNA) levels of CCL2 and its main receptor, CCR2, in the cerebellum post-DHB. Total RNA isolated from the cerebellum was subjected to RT2 qPCR primer assay. Compared to sham controls, exposure to DHB resulted in higher levels of CCL2 mRNA in the cerebellum at 6 h and 7 days post-injury (Fig. 7). The blast-induced increase in CCL2 gene expression was greater at 6 h than at 7 days post-injury. Compared to sham controls, expression of CCR2 mRNA also increased significantly at 6 h post-DHB.

Reverse-transcriptase quantitative polymerase chain reaction analysis showing the effect of blast exposure on expressions of CCL2 and CCR2 genes in cerebellum. mRNA levels are presented as a fold change relative to sham control. Statistical analysis was carried out by one-way analysis of variance followed by Tukey's multiple comparisons (n = 8). *p < 0.05; **p < 0.01. CCL2, chemokine (C-C motif) ligand 2; CCR2, C-C chemokine receptor type 2; mRNA, messenger RNA.
To determine an association between CCL2 protein in CSF or plasma and CCL2 gene expression in brain tissue, we performed a statistical analysis on paired data from 20 animals. The ΔCt (threshold cycle change) method, calculating a difference in threshold cycle between the target and house keep genes, was used to quantify gene expression. 38 The results revealed a strong negative correlation (inverse relationship) between the ΔCt value of CCL2 mRNA in the cerebellum and CCL2 protein in CSF (Fig. 8A; Pearson's r = −0.86; p < 0.0001) or in plasma (Fig. 8B; Pearson's r = −0.76; p < 0.001), respectively. These data indicate that blast-induced elevations in CCL2 gene expression in brain cells may contribute to increased CCL2 protein in CSF.

Association between CCL2 level in CSF or plasma and CCL2 gene expression in the cerebellum. ΔCt is the difference after subtracting Ct of actin mRNA from the Ct of CCL2 mRNA in the same sample to normalize for variation in the amount. (
Discussion
The primary clinical indicators of TBI and its severity are the Glasgow Coma Scale, pupil reactivity, and conventional neuroimaging, including head computed axial tomography and magnetic resonance imaging. 39,40 Unfortunately, these outcome measures are typically insensitive to diffuse axonal injury and mTBI. Although advanced structural neuroimaging techniques have been explored as more-sensitive quantitative measures of abnormalities in the brain related to TBI, they are still under investigation to substantiate the clinical relevance in individual patients. 41,42 Over the past decade, molecular biomarkers derived from acute neuronal, axonal, glial, and endothelial injuries that can be measured in biofluids (e.g., CSF and blood) have received considerable attention as promising candidates for diagnosis and prognosis of TBI. Although numerous candidate biomarkers of structural damage, such as neurofilament light, microtubule-associated protein 2, ubiquitin carboxyl terminal hydrolase L1, glial fibrillary acidic protein, and S100B, have been identified and investigated, most are hampered by low specificity and/or sensitivity when used individually. 42 –46 Biomarkers of secondary and reparative processes, such as interleukin (IL)-6, IL-1β, IL-8, and tumor necrosis factor alpha, have also been evaluated in animal studies 47 and in clinical trials, 48 but verification and validation of their utility, particularly for mTBI, remains a sizeable obstacle.
The major challenges confronted in exploration of biomarkers for bTBI include the development of fidelic, reliable, and predictive mTBI animal models and establishment of “high-throughput” biomarker detection methods. In previous studies, we have characterized neuropathological, neurobehavioral, and neurochemical features of mouse and rat models of bTBI using single and tightly coupled repetitive blast overpressure wave exposures within a compressed air-driven shock tube. 36,37,49 Along with others, 50,51 we have observed neuropathological features that closely correspond to neuroimaging findings described in patients with blast-related mTBIs. 50,52 In particular, cerebellar white matter abnormalities and cerebellar dysfunction described in combat veterans parallel the consistent fiber degeneration and Purkinje cell loss observed in these rodent pre-clinical models. 50,51 Using voxel-wise analysis of diffusion tensor imaging to quantify white matter injury, we further observed a pronounced increase in microstructural damage with a closely coupled second blast exposure. This was particularly prominent in the cerebellum, and suggests that primary bTBI may sensitize the brain to subsequent insult from either blast or impact acceleration. 37 In the present study, we have further evaluated the effects of single and closely coupled repeated blast exposures on levels of CCL2 in CSF, plasma, and brain and determined their association with neurobehavioral and neuroanatomical outcomes. These blast-induced disruptions were preceded by significant acute increases in levels of CCL2 in CSF and brain tissue extracts, which corresponded with the number and intensities of blast overpressure exposures and were also commensurate with the eventual extent of neuromotor impairment and neuropathological abnormalities resulting from these exposures. Tightly coupled repeated blast exposures, which caused appreciably greater functional deficits and neuropathological and biochemical changes than a single blast of identical intensity, caused similarly significantly greater elevations of CCL2 in both CSF and brain tissue. Changes in CCL2 in CSF and in plasma correlated with levels of CCL2 mRNA in cerebellum, which, as noted above, is the brain region most consistently and severely neuropathologically disrupted by repeated blast. Although, clearly, these findings do not reveal a causal involvement of CCL2 in the injury mechanisms triggered by BOP, they nevertheless point to this possibility as well as the utility of further consideration of CCL2 as a biomarker for these events.
Chemokines and their receptors are essential for brain development and the maintenance of homeostasis in the central nervous system (CNS). In the MCP family, CCL2 is the most potent at activating signal transduction leading to monocyte transmigration. 53 CCL2 has been suggested to be a biomarker for acute kidney damage and other autoimmune diseases 54,55 and also has been identified as an important mediator of the initiation and maintenance of pain hypersensitivity. 56 –58 The sustained elevation of CCL2 levels in the CSF of patients with sTBI for 10 days post-injury further points to the potential utility of CSF levels of CCL2 as a diagnostic and prognostic marker of brain injury. 22 In the present study, we have demonstrated that changes in CSF CCL2 levels corresponded with the relative severity of the blast overpressure injuries. Given that CCL2 mRNA and protein expression increased substantially in the brain acutely after blast injury, the concomitant sizeable acute spikes in CCL2 concentrations in the CSF likely originated in the brain, even though possible minor contributions from the circulation attributed to leakage across the blast-damaged blood–brain barrier cannot be completely ruled out.
In a recent series of studies, a brain-wide network of paravascular channels, known as the glymphatic system, has been shown to be responsible for clearance of interstitial solutes from the brain and is principally responsible for transport of biomarkers to the blood by the cervical lymphatics. 59,60 Glymphatic activity is impaired post-TBI, 59 which potentially confounds biomarker appearance in the circulation and consequently compromises the clinical utility of blood-based biomarkers to objectively and consistently predict severity of injury. In addition, TBI-induced impairment of glymphatic function has also been shown to be a key factor contributing to aberrant accumulation of phosphorylated tau post-TBI, promoting tau aggregation and onset of neurodegeneration. 59 Given that elevated CCL2 levels were significantly greater at 6 h than at 1 h post-injury, it would appear that CCL2 production and release in the brain increased subsequent to the primary blast insult. The elevated CSF CCL2 levels, which corresponded with the magnitude of motor coordination impairment, persisted longer than did the CCL2 elevations in plasma and might thus have greater utility as an early-response biomarker for diagnosis and prognosis of blast-induced brain injuries than measurements taken from the circulation.
CCL2 is one of a number of brain chemokines that have been extensively evaluated after various forms of experimental TBI, including fluid percussion injury, 27,28 cortical impact injury, 25,29 blast and head-only blast exposure, 21, 23 and weight drop. 22 Semple and colleagues reported that CCL2 levels increased rapidly in cerebral cortices of mice subjected to weight drop closed head inury, with a maximum 40-fold increase at 4–12 h and a return to near normal levels by 24 h post-trauma. 22 A similar temporal pattern of CCL2 change was recorded in the cerebellum after a single blast exposure in the current study and in CSF after single and double blast exposures. In contrast, significantly greater increases in CCL2 in the cerebellum after double blast exposure persisted through 3 days post-injury. It is noteworthy that head-only exposure of rats to blast overpressure transiently altered expression of several brain cytokines and chemokines, which, with the exception of macrophage inflammatory protein 1 alpha, returned to baseline levels by 24 h post-injury, confirming that these changes in the CNS can occur independently of systemic injury responses. 21
In addition to measuring CCL2, we investigated the stability of CCL2 protein and sensitivity of the ELISA kit. We did not find a significant difference in measurements among the samples, which were stored in −20°C, 4°C, and room temperature (data are not shown), indicating a high stability of CCL2 protein. ELISA detection of CCL2 requires only 4 μL of CSF or 2 μL of plasma and is reliable and reproducible.
Recent studies on glia-neuron cross-talk show that astrocyte-derived CCL2 is an important immediate mediator of neuroprotection against excitotoxicity. 61 Although this immediate inflammatory response after primary injury to the brain initially provides a restorative/reparative function, it can persist beyond its beneficial effect and potentially lead to secondary injuries involving alterations in neuronal excitability, axonal integrity, central processing, and other changes stemming from a cytokine storm. These protracted biochemical changes after the insult reflect progressive and potentially reversible molecular and cellular pathophysiological mechanisms, including chemokine-mediated accumulation of inflammatory cells in the brain parenchyma,, which appears to play a crucial role in the pathogenesis of traumatic injuries 15 and in the evolution of neuroinflammatory diseases. 22,34,35 For decades, TBI has been linked to Alzheimer's disease and new data suggest important additional links between neurotrauma and other neurodegenerative diseases, including Parkinson's disease, amyotrophic lateral sclerosis, multiple sclerosis, and Huntington's disease. 62 –64 Given that these studies expand to explore neurodegeneration and/or neurorestoration after blast TBI, CCL2 warrants consideration as part of the neurobiological linkage.
Conclusion
Cytokines have both beneficial and detrimental effects in the injured brain, contributing to the facilitation of neural regeneration and also to secondary brain damage through neuroinflammatory processes. Chemokines and their receptors are of particular interest in the CNS in response to brain injuries. Acute increase in CCL2 expression in the brain and associated elevation in CSF suggested that CCL2 levels in CSF can be used as a biomarker of bTBI. Given that the whole body is exposed to blast, plasma CCL2 levels can be more appropriately considered alone as an indicator for blast-induced polytrauma. Paralleling its proposed roles in other neurodegenerative disorders, sustained high levels of CCL2 and increases in its receptor expression in the CNS post-blast may contribute to neurodegenerative processes, including CTE, and therefore should be recognized as a potentially important target for therapeutic drug intervention. Thus, chemokine ligands and receptors may be targets for immunomodulatory therapy against bTBI.
Footnotes
Acknowledgments
We thank Dr. James DeMar for useful manuscript corrections and comments and Dr. Yuanzhang Li for valuable suggestions regarding data analysis. Technical guidance and assistance from Stephen VanAlbert and Andrea Edwards are also gratefully acknowledged. This work was supported by Congressionally Directed Medical Research Program awards W81XWH-11-2-0127, W81-XWH-08-2-0018, and W81XWH-08-2-0017. The contents, opinions, and assertions contained herein are private views of the authors and are not to be construed as official or reflecting the views of the Department of the Army or the Department of Defense.
Author Disclosure Statement
No competing financial interests exist.