
Abstract
Glibenclamide is a hypoglycemic drug that is widely used for the treatment of diabetes mellitus type 2 (DM II), but it also plays a protective role following injury to the central nervous system (CNS). However, the precise mechanisms underlying its neuroprotective actions remain to be elucidated. Therefore, the present study evaluated the effects of glibenclamide on the blood–brain barrier (BBB) in a mouse model of traumatic brain injury (TBI). In the present study, 86 adult male C57BL/6 mice were exposed to a controlled cortical impact (CCI) injury and then received glibenclamide (10 μg) for 3 days. Tight junction (TJ) protein levels, BBB permeability, and tissue hemoglobin levels were evaluated following the CCI injury. Additionally, a biaxial stretch injury was applied to cell cultures of bEnd.3 cells using the Cell Injury Controller II system to explore the mechanisms by which glibenclamide inhibits apoptosis-signaling pathways. Compared with the control group, glibenclamide-treated mice exhibited decreases in brain water content (p < 0.05), tissue hemoglobin levels (p < 0.05), and Evans Blue extravasation (p < 0.01) after the CCI injury. Glibenclamide primarily attenuated apoptosis via the JNK/c-jun signaling pathway and resulted in an elevation of stretch injury-induced ZO-1 expression in bEnd.3 cells (p < 0.01).Glibenclamide downregulated the activity of the JNK/c-jun apoptosis-signaling pathway which, in turn, decreased apoptosis in endothelial cells (ECs). This may have prevented the disruption of the BBB in a mouse model of TBI.
Introduction
T
Glibenclamide, which is known as glyburide in the United States, is a widely prescribed drug that has been used for the treatment of diabetes mellitus type 2 (DM II) since the 1960s. 6 Glibenclamide binds to sulfonylurea receptors and stimulates the closure of adenosine triphosphate (ATP)-sensitive potassium channels which, in turn, encourages the secretion of insulin from pancreatic β cells. As a member of the sulfonylurea class of drugs, glibenclamide inhibits sulfonylurea receptor 1 (SUR1) and KATP (SUR1/Kir6.2) channels on β cells in the pancreatic islet. This leads to enhanced insulin release, which is beneficial for patients with DM II. 7 –10 Over the last decade, glibenclamide has received renewed attention because of a series of fundamental studies that reported its protective role following acute central nervous system (CNS) injuries, kidney ischemia reperfusion injury, intestinal ischemia, and reperfusion injury. 11,12 Laboratory investigations have also shown that subjects with various CNS pathologies, including focal cerebral ischemia, 13 –17 spinal cord injury, 18 –21 TBI, 22 –24 subarachnoid hemorrhage, 25,26 neonatal encephalopathy caused by prematurity, 27,28 and metastatic brain tumors, 29 benefited from the administration of glibenclamide. Further, retrospective clinical studies have suggested that DM II patients who experience an ischemic stroke would benefit from continued sulfonylurea drug therapy during hospitalization, and that they have improved outcomes following discharge.
The blood–brain barrier (BBB), which is primarily composed of endothelial cells (ECs), but that also receives structural and functional support from astrocytes, pericytes, and components of the extracellular matrix (ECM), separates circulating blood from neural tissue. ECs are a core anatomical element of the BBB; however, they differ from ECs in other tissues because of the presence of continuous intercellular tight junctions (TJs), which greatly limit both the paracellular and transcellular movement of molecules through the EC layer. This implies that the passage of molecules through the BBB is precisely regulated. Therefore, the unique structural and functional features of the BBB guarantee that neurons and glial cells in the brain continue to function properly because of the maintenance of a stable microenvironment. The breakdown of the BBB and/or the disruption of TJs leads to an increased extravasation of immune cells and the poorly regulated flux of molecules and ions across the BBB. Additionally, a failure of the BBB may occur following CNS injuries such as stroke, trauma, subarachnoid hemorrhage, and gliomas, and this type of failure can also induce secondary injuries. 30
Recent research has revealed that glibenclamide has protective capabilities following cardiovascular events. Further, there is an upregulation of SUR1 in the brain following TBI and stroke, and the blockage of this channel with sulfonylurea compounds, such as tolbutamide or glibenclamide, inhibits the swelling of astrocytes and reduces brain edema after stroke. 18 Similarly, glibenclamide attenuates brain edema and the swelling of astrocytes after acute liver failure, 31 intestinal ischemia, and reperfusion injury. 12 Therefore, the present study investigated the effects of glibenclamide on the BBB in a mouse model of TBI.
Methods
Experimental design
The experimental protocol was approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University in Shanghai, China. The present study included 150 adult male C57BL/6 mice weighing 20–25 g (Shanghai SLAC Laboratory Animal Corporation; Shanghai, China) that were divided into two groups: one group received glibenclamide treatment and the other received a vehicle. The glibenclamide was dissolved in water containing 25% Cremophor® EL (Sigma Aldrich; St. Louis, MO) and 10% dimethyl sulfoxide (DMSO; Sigma Aldrich), and the dose (10 μg) was chosen based on a previous study. 32 Both the glibenclamide and vehicle solutions were intraperitoneally injected immediately after the induction of a controlled cortical impact (CCI) injury, and then administered daily until the animals were euthanized. Unless otherwise indicated, the mice were euthanized and samples were collected for further analysis at 1 day and 3 days after the CCI injury. Flow cytometry (BD Biosciences; San Jose, CA) was used to detect apoptotic cells, and the amount of tissue hemorrhaging was quantified using Drabkin's color reagent (Analisa; Belo Horizonte, Brazil). The whole experimental design and the number of animals used are displayed in Figure 1.
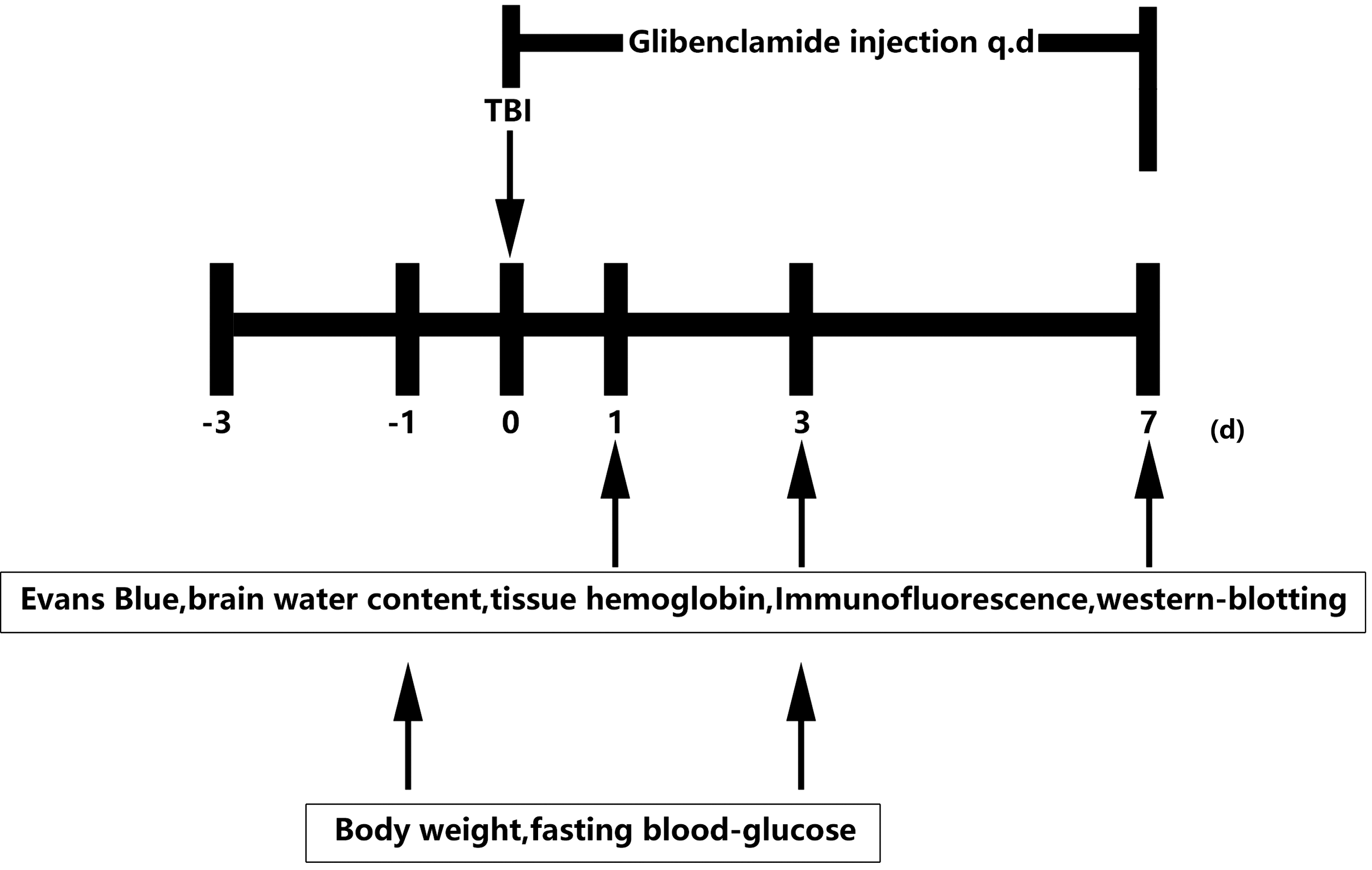
Experimental design. Graph illustrating the experimental design including traumatic brain injury (TBI), glibenclamide administration, Evans blue, brain water content, protein expression, and neurobehavioral assessments.
Surgical procedures and mouse model of CCI injury
Adult male C57BL/6 mice were anesthetized with ketamine (75 mg/kg) and xylazine (10 mg/kg) and then placed on a heated pad to maintain their core body temperature at 37°C. The head was mounted in a stereotaxic frame (Stoelting; Wood Dale, IL), and the surgical site was cleaned with ethanol scrubs. A midline incision (10 mm in length) was made over the skull, the skin and fascia were retracted, and a craniotomy was performed using a 4 mm trephine over the central aspect of the left parietal bone 1 mm lateral to the sagittal suture. Care was taken to keep the dura intact, and mice were excluded from the study if dural integrity was breached.
A TBI was created using a CCI device (PinPoint Precision Cortical Impactor PCI3000, Hatteras Instruments Inc.; Cary, NC) that was rounded, 3 mm in diameter, had a steel impactor tip, and was oriented perpendicular to the cortical surface. An injury of moderate severity was induced using an impact velocity of 1.5 m/sec, a deformation depth of 1.5 mm, and a dwell time of 100 ms. After the injury was induced, sterile cotton was used to control bleeding on the injured cortical surface, the incision was closed with interrupted 6-0 silk sutures, and the animal was placed in a heated cage to maintain its body temperature until regaining full consciousness. Subsequently, all mice were returned to their home cage. Sham animals underwent the exact same procedure as the injured mice, but the impact intervention was not performed. The TBI model and drug treatment were conducted by the same person to minimize variance.
Cell cultures
To investigate the mechanisms underlying the protective actions of glibenclamide, mouse brain microvascular ECs (bEnd.3 cells) were purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco Laboratories; Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, and 100 μg/mL penicillin. The cultures were then transferred to an anaerobic chamber infused with a gas mixture containing 5% CO2 and 95% N2. For the experiments, the cells were seeded at a density of 0.14–0.6 × 105/cm2 on collagen-coated glass cover-slips or BioFlex elastic membrane supports (Flexcell International Corp.; Burlington, NC).
Mechanical cell injury
During preparation for the injury, the cells were grown to confluence on BioFlex six well culture plates with collagen-coated Silastic membranes (Flexcell International Corp). A biaxial stretch was applied to the cell cultures using the Cell Injury Controller II system (Virginia Commonwealth University), which delivers a 50 ms burst of nitrogen gas that produces a downward deformation of the silastic membrane and adherent cells. The bEnd.3 cells were injured by a 7.5 mm membrane deformation and then incubated for 24 h. This degree of stretch is thought to be analogous to the mechanical stress range exerted on the human brain following a rotational acceleration–deceleration injury. 33,34
Measurements of brain edema and brain water content
The water content in the brain was quantified using the wet–dry method, which we have described previously. 35 Briefly, water content was estimated in a 3 mm coronal section of tissue from the ipsilateral cortex (or corresponding contralateral cortex) that was centered on the impact site. The tissue samples were immediately weighed to determine wet weight, and then the samples were dried in an oven at 100°C for 24 h to obtain the dry weight. Tissue water content (%) was calculated as follows: (wet weight-dry weight) / (wet weight × 100).
Evans Blue extravasation
BBB permeability was investigated 3 days after the TBI procedure by measuring the extravasation of Evans Blue (Sigma Aldrich). The dye (2%; 4 mL/kg) was intravenously injected 2 h prior to euthanasia. Following euthanasia, the mice were perfused with phosphate-buffered saline (PBS) through the left ventricle of the heart and, after removal of the intravascular-localized dye, the brain was divided into two hemispheres. Each sample was immediately weighed and homogenized in a solution with 1 mL 50% trichloroacetic acid. The samples were centrifuged at 12,000g for 20 min, the supernatant was diluted with ethanol (1:3), and the absorbance was determined at 620 nm using a spectrophotometer (BioTek, Winooski, VT). The quantity of dye was calculated using a standard curve and expressed as micrograms per gram of brain tissue.
Measurement of tissue hemoglobin levels
Hemoglobin levels in the tissue were used as an index of tissue hemorrhaging. Following the perfusion, a sample of traumatic foci (∼100 mg) was removed from each mouse brain and homogenized in Drabkin's color reagent according to the manufacturer's instructions (Analisa). The samples were centrifuged at 3000g for 15 min, the supernatant was filtered using 0.2 μm filters, and the extravasated blood in the tissue homogenates was quantified at 520 nm with a spectrophotometer (BioTek, Winooski, VT).
Immunostaining
A double-staining procedure using ZO-1 (Invitrogen; Carlsbad, CA)/CD31 (BD Biosciences, San Jose, CA), and occludin (Invitrogen)/CD31 (BD Biosciences) was performed as previously described. 35 Briefly, brain sections were blocked with 10% FBS for 1 h and then incubated with anti-ZO-1 antibodies (1:100 dilution)/anti-CD31 antibodies (1:200 dilution) and anti-occludin antibodies (1:100 dilution)/anti-CD31 antibodies overnight at 4°C. After washing, the brain sections were incubated with the appropriate second antibodies for 1 h at 37°C. Finally, the brain sections were examined with a confocal microscope (Leica; Solms, Germany), and photographs were taken for further analysis.
Western blot analysis
For the Western blot analyses, the samples were lysed in radioimmunoprecipitation assay (RIPA; Millipore; Bedford, MA) supplemented with 1 mmol/L of the PMSF protease inhibitor (Thermo; Waltham, MA), a Halt™ Protease Inhibitor Cocktail (Thermo; Waltham, MA), and a phosphatase inhibitor (Thermo). After denaturing, samples containing the same amount of protein were loaded onto the resolving gel (Promoton; Shanghai, China) for electrophoresis, and the proteins were transferred onto a nitrocellulose membrane (Whatman; Piscataway, NJ). After blocking with 5% nonfat milk, the membranes were incubated with primary antibodies overnight at 4°C at the following dilutions: ZO-1 (1:500), occludin (1:500), JNK, p-JNK (1:1000, Cell Signaling Technology; Beverly, MA), p-c-jun, c-jun (1:500), and β-tubulin (1:1000, Santa Cruz Technology; Santa Cruz, CA). After washing, the membranes were incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature and then reacted with an enhanced chemiluminescence substrate (Pierce; Rockford, IL). The results were recorded using Quantity One image software (Bio-Rad; Hercules, CA), and the relative intensity was calculated with Gel-Pro Analyzer software (Media Cybernetics; Bethesda, MD).
Flow cytometry
bEnd.3 cells were grown on BioFlex six-well culture plates (Flexcell International Corp.). Following the mechanical injury induced by biaxial stretching, the cells were cultured in an incubator at 37°C, humidified in an atmosphere of 5% CO2 for 24 h, treated with 0.25% trypsin without ethylenediaminetetraacetic acid (EDTA), washed twice with cold PBS, and then resuspended in the binding buffer. Next, 100 μL of the solution was transferred to a 1.5 mL centrifuge tube, and a 5 μL mixture of annexin V, Alexa Fluor® 488, and propidium iodide (PI; Component B), was added to the solution. The solution was then gently mixed with the cells, further incubated for 10 min at room temperature under protection from light, and 400 μL of the binding buffer was added to each tube. Apoptotic cells were assessed using flow cytometry within 1 h, and the stained cells and controls were analyzed with an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA).
Statistical analysis
All data are presented as means ± standard errors of the mean. Equality of variance was assessed with a Levene's test, and t tests were used to compare the means. P values <0.05 were considered to indicate statistical significance. GraphPad Prism 6 (GraphPad Software; San Diego, CA) was used to create graphic representations of the data, and all data analyses were performed with SPSS 20.0 for Windows (SPSS Inc., Chicago, IL).
Results
Glibenclamide reduced brain edema in a mouse model of TBI
Mice exposed to the CCI procedure exhibited a significant increase in the water content of the ipsilateral cortical tissue samples (81.3%) compared with the control group (78.2%; p < 0.01 vs. vehicle) at 3 days after TBI (Fig. 2A). There was a significant reduction in the brain water content of mice treated with 10 μg of glibenclamide (79.7%; p < 0.05 vs. vehicle).

Glibenclamide inhibits brain edema after traumatic brain injury (TBI).
Aquaporin 4 (AQP4) plays an important role in the formation of brain edema. 36 To assess the biological effects of glibenclamide on the formation of brain edema after TBI, mice treated with 10 μg of glibenclamide were euthanized 3 days after the TBI procedure. Western blot analyses revealed that AQP4 protein levels increased after TBI but were significantly lower in the glibenclamide-treated group compared with the vehicle group (Fig. 2B–D). Taken together, these results indicate that the inhibition of SUR1 by glibenclamide reduced cerebral edema following TBI and that this, at least in part, occurred via the downregulation of AQP4 expression.
Further, to investigate whether glibenclamide would influence glucose levels after TBI, the fasting blood glucose (FBG) levels and body weight of the mice were assessed prior to and 1 day and 7 days after the TBI procedure. The results indicated that glibenclamide did not have an effect on either FBG levels or body weight (Table 1).
Data are presented with mean-standard error of the mean. Table showed vein blood glucose levels and body weight at 1 day before TBI and at 3 days after TBI in glibenclamide-(Gliben) and vehicle-treated mice (n = 4 per group).
Glibenclamide reduced BBB disruption in a mouse model of TBI
To evaluate BBB permeability after TBI, Evans blue extravasation was evaluated. Glibenclamide treatment remarkably decreased EB leakage in the ipsilateral cortex (Fig. 3A), indicating that TBI-induced BBB disruption was alleviated by SUR1 inhibition (Fig. 3B, p < 0.01 vs. vehicle group).We quantified the amount of extravasated blood present in contused tissues at 1 day after injury (Fig. 3C). Three groups of mice were studied: control, vehicle-treated, and glibenclamide-treated groups. In the vehicle-treated group, the amount of extravasated blood present at 24 h was significantly greater than in the glibenclamide-treated group (Fig. 3D, p < 0.05)
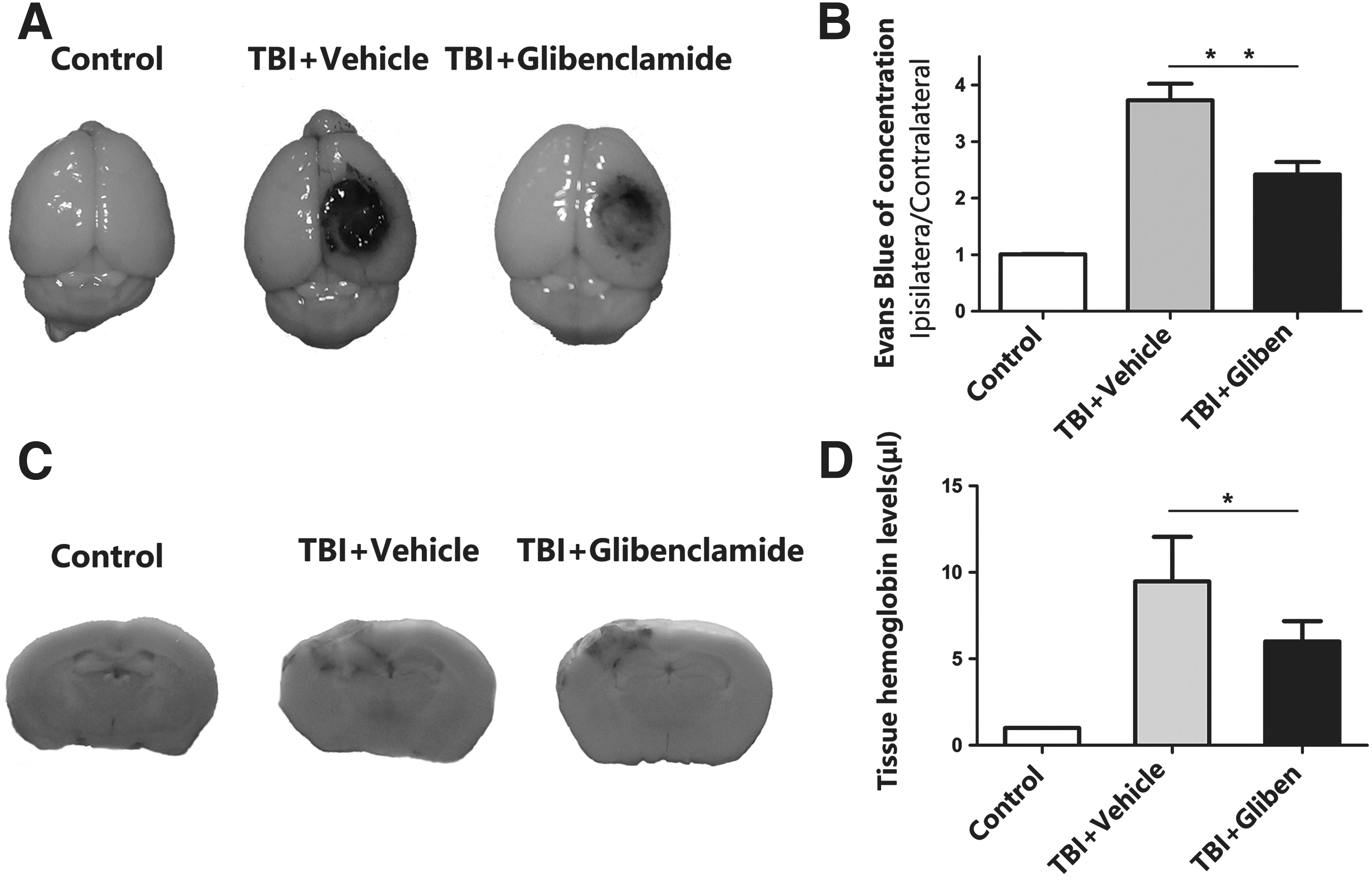
Glibenclamide rearrangement and lessened Evans blue extravasation and tissue hemorrhage level.
To evaluate the integrity of BBB after glibenclamide treatment, a double-staining procedure using ZO-1/CD31 and occludin/CD31 was performed 3 days post-TBI to assess the distribution of TJs in situ based on changes in gap formation and rearrangement. Occludin and ZO-1 were continuously located on the margin of the ECs in the control group, whereas fewer gaps were formed in the glibenclamide-treated group (Fig. 4A). Additionally, Western blot analyses revealed that glibenclamide-treated mice exhibited overexpression of ZO-1 and occludin (Fig. 4B, C; p < 0.5 vs. vehicle group).
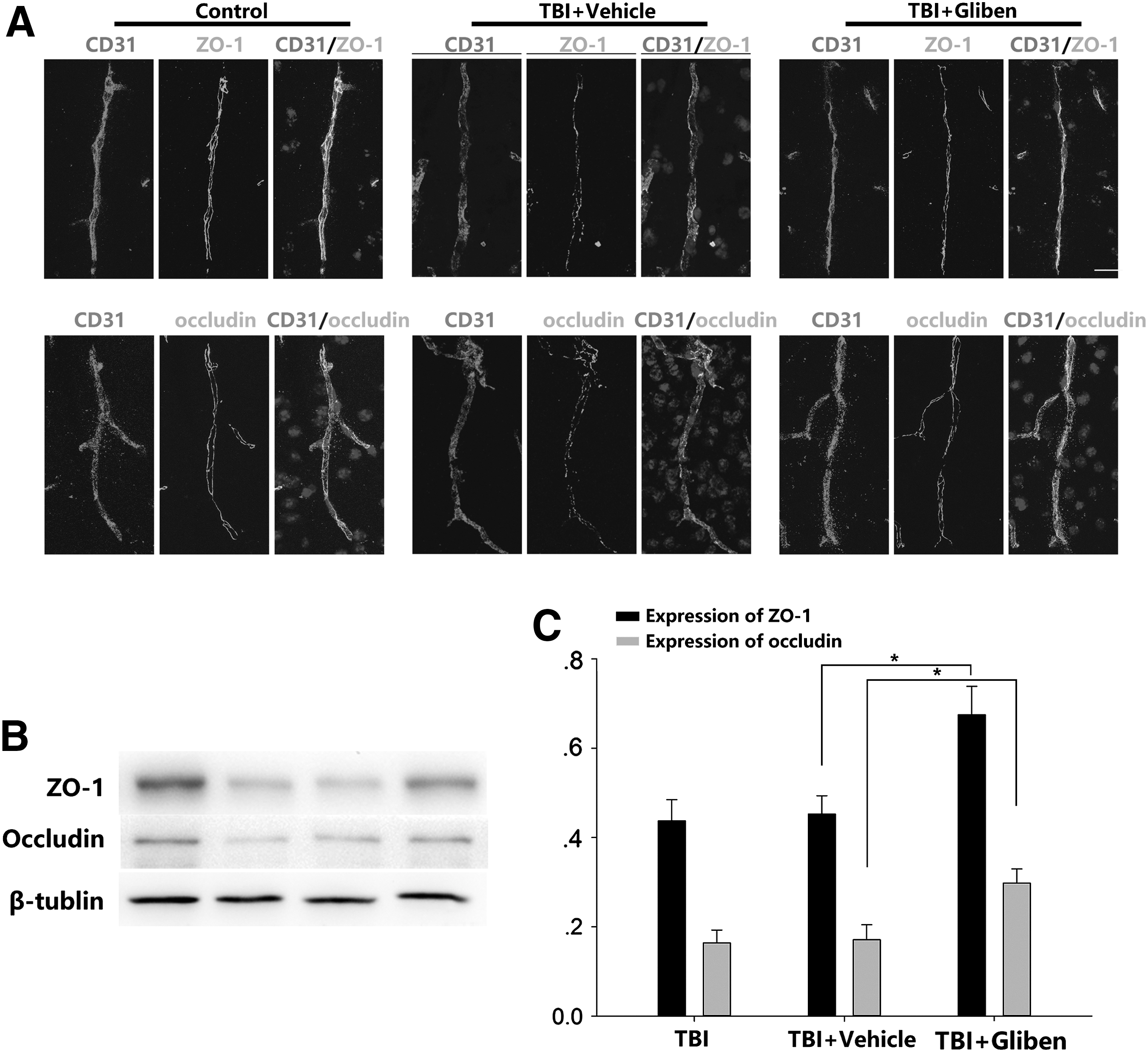
Glibenclamide promoted ZO-1 and occludin after traumatic brain injury (TBI).
Glibenclamide treatment can inhibit loss of ZO-1 and reduce early-stage apoptosis induced by stretch injury on bEnd.3 cells
To assess the ZO-1 level, immunocytochemistry was used from fixed bEnd.3 cells, and protein samples were collected for Western blot at 24 h after stretch injury. ZO-1 was expressed constituently at high levels in ECs, and after stretch injury, its expression was lowered hugely (Fig. 5A, B). Western blot result indicated that ZO-1 was increased after glibenclamide treatment compared with the stretch injury group (Fig. 5B, C; p < 0.05 vs. glibenclamide group).
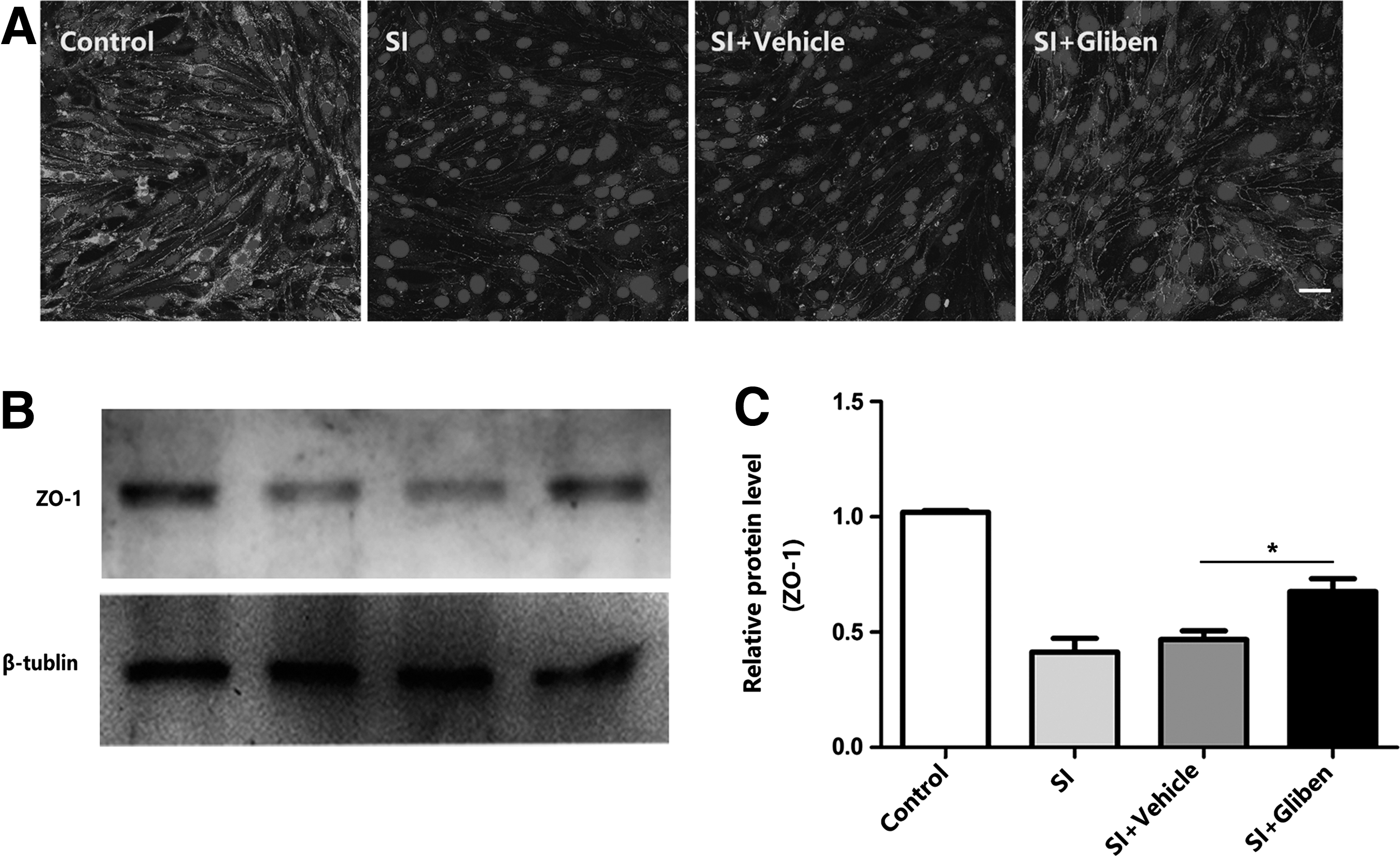
Glibenclamide inhibits loss of tight junction proteins in endothelial cells after stretch injury (SI). bEnd.3 cells were pretreated with vehicle or glibenclamide (10 μM) for 40 min before stretch injury.
Flow cytometry-based assays can be used to determine cell apoptosis by determining the levels of annexin V-positive cells. In the present study, 10 μM of glibenclamide significantly decreased the numbers of Annexin V-positive cells, which are undergoing early-stage apoptosis. These findings suggest that the stretch injury induced early-stage apoptosis (Fig. 6A, B), but that these processes were attenuated by glibenclamide.
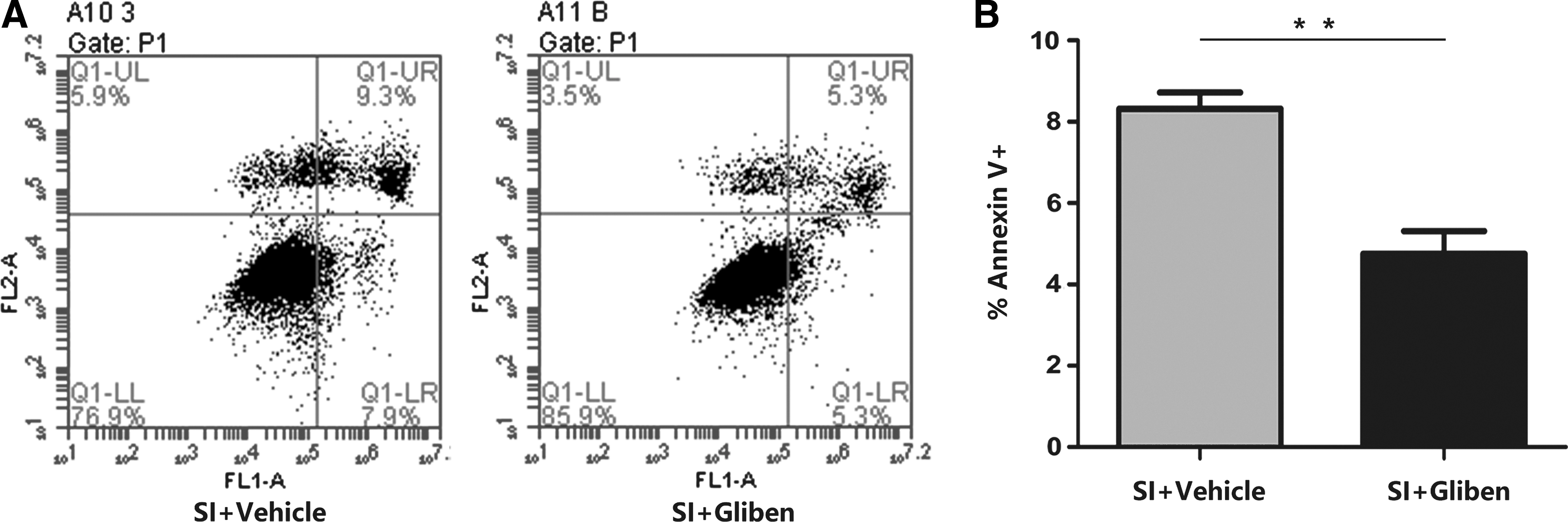
Glibenclamide inhibits apoptosis in endothelial cells after stretch injury (SI). bEnd.3 cells were pretreated with vehicle or glibenclamide (10 μM) for 40 min before stretch injury.
Glibenclamide modulated the stretch injury-induced expression of pro-apoptotic proteins and caspase-3 activation
JNK modulates the expression of pro-apoptotic and anti-apoptotic members of the Bcl-2 family, which are interrelated with the mitochondrial intrinsic apoptotic pathway. 37 Therefore, the expressions of Bcl-2 and BAX were examined in the present study to determine the effects of stretch injury on JNK/c-jun pathway (Fig. 7A, B). Following the stretch injury, the expression of the pro-apoptotic BAX protein significantly increased, whereas that of the anti-apoptotic Bcl-2 protein was markedly reduced (Fig. 8 A, B). However, glibenclamide treatment (10 μM) reversed the stretch injury-induced pro-apoptotic signaling events in the ECs (Fig. 8 A, B) as evidenced by significant reductions in the expression of the BAX protein (Fig. 8 A, B) and significant elevations in the expression of the Bcl-2 protein (Fig. 8 A, B) relative to the vehicle group. Additionally, the level of the cleaved (activated) forms of caspase-3 exhibited an increase 6 h after the stretch injury; however, glibenclamide significantly decreased these levels compared with those of the vehicle controls (Fig. 8 A, B). Therefore, the present results indicate that glibenclamide inhibited apoptotic cell death in ECs following stretch injury.

Effects of glibenclamide on the stretch injury (SI)-induced apoptosis based on the JNK pathway.
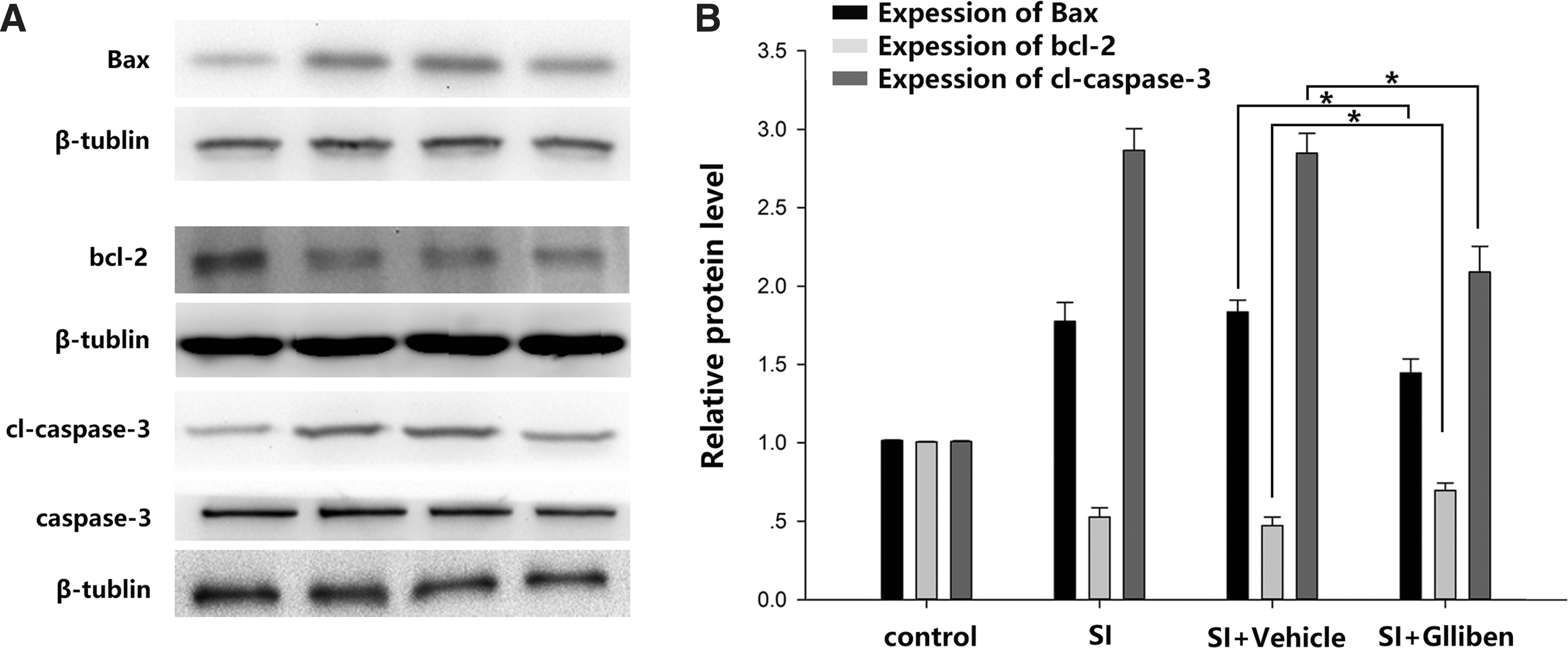
Effects of glibenclamide on the anti-apoptosis and pro-apoptosis proteins in bEnd.3 cells.
Discussion
Glibenclamide is an ATP-sensitive potassium channel (KATP) blocker that is the first-line sulfonylurea pharmacotherapy option for the treatment of DM II in the United States. 38 Previous studies have shown that low doses of glibenclamide can inhibit SUR1 without affecting blood sugar levels. 39 The present study demonstrated that glibenclamide protected the mouse brain against TBI-induced damage via the attenuation of the breakdown of the BBB. The in vivo experiments conducted in the present study showed that the glibenclamide-induced protection of the BBB reduced brain edema and tissue hemorrhaging. These effects were potentially mediated by the protection of ECs via changes in the activity of the JNK/c-jun pathway and its regulation of pro-apoptotic and anti-apoptotic proteins. Further, glibenclamide upregulated ZO-1 in cultured ECs following stretch injury and markedly inhibited the apoptosis of bEnd.3 cells as assessed by flow cytometry.
The JNK/c-jun pathway and its associated proteins play important roles in the stimulation of apoptotic signaling, which can be activated by oxidative stress and other adverse environmental factors. 40 The activation of JNK has been implicated in the apoptosis of ECs following exposure to various chemical and biological agents, 41,42 and JNK is a key contributor to the regulation of the functions of various pro- and anti-apoptotic proteins under pathological conditions.In conjunction with reactive oxygen species (ROS), JNK proteins promote apoptosis via the stimulation of the activities of pro-apoptotic proteins and the inhibition of anti-apoptotic proteins.
Stress-induced apoptosis is associated with the downregulation of the anti-apoptotic Bcl-2 protein and/or with the upregulation of the pro-apoptosis BAX protein. 43 The occurrence of cellular damage has been observed in stretch injury-treated astrocytes and neurons, using many techniques. 34 In the present study, exposure to stretch injury elevated the expression of BAX and reduced the expression of Bcl-2, suggesting that stretch injury stimulates apoptosis via regulation of the JNK/c-jun signaling pathway. However, treatment with glibenclamide reversed the levels of BAX and Bcl-2 in the ECs, which suggests that this drug may be protective against stretch injury-induced damage via the regulation of the JNK/c-jun signaling pathway.
The JNK/c-jun signaling pathway also partly initiates extrinsic apoptotic pathways via the activation of death receptors. 37 Previous studies have shown that the JNK/c-Jun/Ap1 pathway may increase the expression of pro-apoptotic genes, such as tumor necrosis factor-α (TNF-α), FasL, and BAK. Further, the antagonism of SUR1 may protect ECs against oxidative stress. 25 In the present study, there were increased expressions of p-JNK1/2, p-c-Jun, TNF-α, and BAX in bEnd.3 cells following stretch injury, which implies that the activation of the JNK/c-jun signaling pathway is crucial for the induction of damage to the ECs after TBI. However, treatment with glibenclamide significantly attenuated the expression of these proteins, which indicates that the neuroprotective actions of this drug in the BBB may be a result of changes in activity within the JNK signaling pathway.
The apoptotic process is also highly regulated by various forms of caspase. During apoptosis, the caspase pathway is a downstream target of the JNK/c-jun signaling pathway, and the convergence of intrinsic and extrinsic pathways activates caspase-3, which plays an important role in the cleavage or degradation of critical proteins. 44 A number of studies have reported that the increased expression of caspase-3 is related to apoptosis in a variety of organisms 45 and that apoptosis follows the increased expression of caspase-3 mRNA or protein in cells and organisms after exposure to various environmental factors. 46,47 In the present study, stretch injury elevated caspase-3 activity in bEnd.3 cells, but glibenclamide pretreating glibenclamide for 40 min before stretch injury markedly decreased the injury-induced caspase-3 activity in these ECs. These results suggest that glibenclamide may protect ECs against TBI-induced apoptosis by activating the JNK signaling pathway.
Conclusion
In conclusion, the present study examined the protective effects of glibenclamide on BBB integrity after TBI and showed that glibenclamide attenuated the disruption of the BBB and improved functional recovery following the injury. Additionally, the present findings suggest that glibenclamide protected ECs against stretch injury-induced apoptosis by regulating activity within the JNK-signaling pathway. Taken together, these findings indicate that glibenclamide may be a promising potential therapeutic intervention for the prevention of disruption to the BBB after TBI.
Footnotes
Acknowledgments
This work was supported by grants from National Nature and Science Foundation of China (no. 81271383,81471245, 81501048) and the Shanghai Science and Technique Committee (no.13411951401).
Author Disclosure Statement
No competing financial interests exist.