
Abstract
Traumatic brain injury (TBI) is a highly complex multi-factorial disorder. Experimental trauma involves primary and secondary injury cascades that underlie delayed neuronal dysfunction and death. Mitochondrial dysfunction and glutamatergic excitotoxicity are the hallmark mechanisms of damage. Accordingly, a successful pharmacological intervention requires a multi-faceted approach. Guanosine (GUO) is known for its neuromodulator effects in various models of brain pathology, specifically those that involve the glutamatergic system. The aim of the study was to investigate the GUO effects against mitochondrial damage in hippocampus and cortex of rats subjected to TBI, as well as the relationship of this effect with the glutamatergic system. Adult male Wistar rats were subjected to a unilateral moderate fluid percussion brain injury (FPI) and treated 15 min later with GUO (7.5 mg/kg) or vehicle (saline 0.9%). Analyses were performed in hippocampus and cortex 3 h post-trauma and revealed significant mitochondrial dysfunction, characterized by a disrupted membrane potential, unbalanced redox system, decreased mitochondrial viability, and complex I inhibition. Further, disruption of Ca2+ homeostasis and increased mitochondrial swelling was also noted. Our results showed that mitochondrial dysfunction contributed to decreased glutamate uptake and levels of glial glutamate transporters (glutamate transporter 1 and glutamate aspartate transporter), which leads to excitotoxicity. GUO treatment ameliorated mitochondrial damage and glutamatergic dyshomeostasis. Thus, GUO might provide a new efficacious strategy for the treatment acute physiological alterations secondary to TBI.
Introduction
T
Two major pathophysiological processes contribute to brain injury post-trauma: the primary injury, in which damage is caused as a direct result of the mechanical impact; and the secondary injury, which is initiated immediately after brain trauma attributed to further cellular damage from the effects of primary injuries. The latter continues to develop over a period of hours or days. 4 During this second stage, the damage is primarily attributed to neurotransmitter release, calcium overload, free-radical–mediated damage, proapoptotic gene activation, and mitochondrial dysfunction. 5,6
As a principal excitatory neurotransmitter, glutamate plays an essential role in brain plastic processes, such as learning/memory, development, and aging. 7 On the other hand, overstimulation of the glutamatergic system may lead to a excitotoxicity. 8 Glutamate uptake is a crucial process for maintaining extracellular glutamate concentrations below toxic levels. This is achieved through specific excitatory amino acid transporters (glutamate aspartate transporter [GLAST] and glutamate transporter 1 [GLT-1]) that are mainly present in astrocytes. These transporter proteins are modulated by the cell redox status 9 ; thus, increased reactive oxygen species (ROS) production and calcium overload in mitochondria may result in glutamate uptake impairment and cellular death. 10
Mitochondria, an important organelle for cellular bioenergetics, are sensitive to changes in the physiological state of cells and play a critical role in secondary injury post-trauma. 11 In particular, mitochondria are responsible for the functional and structural damage post-TBI. 12 –14 Dysfunctions in this organelle post-TBI have been linked to the impairment of brain mitochondrial electron transfer chain and energy transduction attributed to the overloading of mitochondrion-associated calcium, 12 oxidative damage, and disruption of synaptic homeostasis, 15 ultimately ensuing with cell death. 16
Several therapeutic interventions that target mitochondria have shown promising results by reducing overall neuronal tissue damage as well as enhancing neurological outcome post-TBI. 17 –19 In addition, pharmacological agents that also focused on the glutamatergic system have been shown to be neuroprotective. 20,21 Neurodegenerative and neurotoxic models have demonstrated that guanosine (GUO) plays an important role in the central nervous system. 22 This concept led us to posit that it may act through these signaling pathways, attenuating excitotoxicity and mitochondrial damage. GUO has been implicated in neuroprotection through modulation of the glutamatergic system, counteracting glutamate excitotoxicity in vitro 23 –25 and in vivo. 22,26 Among these actions, GUO has shown efficacy both in a stroke model 27 and acute spinal cord injury in rats. 28
Given these observations and the lack of efficacious trauma treatments, the aim of this study was to investigate whether GUO might protect mitochondrial damage induced by TBI in hippocampus and cortex of rats and to verify the relationship of this effect with modulation of glutamatergic transmission.
Methods
Chemical reagents
2′,7′-dichlorofluorescein diacetate (DCFH-DA), methyltetrazolium bromide (MTT), nicotinamide adenine dinucleotide (NADH), 2,6 dichloroindophenol (DCIP), safranine-O, o-phthalaldehyde (OPT), N-ethylmaleymide, and GUO were purchased from Sigma-Aldrich (St. Louis, MO). [ 3 H]-glutamic acid was purchased from Amersham Biosciences (Amersham, UK). All other chemicals were of analytical grade and were obtained from standard commercial suppliers.
Animals
Male adult Wistar rats (280–320 g) were obtained from a local breeding colony. The animals were kept in a separate animal room, on a 12-h light/dark cycle, in an air-conditioned room (22 ± 2°C, 45–65% humidity). Commercial diet (GUABI, RS, Brazil) and tap water were supplied ad libitum. All the animals were acclimatized to laboratory conditions for 1 week before start the experiment. This study was carried out in strict accord with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Federal University of Santa Maria, Brazil (Permit Number: 153/2014).
Fluid percussion brain injury and experimental design
The fluid percussion brain injury (FPI) model was carried out as previously described. 29 In brief, animals were anesthetized with a single intraperitoneal (i.p.) injection of Equithesin (6 mL/kg), a mixture containing sodium pentobarbital (58 mg/kg), chloral hydrate (60 mg/kg), magnesium sulfate (127.2 mg/kg), propylene glycol (42.8%), and absolute ethanol (11.6%), and placed in a rodent stereotaxic apparatus. A 3-mm-diameter burr hole was drilled on the right convexity, 2 mm posterior to the bregma and 3 mm lateral to the midline, assuring the dura mater remained intact. A plastic injury cannula was placed over the craniotomy with dental cement. When dental cement had hardened, the cannula was filled with chloramphenicol, closed with a proper plastic cap, and the animal removed from the stereotaxic device and returned to its home cage. After 24 h, animals were anesthetized (isoflurane 1%) and the injury cannula was attached to the fluid percussion device. During surgery and FPI, body temperature (37°C) was monitored rectally and maintained with a heating pad and an overhead incandescent bulb. The TBI was produced by a fluid percussion device developed in our laboratory. A brief (10–15 ms) transient pressure fluid pulse (1.46 ± 0.09 atmospheres) impact was applied against the exposed dura causing apnea (30–70 sec), unconsciousness (7–10 min) measured through the righting reflex restoration, 30 and mortality post-TBI was 27.78%. Based on the data, we can classify the FPI protocol as a moderate TBI. 30,31 Pressure pulses were measured extracranially by a transducer (FLUID, Fluid Control hydraulic automation; Belo Horizonte, MG, Brazil) and recorded on a storage oscilloscope (Tektronix TDS 210; Tektronix, Beaverton, OR).
Sham-operated and sham-GUO animals underwent an identical procedure, with the exception of FPI. The animals were randomly divided into four groups: Sham-saline; Sham-GUO; TBI-saline; and TBI-GUO. The GUO dose was chosen based on previous in vivo studies of excitotoxicity, which demonstrated the optimal effect of GUO at 7.5 mg/kg. 26,32 The animals received a single dose of guanosine (7.5 mg/kg dissolved in 0.9% saline) or saline 15 min post-TBI (Fig. 1) by i.p. route. After 3 h, the animals were euthanized by decapitation, the brain was removed, and structures (hippocampus and cerebral cortex) were separated. Samples were used for mitochondrial isolation and for slices preparation, as described below.

Representative illustration of experimental procedure. TBI, traumatic brain injury.
Mitochondrial isolation
Mitochondria were isolated by conventional differential centrifugation as described elsewhere, with some modifications. 30,31 At the end of the TBI protocol described above, the brain was rapidly removed to an ice-cold “isolation buffer” (0.32 M of sucrose, 1 mM of ethylene diamine tetraacetic acid (EDTA), 1 mM of ethylene glycol tetraacetic acid, and 10 mM of Tris-HCl, at pH 7.4) kept at 4°C throughout the isolation procedure, and brain structures (hippocampus and cortex) were separated. Tissue was manually homogenized during two cycles of 10 sec in a Teflon glass potter. The homogenate was centrifuged at 1330g for 5 min in a Hitachi Himac SCR20B RPR 20-2 rotor (Hitachi, Tokyo, Japan). The supernatant was carefully removed and centrifuged at 21,200g for 10 min. The pellets obtained were resuspended in buffer isolation with 15% Percoll. The discontinuous density gradient was prepared manually by layering 3-mL fractions of the resuspended pellet on two pre-formed layers consisting of 3.5 mL of 23% Percoll above 3.5 mL of 40% Percoll. Tubes were centrifuged for 5 min at 30,700g with slow brake deceleration. The material equilibrating near the interface between the 23% and 40% Percoll layers was gently diluted 1:4 with isolation buffer and then centrifuged at 16,700g for 10 min. A firm pellet was obtained and gently resuspended in the isolation buffer and supplemented with 0.2 mg/mL of fatty-acid–free bovine serum albumin (BSA). After centrifugation at 6900g for 10 min, the supernatant was rapidly decanted and the pellet resuspended in the same buffer using a fine Teflon pestle.
Mitochondrial function and redox state
Estimation of reactive oxygen species production
Mitochondrial generation of ROS was determined spectrofluorimetrically with the DCFH-DA assay. 33 Briefly, mitochondrial samples of brain structures (150 μg of protein/mL) were incubated with “isolation buffer III” and the respiratory substrates, glutamate/malate (5 mM) and succinate (5 mM). The reaction was started with the DCFH-DA (1-μM) addition, and the medium was kept at constant stirring during the assay period. Fluorescence analysis was performed at 488 nm for excitation and 525 nm for emission, with slit widths of 3 nm.
Mitochondrial membrane potential determination
Mitochondrial membrane potential (ΔΨm) determination was estimated by fluorescence changes in safranine-O assayed. 34 Briefly, mitochondrial samples of brain structures (250 μg of protein/mL) were incubated with “isolation buffer III”, safranine-O (10 μM), and the respiratory substrates, glutamate/malate (5 mM) and succinate (5 mM). In the end-of-period assay, 2,4 dinitrophenol (0.1 mM) was added to analyze the maximal mitochondrial uncoupling. Fluorescence analysis was performed at 495 nm for excitation and 586 nm for emission, with slit widths of 3 nm. ΔΨ m is presented as arbitrary fluorescence units per second (AFU/s).
Glutathione reduced and oxidized levels
Mitochondrial reduced glutathione (GSH) and oxidized glutathione (GSSG) levels were measured by the fluorimetric method, with some modifications. 35 Mitochondria samples (500 μg of protein/mL) were resuspended in sodium-phosphate buffer and phosphoric acid (H3PO4) 4.5%, and were centrifuged at 100,000g for 30 min (HITACHI CP 80WX Ultracentrifuge; Hitachi). For GSH determination, the supernatant was added to phosphate buffer and OPT (1 mg/mL). Fluorescence was measured at 420 nm for emission and 350 nm for excitation, with slit widths of 3 nm. For GSSG determination, the supernatant resulting from the centrifugation was added to N-ethylmaleymide (40 mM) and incubated at room temperature for 30 min. After the incubation, it was added to NaOH solution (100 mM) and OPT. GSH and GSSG levels were determined from comparisons with a linear GSH or GSSG standard curve, respectively. Results are expressed as GSH/GSSG ratio.
Manganese superoxide dismutase activity
Aliquots (100-μL) of isolated mitochondria were added to a medium containing sodium bicarbonate-carbonate buffer (50 mM; pH 10.2), EDTA (2 mM), KCN (1 mM), and adrenaline (0.4 mM). The kinetic analysis of superoxide dismutase (SOD) was started after adrenaline addition, and the color reaction was measured at 480 nm. 36
Dehydrogenase activity
Dehydrogenase activity assay was carried out according to Bernas and Dobrucki, with minor modifications. 37 Samples were incubated in buffer containing glutamate/succinate (5 mM each) and MTT (0.5 mg/mL) for 30 min at 37°C, and MTT reduction reaction was stopped by the addition of 1 mL of dimethyl sulfoxide. Formazan levels were determined spectrophotometrically, reported as the difference of absorbance between 570 and 630 nm. Individual samples were expressed as a percent of the mean control value in the experiment.
Mitochondrial complex I assay
The activity of complex I (NADH dehydrogenase) was performed according to Bottje and colleagues. 38 Briefly, activity was measured by oxidation of NADH. Mitochondria samples were added to a solution containing 35 mM of potassium phosphate buffer (pH 7.4) and 1.3 mM of DCIP in a final volume of 1 mL. The reaction was initiated with the addition of 0.15 mM of NADH. Absorbance at 600 nm was monitored for 3 min to follow the rate of oxidation of NADH, and activity was determined using an extinction coefficient of 6.22 mM−1 cm−1.
Mitochondrial complex II assay
Activity of complex II (succinate: ubiquinone oxidoreductase) was determined through reduction of DCIP by succinate. 39 The reaction mixture consisted of 50 mM of potassium phosphate buffer (pH 7.0), 1 mM of KCN, 0.05 mM of DCIP, 20 μM of rotenone, 16 mM of succinate, and 0.1 mg prot/mL of mitochondria. Absorbance changes were followed at 600 nm, using an extinction coefficient of 19.1 mM–1 cm–1 for DCIP.
Swelling
Measurement of mitochondrial swelling was performed in a RF-5301 Shimadzu spectrofluorometer (Shimadzu Corporation, Kyoto, Japan) at 600 nm (slit 1.5 nm for excitation and emission). 40 Briefly, mitochondrial samples (100 μg of protein/mL) were incubated with reaction buffer (250 mM of sucrose and 10 mM of Tris-HCl, pH 7.4), inorganic phosphate, CaCl2 (0.1 mM), and with the respiratory substrates, glutamate/malate (5 mM) and succinate (5 mM). Data for mitochondrial swelling are expressed as arbitrary absorbance units per second (AU/s).
Functionality of glutamatergic system
[3H]-glutamate uptake assay
After decapitation, the hippocampus and cortex were immediately dissected on ice (4°C). The slices (0.4-mm thickness) were rapidly obtained using a McIlwain Tissue Chopper and immersed at 4°C in Hank's balanced salt solution (HBSS) buffer (pH 7.2). Subsequently, slices were pre-incubated with HBSS at 37°C for 15 min. Briefly, the incubation was started by the addition of 0.33 μCi/mL of [3H]-glutamate. The reaction was stopped after 7 (cortex) or 5 min (hippocampus) with two ice-cold washes using 1 mL of cold HBSS. After washing, 0.5 N of NaOH were immediately added to the slices and they were stored overnight. Sodium-independent uptake was determined using choline chloride instead of NaCl, which was subtracted from the total uptake to obtain the Na+-dependent uptake. 23 Incorporated radioactivity was determined with a Packard scintillator (Microbeta 2 ; PerkinElmer, Waltham, MA). Protein content was determined using BSA as a standard. 41
Expression of glutamate transporters
Western blotting was performed according to Franco and colleagues. 42 The hippocampus and cortex were homogenized at 4°C in a buffer (pH 7.0) containing 50 mM of Tris, 1 mM of EDTA, 0.1 mM of phenylmethyl sulfonyl fluoride, 20 mM of Na3VO4, 100 mM of sodium fluoride, and protease inhibitor cocktail (Sigma-Aldrich). The homogenates were centrifuged at 1000g for 10 min at 4°C, and the supernatants were collected. After total protein determination, 41 using BSA as a standard, β-mercaptoethanol was added to samples in a final concentration of 8%. Then, samples were frozen at −80°C for further analysis. Proteins were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Then, membranes were incubated with specific primary antibodies of glutamate transporters anti-rabbit GLAST (1:1000; Abcam, Cambridge, MA), anti-mouse GLT-1 (1:1000; Thermo Fisher Scientific, Waltham, MA), and β-actin (1:5000; Sigma-Aldrich) for the determination of specific protein targets. Nitrocellulose membranes were provided by GE Healthcare (São Paulo, Brazil). Anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG produced in goat, used as secondary antibody, were purchased from Sigma-Aldrich (São Paulo, Brazil). Densitometric analyses were performed by Quantity One Bio-Rad software (Bio-Rad Laboratories, Hercules, CA), and data were normalized to β-actin as protein control and represented as the percentage relative to sham group.
Protein determination
Protein was measured by the Coomassie blue method, according to Bradford using, BSA as a standard (1 mg/mL). 41
Statistical analysis
Ex vivo data were analyzed using a two-way analysis (GUO × TBI) of variance, followed by post-hoc comparisons using Newman-Keuls' multiple test, when appropriate. Main effects are presented only when the second-order interaction was not significant. Results are expressed as the mean ± standard error of the mean. Differences between groups were considered statistically significant when p < 0.05.
Results
Estimation of mitochondrial membrane potential in mitochondria
ΔΨm showed a significant TBI and GUO interaction in hippocampus (F (1.16) = 6.14; p < 0.01; Fig. 2A) and cortex (F (1,16) = 13.03; p < 0.01; Fig. 2B), corroborating the increased ROS generation. TBI caused a significant decrease in ΔΨm . Moreover, the GUO treatment restored the ΔΨm to levels indistinguishable from the sham-operated animals in both cerebral structures. This disruption in mitochondrial function is consistent with changes in redox status.

Effect of GUO treatment (7.5 mg/kg intraperitoneally) on mitochondrial membrane potential (ΔΨm) in hippocampus and cortex of rats submitted to TBI. Mitochondrial membrane potential (
Mitochondrial redox state
Optimal mitochondrial function is vital for maintain homeostasis in neural cellular metabolism. Our results corroborate with these hypothesis; the analysis of DCFH levels revealed a significant TBI and GUO interaction in hippocampus (F (1.16) = 8.01; p < 0.01; Fig. 3A) and cortex (F (1,16) = 7.54; p < 0.001; Fig. 3B) of rats. The TBI group showed a significant increase in ROS production in both brain regions in comparison to the sham group, and treatment with GUO protected against this effect.

Effect of GUO treatment (7.5 mg/kg intraperitoneally) on mitochondrial redox status in hippocampus and cortex of rats submitted to TBI. ROS levels (
Altered GSH levels, and of manganese superoxide dismutase (MnSOD), activity has been implicated as potential contributors to oxidative damage and alteration in mitochondrial redox system post-trauma. Evaluation of GSH/GSSG levels demonstrated a significant main effect of TBI in hippocampus (F (1.16) = 6.34; p < 0.05; Fig. 3C) and cortex (F (1.16) = 10.33; p < 0.05; Fig. 3D). The results showed a reduction in GSH levels caused by TBI; treatment with GUO ameliorated this effect in both structures. Moreover, analysis of MnSOD activity showed a significant interaction between TBI and GUO in hippocampus (F (1.16) = 8.64; p < 0.01; Fig. 3E) and cortex (F (1.16) = 7.25; p < 0.01; Fig. 3F) of rats. Trauma caused an inhibition of MnSOD activity and GUO treatment protected against this effect in all structures, showing indistinguishable activity levels from the sham group.
Dehydrogenase activity and mitochondrial complex I and II assays
The previous results showed increased ROS production associated with a loss of membrane potential that might reduce electron flow and adversely affect mitochondrial function post-TBI. Analysis of dehydrogenase activity displayed a significant TBI and GUO interaction in hippocampus (F (1.16) = 6.50; p < 0.001; Fig. 4A) whereas in cortex only a main effect of TBI (F (1.16) = 37.52; p < 0.001; Fig. 4B). The results showed decreased mitochondrial dehydrogenase activity in both brain structures of the TBI group. Treatment with GUO maintained normal enzyme activity in the hippocampus in comparison to the sham group. In contrast, it only partially restored the dehydrogenase activity in the cortex.
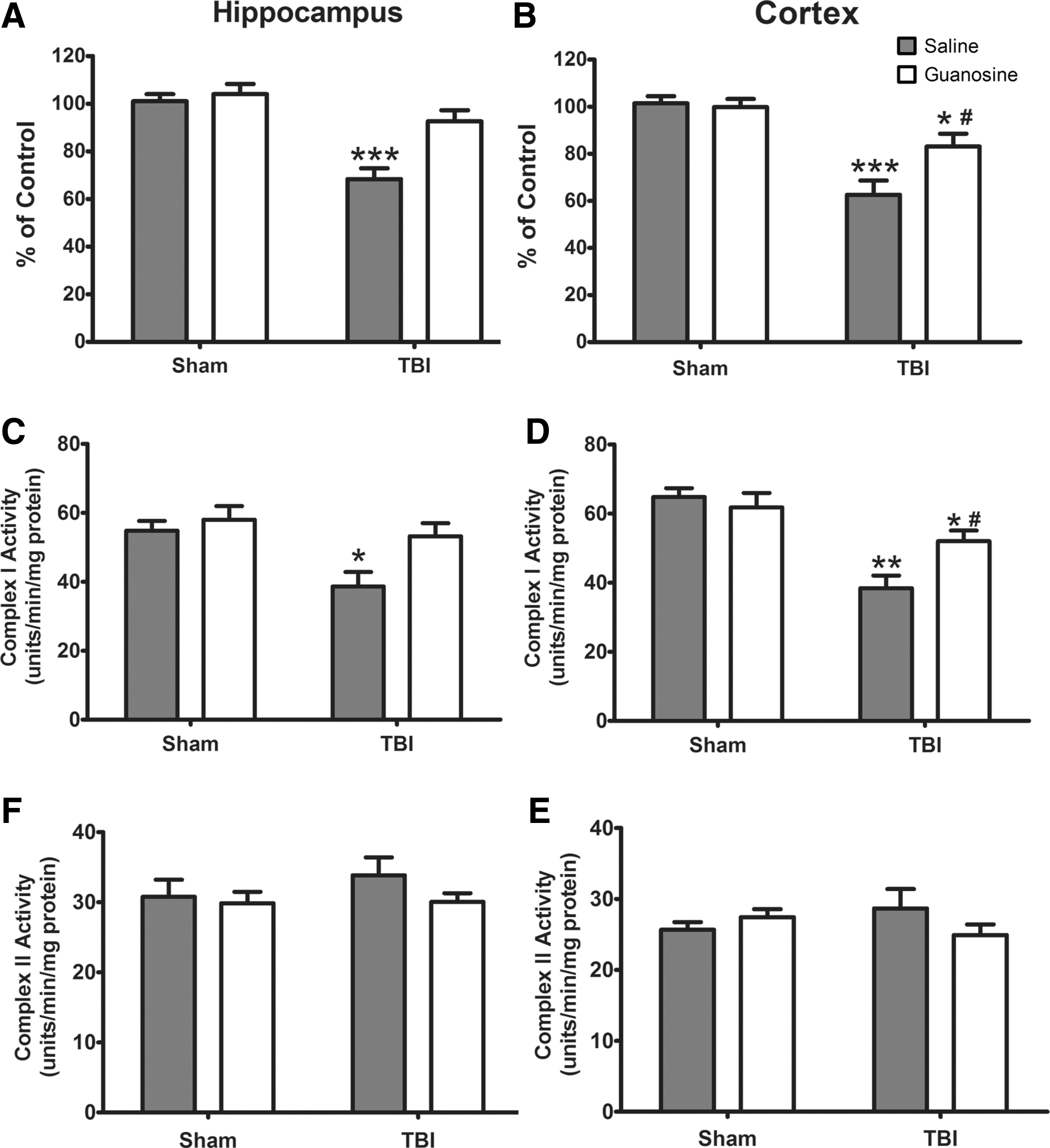
Effect of GUO treatment (7.5 mg/kg intraperitoneally) on dehydrogenase and mitochondrial complex I and II activities in hippocampus and cortex of rats submitted to TBI. Dehydrogenase activity (
Analysis of mitochondrial complex I activity revealed a significant main effect of TBI in hippocampus (F (1.16) = 6.78; p < 0.01; Fig. 4C) and cortex (F (1.16) = 5.32; p < 0.01; Fig. 4D). The complex I activity was inhibited by TBI in both structures and GUO treatment fully restored the activity in the hippocampus and partially in the cortex. The complex II activity was not inhibited in either hippocampus (Fig. 4E) or cortex (Fig. 4F).
Mitochondrial swelling
Analysis of maximal calcium-buffering capacity revealed a significant interaction between TBI and GUO in hippocampus (F (1.16) = 6.10; p < 0.01; Fig. 5A) whereas in cortex there was a significant main effect of TBI (F (1.16) = 17.19; p < 0.01; Fig. 5B). Mitochondrial swelling was noted in the hippocampus and cortex of the TBI group, with GUO treatment preventing this effect in the hippocampus. In the cortex, it blunted the mitochondrial swelling in relation to TBI, but could not restore it to sham levels. These data confirm the hypothesis that excessive Ca+2 uptake into mitochondria is intimately related to functional impairment of this organelle. The alterations found on disruption of Ca2+ homeostasis associated with mitochondrial dysfunction led us to evaluate the glutamatergic system functionality.
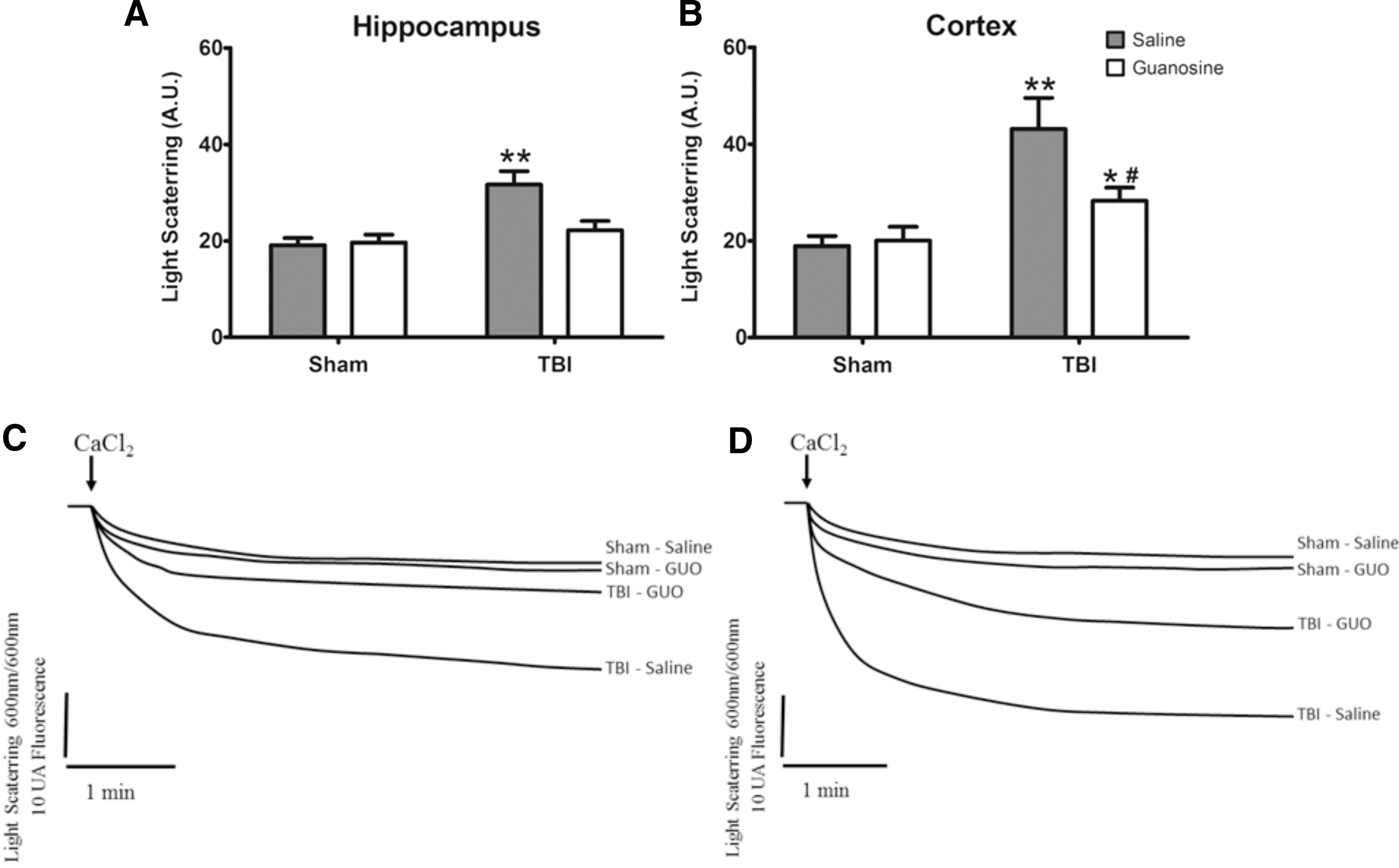
Effect of GUO treatment (7.5 mg/kg intraperitoneally) on mitochondrial swelling in hippocampus and cortex of rats submitted to TBI. Mitochondrial swelling (
Glutamatergic system evaluation
Analysis of glutamate uptake showed a significant TBI and GUO interaction in hippocampus (F (1.16) = 6.37; p < 0.01; Fig. 6A), whereas in cortex there was a significant main effect of TBI (F (1.16) = 38.46; p < 0.001; Fig. 6B). TBI led to decreased glutamate uptake in the cerebral structures of rats when compared to the sham group. Treatment with GUO fully reversed this alteration in the hippocampus and partially in the cortex.
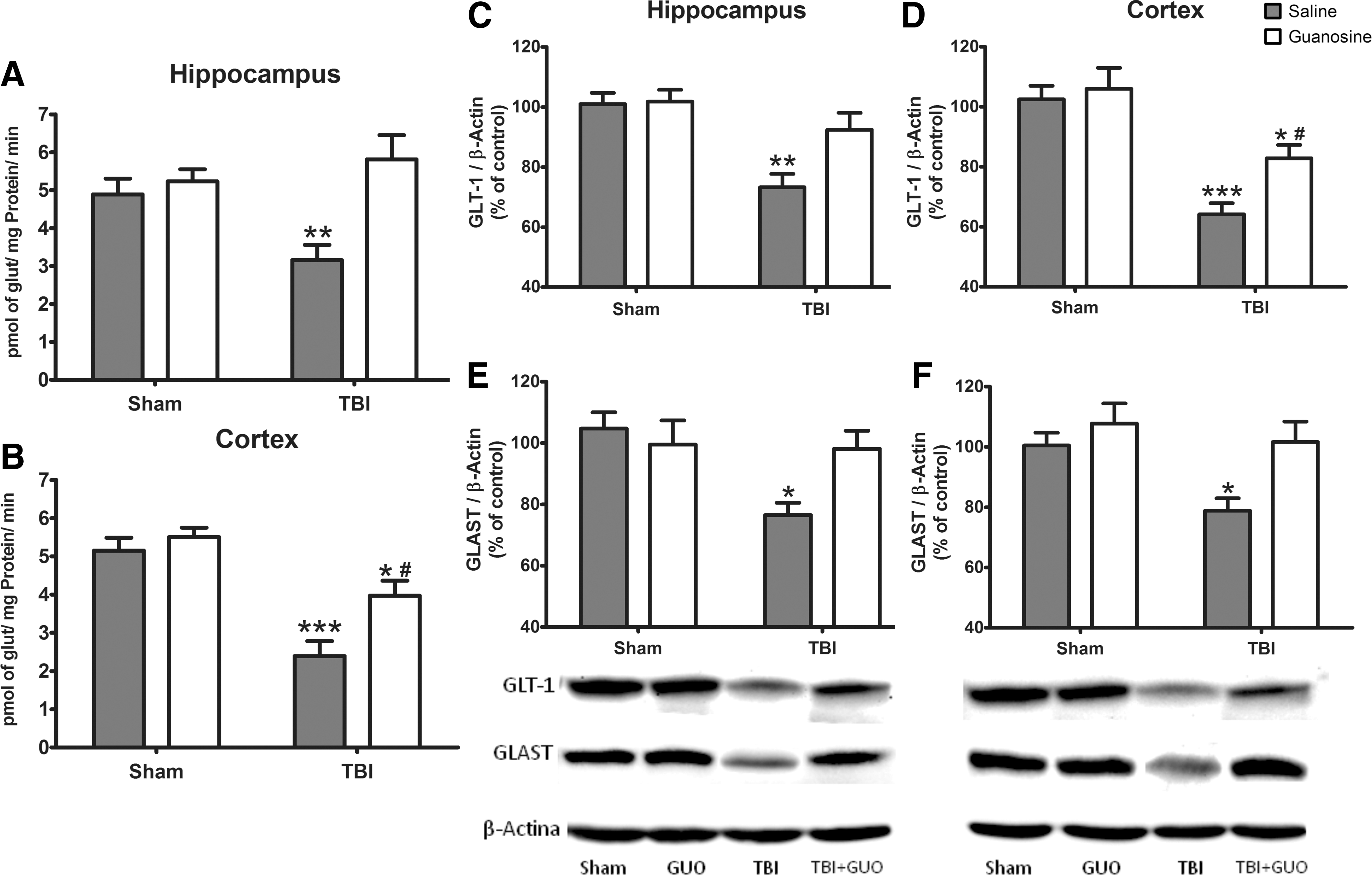
Effect of GUO treatment (7.5 mg/kg intraperitoneally) on glutamatergic system functionality in hippocampus and cortex of rats submitted to TBI. Glutamate uptake (
Analysis of GLT-1 levels showed a main effect of TBI in hippocampus (F (1.16) = 16.56; p < 0.01; Fig. 6C) and cortex (F (1.16) = 32.93; p < 0.001; Fig. 6D) of rats. TBI led to a reduction in GLT-1 levels in both cerebral structures when compared to the sham group. GUO fully reversed this reduction in the hippocampus and partially restored this effect in the cortex. The results on GLAST levels demonstrated a significant TBI and GUO interaction in hippocampus (F (1.16) = 5.67; p < 0.05; Fig. 6E) and cortex (F (1.16) = 5.46; p < 0.05; Fig. 6F). The data revealed diminished expression of GLAST caused by TBI in both cerebral structures and protective effect by GUO treatment.
Discussion
TBI is a major public health concern and a leading cause of disability worldwide. 2 Though some of the pathophysiological processes associated with TBI have been deciphered, 1,13 there are limitations to the therapeutic strategies available for counteracting its neuropathological sequelae. Here, we show, for the first time, that GUO, an endogenous guanine nucleoside, can mitigate the secondary damage associated with in vivo TBI. 25,26,43,44 Specifically, we demonstrate that GUO treatment post-TBI restores glutamatergic function and the maintenance of mitochondrial activity.
Several factors may be responsible for the association between mitochondrial dysfunction and glutamatergic excitotoxicity in the secondary cascade of trauma. Post-TBI, high levels of glutamate are observed, resulting in the excessive activation of N-methyl-D-aspartate receptor (NMDA). 5,45,46 Consequently, mitochondrial exposure to excessive glutamate levels rapidly depolarizes and increases ROS production. 13,47 –49 This dysfunction appears to be dependent upon the sequestration of Ca+2 by mitochondria, 47,48,50 which sequester and release Ca+2 through the mitochondrial membrane. Our results showed that the large increase in ROS 3 h post-TBI was correlated with a decreased ΔΨm. GUO treatment maintained the ΔΨm and decreased ROS production in both the hippocampus and cortex. Corroborating these observations, Dal-Cim and colleagues have previously demonstrated that GUO prevented the disruption of mitochondria membrane potential induced by oxygen glucose deprivation (OGD) in hippocampal slices, 43 as well as protected neuroblastoma cells from oxidative damage induced by impairment of mitochondrial activity. 44
Oxidative stress is one of the first markers of trauma being routinely observed 30 min to 1 h post-injury and is corroborated by other mitochondrial dysfunction markers 3 h post-TBI. 13,15 In agreement, we observed a significant decrease in the GSH/GSSG ratio and inhibition of MnSOD activity in animals submitted to TBI; treatment with GUO maintained GSH levels and enzyme activity indistinguishable from the sham-operated animals. These events can lead to an impaired mitochondrial redox system and oxidative phosphorylation. 12,13
Inhibition of the mitochondrial respiratory complexes 13,51 and decreased electron flow in the mitochondrial electron transport system were observed in various models of trauma. 13,15,52 Consistent with these observations, we noted a decrease in mitochondrial viability accompanied by inhibition of complex I activity in animals submitted to trauma. Notably, GUO treatment fully prevented these effects in the hippocampus and partially restored mitochondrial enzyme activity in the cortex. Studies have reported that mitochondrial complex I is a selective target for peroxynitrite-mediated oxidative damage 13,53 besides contributing to the inhibition of the mitochondrial redox system.
Evidences indicate that inducible nitric oxide synthase (iNOS) isoform catalyze substantial synthesis of nitric oxide (NO) in TBI, 54,55 thus reacting with superoxide ions (O2•−) and leading to formation of peroxynitrite (ONOO−), which inhibits neuronal respiration. 13,15,56 Consequently, excessive NO formation may activate NMDA receptors that stimulate Ca2+ entry and induce neuronal death (through the mitochondrial damage), further increasing glutamate excitotoxicity. 55,57 The neuroprotective effect of GUO on these parameters may be associated with decreased NO production. Analysis of slices under OGD conditions and glutamate-induced cell death are consistent with this hypothesis, showing a reduction in iNOS levels by GUO through activation of the phosphatidylinositol-3 kinase (PI3K) pathway. 43,58,59
Ca2+ homeostasis represents one of the key mechanisms of TBI, linking the glutamatergic excitotoxicity and mitochondrial dysfunction. 49,51,60 This organelle precisely regulates intracellular homeostasis by sequestering and releasing Ca2+, using several mechanisms that can establish a Ca2+ cycle and threaten cellular survival. 60,61 However, excessive amounts of this ion are detrimental and have been shown to result in significant mitochondrial swelling. 15,50,51 Damage to the bioenergetic integrity of mitochondria led to opening of the mitochondrial permeability transition pore and release of cytocromo c, resulting in a significant neuronal death. 5,40,51 The literature corroborates altered Ca2+ homeostasis within 30 min post-injury, followed by mitochondrial demise 3 h later. 13,15 Our results show pronounced alterations in mitochondrial swelling 3 h post-trauma, an effect that is completely reversed in the hippocampus and partially restored in the cortex. These protective effects are likely attributed to the modulation of large conductance of Ca2+-activated K+ channels (BK) by GUO. 43,59
Several neuroprotective effects of GUO against excitotoxicity have been documented, likely attributed to the modulation of the glutamatergic system. 24,26 In agreement, we showed that TBI decreased glutamate uptake in hippocampal and cortical slices. Moreover, the levels of GLT-1 and GLAST were reduced post-TBI in both brain structures. We observed that GUO was fully able to recover the glutamate uptake and avoided the reduction in glutamate transport levels in both brain structures. These neuroprotective effects might be linked to modulation of glutamate uptake and glutamate transporter activity, given that blockers of these carriers inhibited the GUO effect. 24 It is reported that glutamate transporters are modulated by the cell redox status, and an increased ROS production may result in reverse activity of GLT-1 and GLAST, 9 which contributes to the glutamate uptake impairment. Up until now, GUO normalized the activity of the glutamatergic system, reduced ROS production and alterations in the redox system, and restored the mitochondrial activity. Further, it promoted Ca2+ homeostasis, thus mitigating mitochondrial swelling.
The molecular targets and signaling pathways recruited by GUO to modulate its neuroprotective effects remain under evaluation, with GUO being recently classified as an “orphan neuromodulator.” 63 This concept led us to hypothesize various cellular mechanisms for its action. With regard to BK channels, strong evidence shows that the activation of adenosine A1 receptor (A1) in neurons may preferentially regulate potassium channel conductance. 62,64 In turn, A1 can also control NMDA receptors as well as inhibit post-synaptically located voltage-sensitive calcium channels. 63 Thus, we posit that GUO may exert its neuroprotection through modulation of A1 receptor activity, promoting the restoration of ionic gradient and the regulation of glutamatergic transmission. 22,43,59 Notably, GUO was recently shown to have a modulatory effect on the glutamatergic system, which was mediated by activation of PI3K. 43,58,59 In addition, modulation of glutamate uptake and regulation of glutamate transporters activity/expression are linked to the PI3K pathway, 65,66 which is modulated by A1. 67
It is suggested that extracellular GUO may contribute to cell signaling through an indirect mechanism involving the adenosinergic system, given that it led to accumulation of extracellular adenosine. 22,66 Indeed, we cannot exclude the possibility that GUO acts through a selective receptor and secondarily modulates the activity of adenosine receptors, BK channels, and the PI3K pathway (Fig. 7).
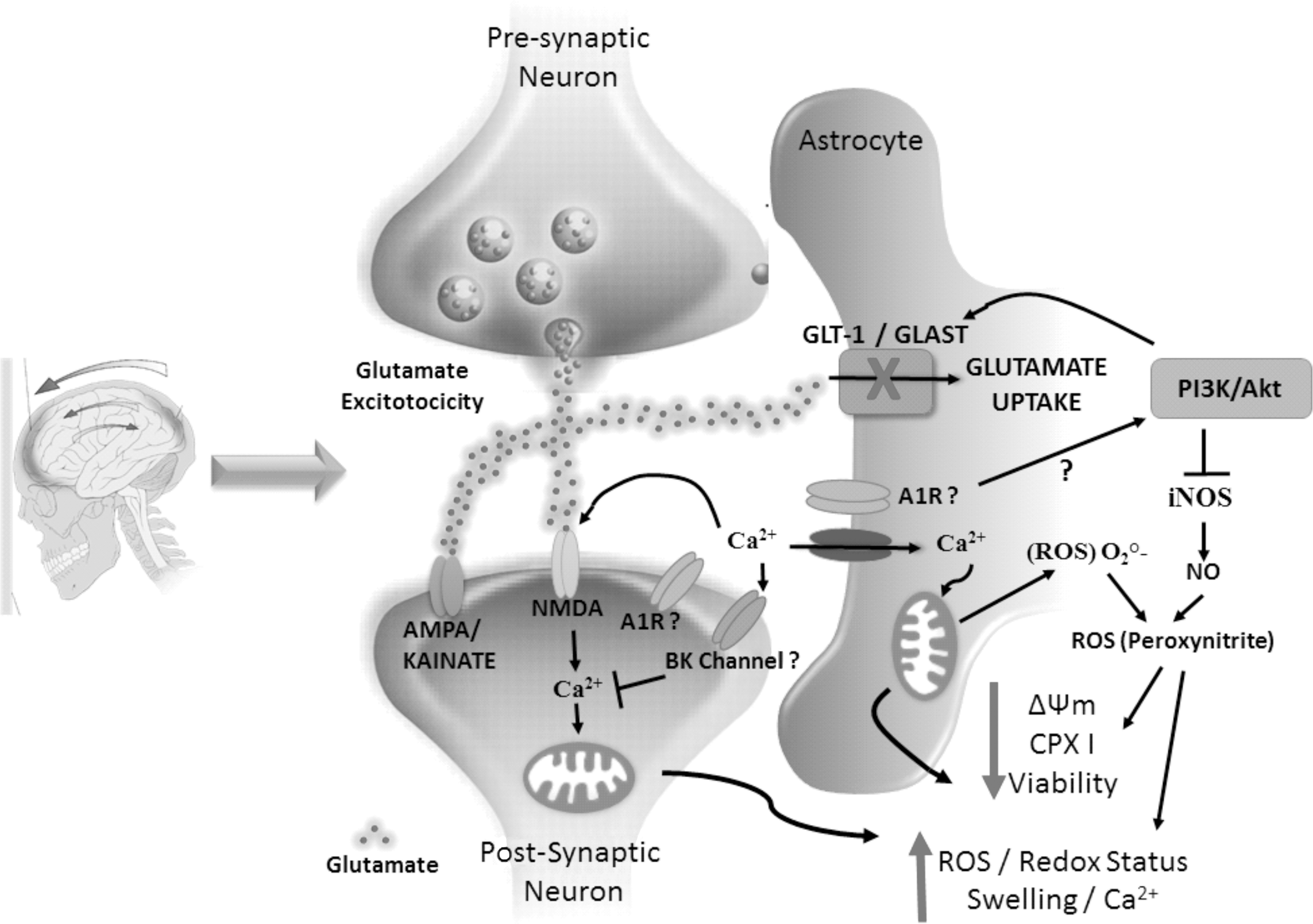
Schematic illustration of GUO effects against neurotoxicity and mitochondrial impairment induced by TBI in rats. GUO was able to block the injuries caused by the trauma. The signal (“?”) indicates a possible mechanism of action involved. A1R, adenosine A1 receptor; Akt, protein kinase B; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BK, Ca2+-activated K+ channels; CPX I, mitochondrial complexin I; GLAST, glutamate aspartate transporter; GLT-1, glutamate transporter 1; GUO, guanosine; iNOS, inducible nitric oxide synthase; NMDA, N-methyl-D-aspartate receptor; NO, nitric oxide; PI3K, phosphoinositide 3-kinase; ROS, reactive oxygen species; TBI, traumatic brain injury.
In conclusion, we noted that after the primary insult (mechanical trauma), changes in excitatory amino acids, increase of oxidative stress, and mitochondrial dysfunction occur, which are believed to be the major factors that contribute to the progressive neuropathology observed in TBI. This excessive activation of NMDA receptors, 7,10,61 depolarization, and ROS production leads to a subsequent influx of Ca2+. At this point, we have a chain reaction where we observe the glutamatergic excitotoxicity acting as a trigger, thus the homeostasis of calcium begins its imbalance leading to mitochondria to a toxic stimulus.
Thus, GUO treatment in a TBI rat model produced a neuroprotective effect attributed to the modulation of the glutamatergic system, optimal maintenance of the redox system, and inhibition of intracellular Ca2+ alterations in mitochondria. Although its neuroprotective mechanism has yet to be fully understood, the properties of this purinergic nucleoside merit further consideration of its efficacy in ameliorating neurological injuries associated with TBI and altered glutamatergic function. It was essential to evaluate whether the drug was promising in these early stages of TBI. After these preliminary findings, we also consider it fundamental to analyze the long-term alterations related to a clinical context.
Footnotes
Acknowledgments
This study was supported by INCT for excitotoxicity and neuroprotection—MCT/CNPq. J.L.F., L.F.R., M.R.F., and F.A.A.S. received a fellowship by CNPq; F.D., R.R.G., G.S.S., and N.F.M received a fellowship by CAPES. Additional support was given by FAPERGS/PRONEM 11/2029-1.
Author Disclosure Statement
No competing financial interests exist.