
Abstract
Traumatic brain injury (TBI) has chronic and long-term consequences for which there are currently no approved pharmacological treatments. We have previously characterized the chronic neurobehavioral and pathological sequelae of a mouse model of repetitive mild TBI (r-mTBI) through to 2 years post-TBI. Despite the mild nature of the initial insult, secondary injury processes are initiated that involve neuroinflammatory and neurodegenerative pathways persisting and progressing for weeks and months post-injury and providing a potential window of opportunity for therapeutic intervention. In this study we examined the efficacy of a novel anti-inflammatory compound, anatabine, in modifying outcome after TBI.
Our model of r-mTBI involves a series of five mild impacts (midline impact at 5 m/sec, 1 mm strike depth, 200 msec dwell time) with an interval of 48 h. Anatabine treatment was administered starting 30 min after injury and was delivered continuously through drinking water. At 6 months after TBI, anatabine treatment improved spatial memory in injured mice. Nine months after TBI, a cohort of mice was euthanized for pathological analysis that revealed reductions in astroglial (glial fibrillary acid protein, GFAP) and microglial (ionized calcium-binding adapter molecule 1, IBA1) responses in treated, injured animals. Treatments for the remaining mice were then crossed-over to assess the effects of late treatment administration and the effects of treatment termination. Nine months following crossover the remaining mice showed no effect of injury on their spatial memory, and whereas pathological analysis showed improvements in mice that had received delayed treatment, corpus callosum IBA1 increased in post-crossover placebo r-mTBI mice.
These data demonstrate efficacy of both early and late initiation of treatment with anatabine in improving long term behavioral and pathology outcomes after mild TBI. Future studies will characterize the treatment window, the time course of treatment needed, and the dose needed to achieve therapeutic levels of anatabine in humans after injury.
Introduction
I
TBI is a physical injury to the central nervous system (CNS) that can result in the initiation of downstream secondary injury mechanisms in the hours, days, and weeks following injury. 4 –7 After human TBI, neuropathology studies have shown evidence of inflammation and progressive white matter degeneration following just a single moderate or severe TBI, 8 with imaging studies showing evidence of persistent neuroinflammation and reduced radial diffusivity and fractional anisotropy, even after mild injury. 9 –11 Consistent with these findings, animal studies have shown persistent inflammation and axonal damage in the corpus callosum of mice exposed to repetitive mild TBI (r-mTBI) at chronic time-points after injury, 12,13 with white matter disruption and an increase in radial diffusivity 60 days after r-mTBI in rats. 14
Pro-inflammatory genes and nuclear factor-kappa B cells (NF-κB) have been reported to remain activated for up to 1 year in a rat model of closed head injury (CHI). 15,16 In mice, chronic microglial activation in combination with progressive lesion expansion has been seen after moderate cortical injury up to 1 year after TBI and up to at least 1 month after mild CHI. 17,18 Our strategy has been to target neuroinflammation in the hope of reducing secondary injury and improving the neuropathological and behavioral outcomes after TBI.
Anatabine is a naturally occurring minor alkaloid that is present in solanaceous plants such as potatoes, tomatoes, and eggplants 19 that inhibits NF-κB activation in HEK293 and SH-SY5Y cells in a concentration-dependent manner and in amyloid precursor protein (APP) over-expressing 7W CHO cells. 20,21 In in vivo studies of the effects of anatabine in the experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis, anatabine improved the clinical score and reduced hindlimb paralysis. 22 Given its excellent bioavailability and anti-inflammatory action, anatabine might have utility as a therapeutic agent after TBI. This study explores the behavioral and neuropathological outcomes of anatabine in a mouse model of closed, mild TBI.
Methods
All mice were male wild type C57BL/6J (Jackson Laboratories) male mice 10 weeks old at the time of injury. Forty-eight mice were divided into groups of 24 mice receiving repetitive injury (five hits with an inter-mTBI injury interval of 48 h), 24 receiving repetitive sham injury (anesthesia alone), and 12 mice from each group assigned to anatabine treatment, 12 to placebo. The injury protocol was performed as previously described. 12 Briefly, mice were anesthetized with 1.5 L/min O2 and 3% isoflurane delivered continuously by a nose cone. The animal was mounted in a stereotactic frame in a prone position secured by ear and incisor bars. Anesthesia controls were matched for the time spent under anesthesia by the r-mTBI mice. An electromagnetic impactor (Impact One™ Stereotaxic Impactor; Leica, Richmond, IL) generated a midline mTBI on the mouse scalp with a 5.0 mm diameter flat face tip, at a 5 m/sec strike velocity, with a 1.0 mm strike depth, and a 200 msec dwell time. Sham injured animals underwent the same procedures and anesthesia duration as the r-mTBI mice on each occasion, to control for the effects of repeated anesthesia.
Thirty min after each injury or sham procedure, mice received an intraperitoneal (IP) injection of either phosphate buffered saline (PBS) or anatabine (2mg/kg) (provided by Rock Creek Pharmaceuticals). This dose was administered via IP to control the timing and dose of the first administration after injury as the mice do not immediately return to drinking from their water bottles after surgery. After the first dose, for the anatabine treatment group, normal water was substituted with anatabine-treated water, and was continuously re-filled for continuous treatment over 9 months (without the need for further IP injections) at an estimated dose of 20mg/kg/day (placebo mice received regular water). Water intake was estimated given previous research showing that C57BL/6J mice consume water at a rate of approximately 7.4 mL/30 g of weight. 23 At 9 months post-TBI, the treated and untreated groups were switched. Previously untreated mice began receiving anatabine in their water at an estimated dose of 20mg/kg/day, whereas mice previously treated with anatabine began receiving regular water alone.
Experimenters were blinded to group assignments during all neurobehavioral testing. Rotarod testing was performed as previously described with modifications. 12 Briefly, baseline testing preceded the administration of r-mTBI or r-sham procedures, but only one day of baseline testing was performed. One day prior to the start of the injury or sham procedure, mice were pre-trained and baseline tested for rotarod performance. All mice were given three trials to acclimatize to the rotarod at a constant speed of 5 revolutions per min (RPM). For all subsequent tests, the rotarod was set to an accelerating speed of 5 to 50 RPM over a period of 5 min. Three trials were given with a 3 min rest period between trials. Latency to fall was recorded and averaged over each day. Baseline testing occurred one day prior to the start of the r-mTBI or r-sham procedure, and one day after the administration of the last r-mTBI or r-sham procedure rotarod testing was resumed. Mice were then tested every other day after surgery starting on day 1 and ending on day 7. This testing was repeated again 6 months after TBI. Post-surgery testing occurred at an accelerating speed of 5 to 50 RPM over a period of 5 min. Three trials were given with a 3 min rest period between trials. Latency to fall and the speed of the rotarod at the moment of the fall were recorded.
Following rotarod, Barnes maze testing was administered as previously described immediately after the first rotarod test and again at 6 months after TBI. 12 The Barnes maze was 1.2 m in diameter and had 18 holes around the perimeter. Each mouse was given 1.5 min to explore the maze and enter the goal box. Each mouse received 30 sec in the goal box at the end of the trial period. Each mouse received four trials starting in front of each of four randomly selected cardinal holes for a period of 6 days. On the 7th day the goal box was removed and the mouse was given a 60 sec probe trial starting in the center of the table. Each mouse's orientation and movement was recorded by Noldus Ethovision XT software. Rotarod testing was performed acutely in the first week after the final injury. Barnes maze testing was performed on days 8 to 15 after the final injury, then at 6 months after TBI the Barnes maze protocol was repeated (both acquisition and probe trials) as described above. Following the crossover of treatment, Barnes maze acquisition and probe testing was again repeated at 12 and 18 months post-TBI (3 and 9 months after treatment crossover).
Pathology was assessed at 9 and 18 months post-TBI. Four mice per group were euthanatized for neuropathological analyses 9 months after TBI/sham procedures. All remaining mice were euthanized 18 months after TBI, with four per group processed for neuropathological analyses at this final time-point. All animals were deeply anesthetized with isofluorane before being intracardially perfused by gravity drip with a heparinized PBS solution pH 7.4 for 3 min, followed by an overnight fixation of brain samples in 4% paraformaldehyde and paraffin embedding. Separate series of 5- to 6-μm-thick sections were cut throughout the extent of the cortex and hippocampus and associated areas using a microtome (2030 Biocut, Reichert/Leica) and mounted on positively charged glass slides (Superfrost Plus, Fisher). Sections were stained in entire batches with antibodies (cell markers) raised against: glial fibrillary acid protein (GFAP) (rabbit anti-GFAP, 1:10,000, Dako) for astrocytosis, ionized calcium-binding adapter molecule 1 (IBA1) for microglia, APP for axonal injury, phosphorylated signal transducer and activator of transcription 3 (p-STAT3) (rabbit anti-pSTAT3, 1:500), or Luxol© fast blue (LFB) for myelin.
Sections were deparaffinized in Histo-Clear (National Diagnostics) and rehydrated in 95% and 80% ethanol before the immunohistochemical procedure. Sections were then rinsed in water and treated with 0.3% hydrogen peroxide for 5 min to block endogenous peroxidase. After rinsing, sections were heat treated in target retrieval solution for 8 min using a pressure cooker for antigen retrieval. Sections were incubated overnight with primary antibodies, followed by rinsing with PBS, and were then stained with a complementary secondary antibody (from the Vectastain Elite ABC Kit or Vector Mouse on Mouse [M.O.M.™] Kit) for 1 h, and finally they were incubated with avidin-biotin-horseradish peroxidase solution (Vectastain Elite ABC Kit, Vector Laboratories) for 1 further h
A solution of 3,3'-diaminobenzidine (DAB) chromogen and hydrogen peroxide was used to develop the sections. Chromogen development was kept at a constant 5 min to limit variability for quantification. The reaction was terminated by rinsing sections in distilled water. Finally, sections were dehydrated using 80% and 95% ethanol followed by Histo-Clear and were then coverslipped. Immunoreacted sections were viewed using an Olympus (BX63) light microscope and photos were taken using an Olympus DP72 camera.
For GFAP and IBA1 staining, immunoreactivity for cell markers was measured quantitatively using optical segmentation. Three non-overlapping 100 μm2 regions were selected for each section (n = 4). Quantification was performed by blind assessment using ImageJ (with each slide analyzed blind with respect to marker or animal group). Images were separated into individual color channels using the color deconvolution plug-in (hematoxylin counter stain and DAB chromogen). Each stain was quantified within the cortex, hippocampus, and corpus callosum. The area of positive immunoreactivity was calculated for each section and expressed as a percentage of the total area. For APP and p-STAT3 staining, the total number of positive profiles were quantified throughout the corpus callosum on each section. For LFB thickness was measured along the center of the body of the corpus callosum.
Statistical analysis
Statistical analysis was performed using JMP 12.0 (SAS; Cary, NC). Normality tests were conducted using the Shapiro-Wilk W test and the data were transformed using square root or natural log transformation when not normally distributed. If the distribution was still not normal after transformation, non-parametric methods were used. Normally distributed data were analyzed using analysis of variance (ANOVA; with repeated measures where appropriate) and t tests. P values of p < 0.05 were considered as statistically significant and marked with an asterisk in the figures.
Results
Behavioral analyses
At the acute evaluation of the rotarod testing, placebo-treated sham mice had a higher fall latency than placebo-treated r-mTBI mice on days 1 through 7 (p < 0.01) (Fig. 1). Although anatabine-treated sham mice had a higher fall latency than anatabine-treated r-mTBI mice on day 1 (p < 0.01), on days 3, 5, and 7 there were no differences between the anatabine-treated sham and TBI mice. Anatabine-treated sham mice were not different from placebo-treated shams on day 1, but by day 7 there was a difference between the two shams (p < 0.05). Upon re-testing at 6 months post-TBI, there were no differences between any group's fall latency on any day of testing (data not shown).
Rotarod testing showed a significantly higher fall latency of placebo-treated sham mice compared with placebo-treated repetitive mild traumatic brain injury (r-mTBI) mice throughout the first week after the completion of the injury procedure (p < 0.05). Anatabine-treated sham animals performed significantly better than r-mTBI mice on day 1 after injury, but performed at similar levels to r-mTBI mice on all subsequent days. Bars represent standard error.
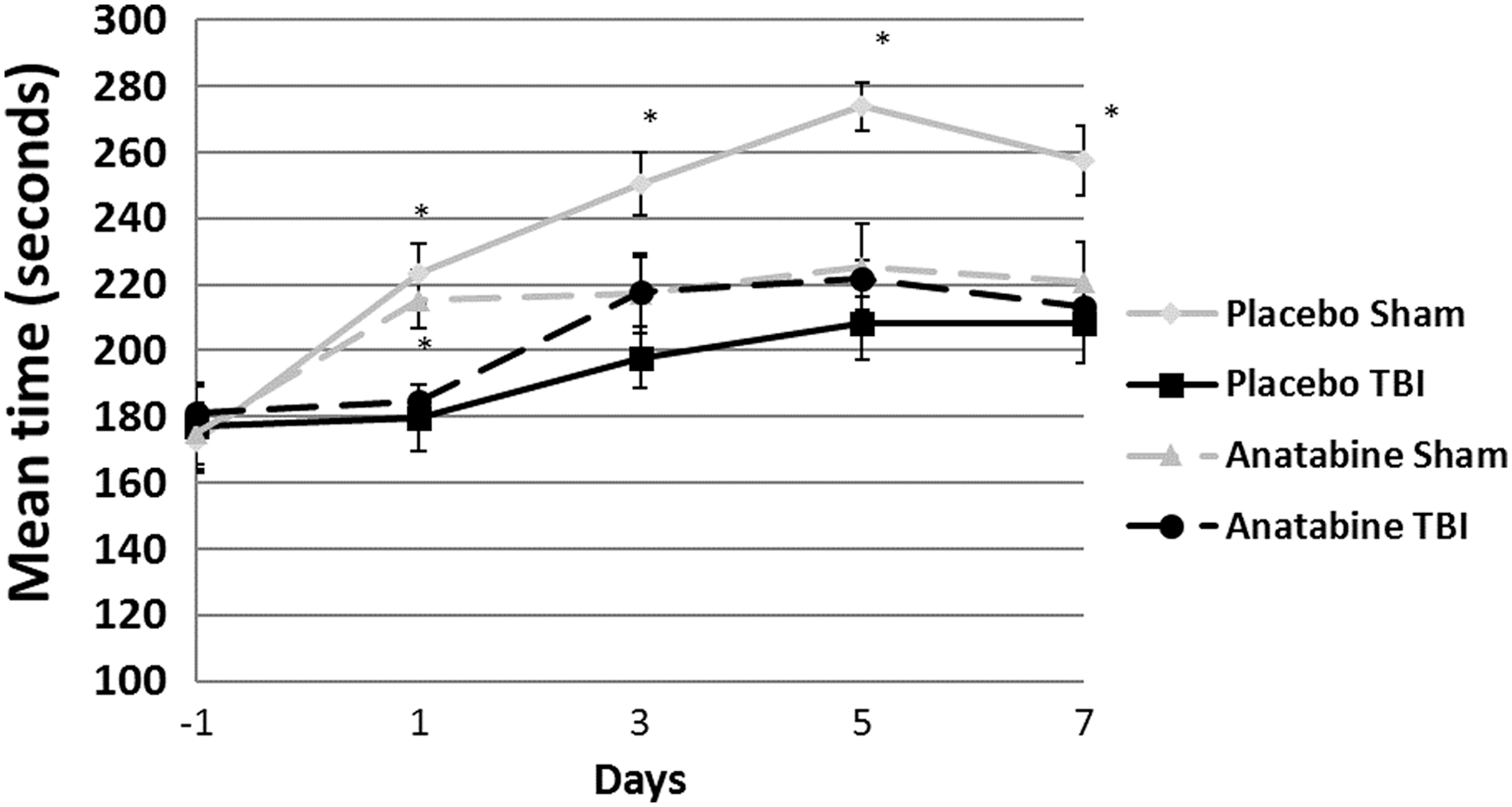
Barnes maze testing was performed on days 8 through 14 after TBI (Fig. 2a) and again 6 months post-TBI. The Barnes maze is a 1.8 m diameter board, with 18 holes positioned around the perimeter, situated in a room rich in visual cues. Acquisition trial testing showed an effect of injury on spatial learning (p < 0.05) but not treatment (Fig. 2c). Treatment with anatabine did not show any effect on the probe trial latency at the acute time-point (Fig. 2a).

(
At the 6-month post-TBI probe trial, anatabine-treated r-mTBI mice showed an improved latency to the target hole compared with placebo-treated r-mTBI mice (p < 0.05) indicating a positive effect of anatabine treatment (Fig. 2b). At this time-point, the anatabine-treated r-mTBI mice located the target hole as quickly as their respective shams (11.0 and 9.9 sec respectively, p > 0.05) as well as placebo-treated shams (11 and 10.6 sec respectively, p > 0.05), showing no effect of injury on their probe trial performance.
Post-crossover behavioral data
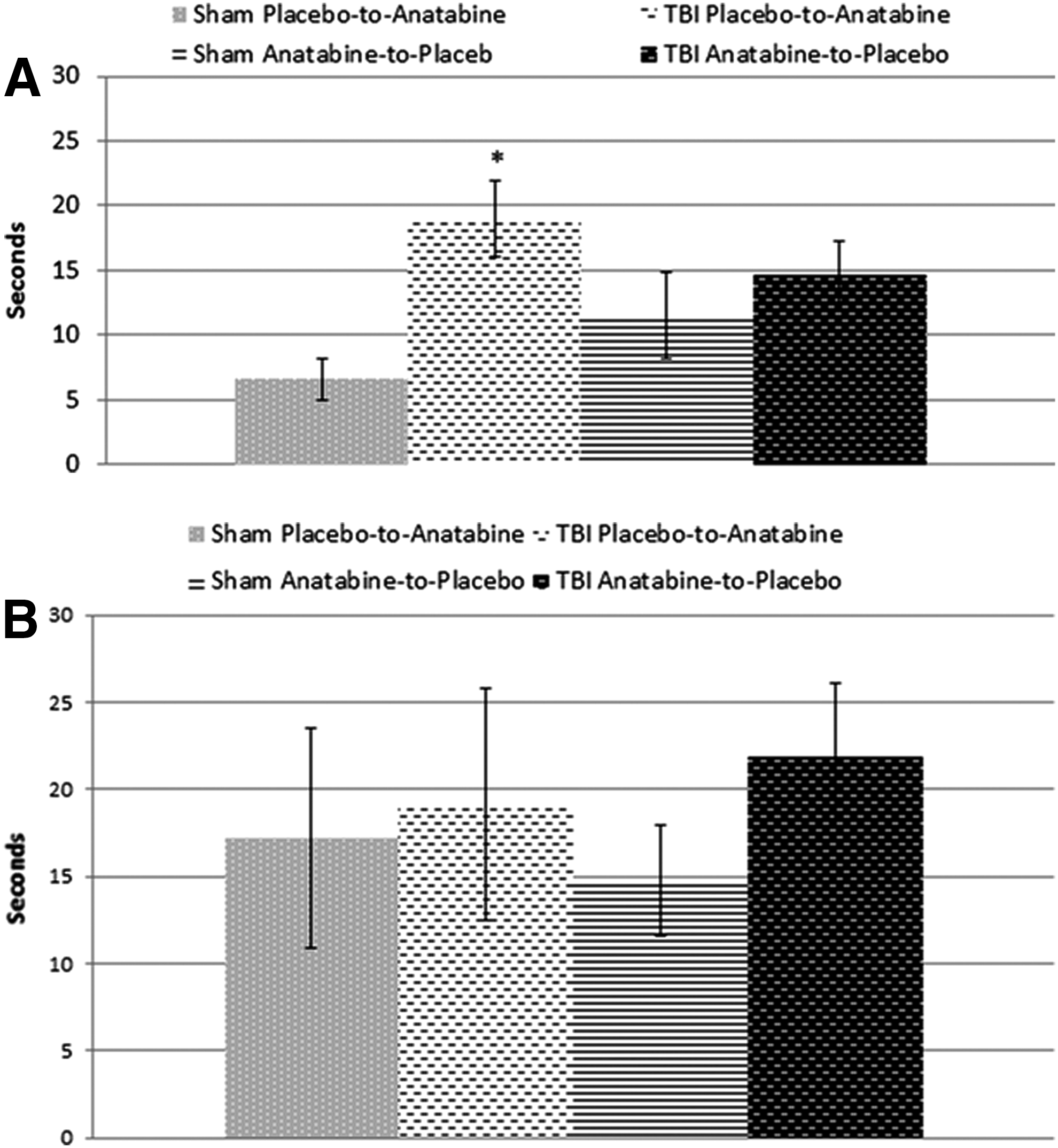
At the 12-month post-TBI probe trial (3 months post-crossover), anatabine-treated r-mTBI mice that were originally treated with regular water showed no improvement compared with their shams in spite of treatment with anatabine (p < 0.05) (Fig. 3a). Mice in the r-mTBI group originally treated with anatabine continued to show no differences compared with their respective sham group despite the cessation of treatment 3 months prior (p > 0.05).
(
Six months later, at the 18-month post-TBI time-point (9 months post-crossover), there were no differences between r-mTBI mice and sham mice in either the original treatment or delayed treatment groups, nor were there any differences between the two r-mTBI groups (p > 0.05) (Fig. 3b).
Neuroinflammatory response
At 9 months after TBI, evaluation of astrocytosis showed that hippocampal GFAP staining was increased in the placebo-treated r-mTBI compared with sham animals (2.9% area and 1.5% area respectively, p < 0.05) (Fig. 4) but not in mice treated with anatabine compared with either sham group (p > 0.05). However, TBI-induced astrocytosis in the corpus callosum was increased in both placebo- and anatabine-treated mice compared with their respective shams (p < 0.01).
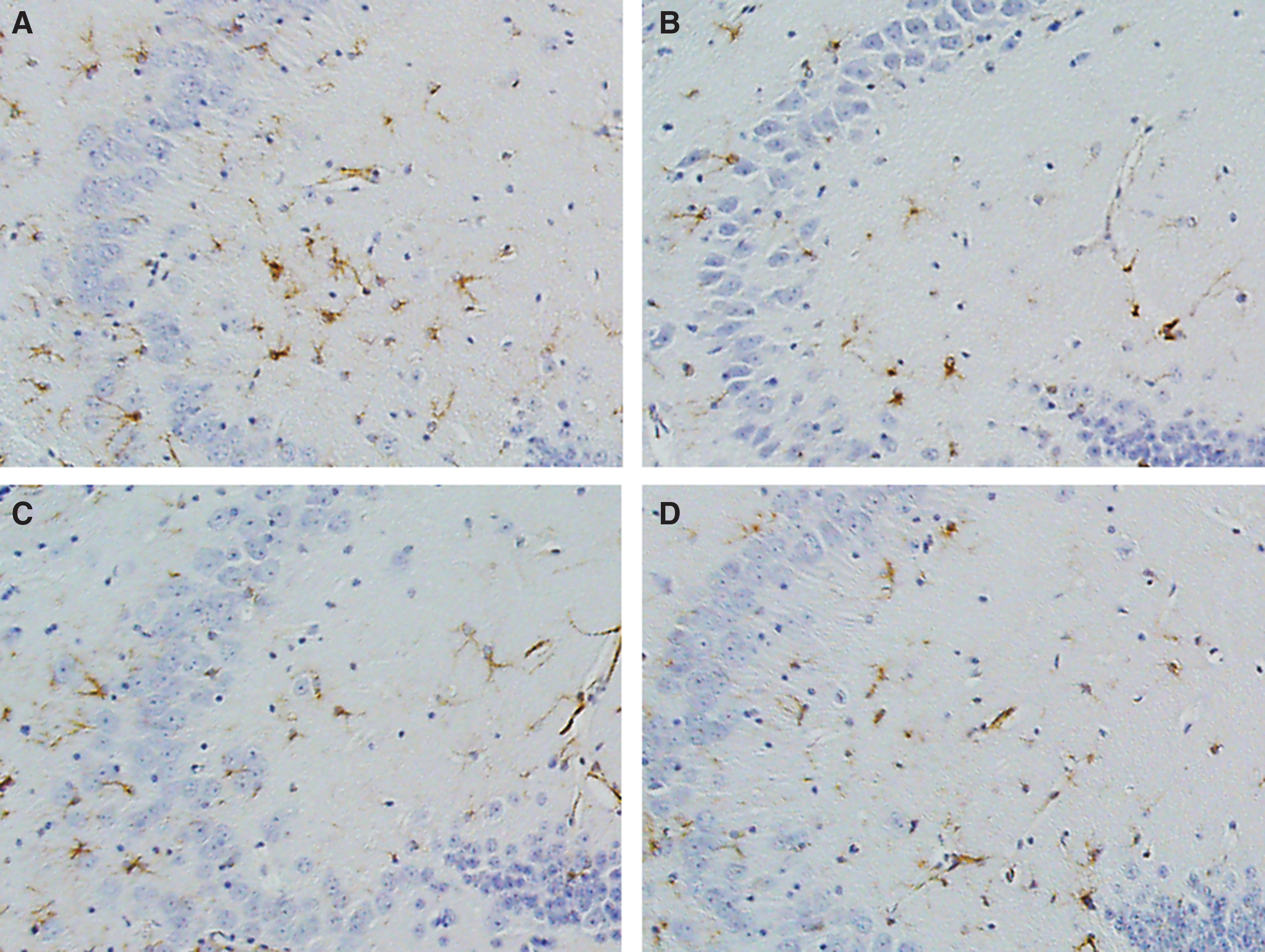
Hippocampal glial fibrillary acid protein (GFAP). (

At 18 months post-injury, 9 months after the treatment crossover, GFAP staining in the hippocampus was increased for all groups. Animals treated with anatabine after the crossover continued to show an increase in GFAP staining within the hippocampus after r-mTBI (p < 0.05). Interestingly, the situation was reversed in the late placebo-treatment group, with late placebo-treated sham animals showing more GFAP staining than their r-mTBI counterparts (p < 0.05).
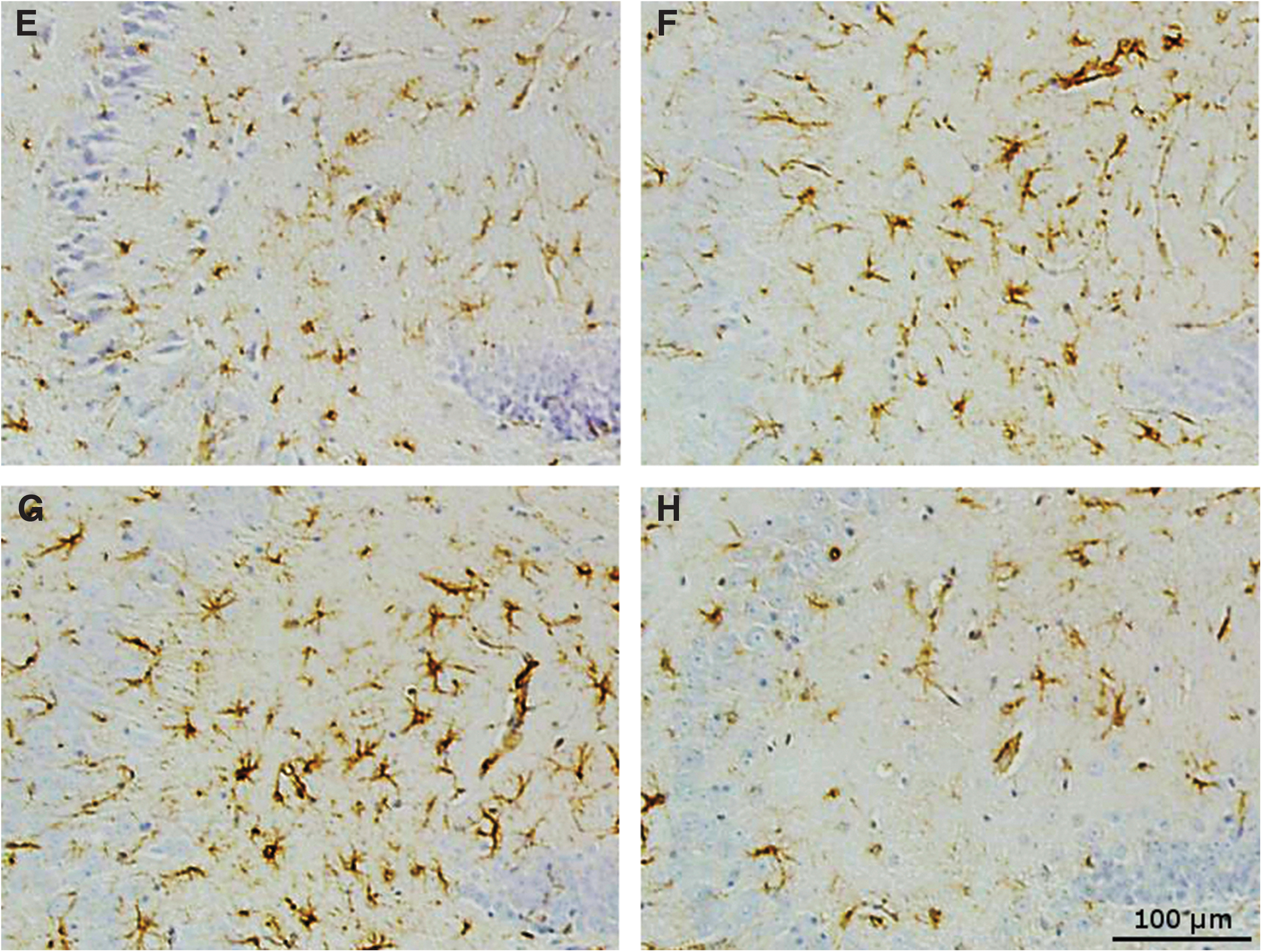
Microglia staining by IBA1 in the hippocampus showed that at 9 months post-injury there was a small increase in the placebo-treated r-mTBI mice compared with sham mice (1.8% area and 1.3% area respectively, p < 0.05) (Fig. 5), which was not seen in anatabine-treated r-mTBI mice compared with their respective shams (1.3% and 1.4% area respectively, p > 0.05). In the corpus callosum the anatabine-treated r-mTBI exhibited less IBA1 staining than placebo-treated r-mTBI mice (p < 0.05), but still showed an increased level of microglial activity compared with sham controls (3.4% and 1.5% area respectively, p < 0.01).
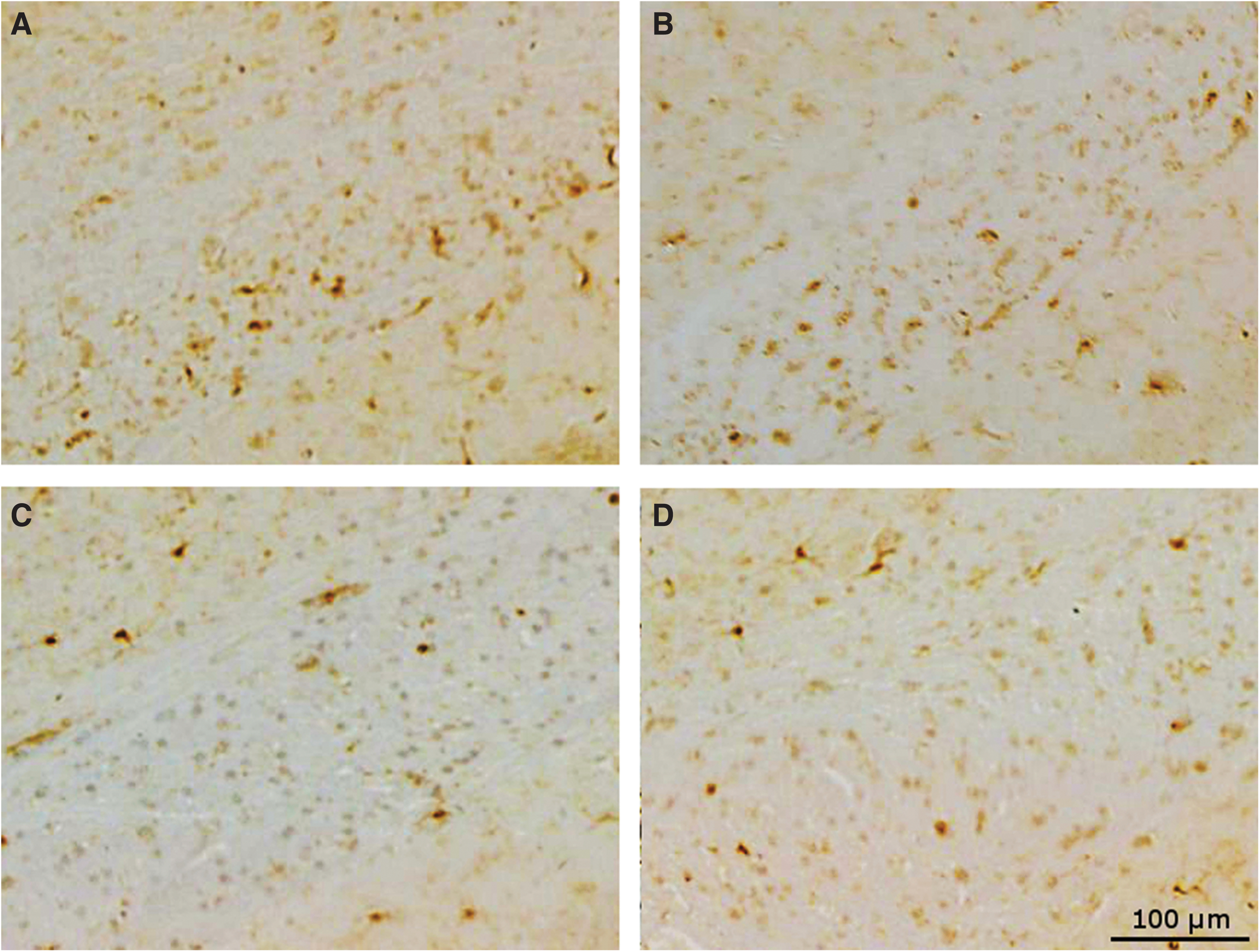
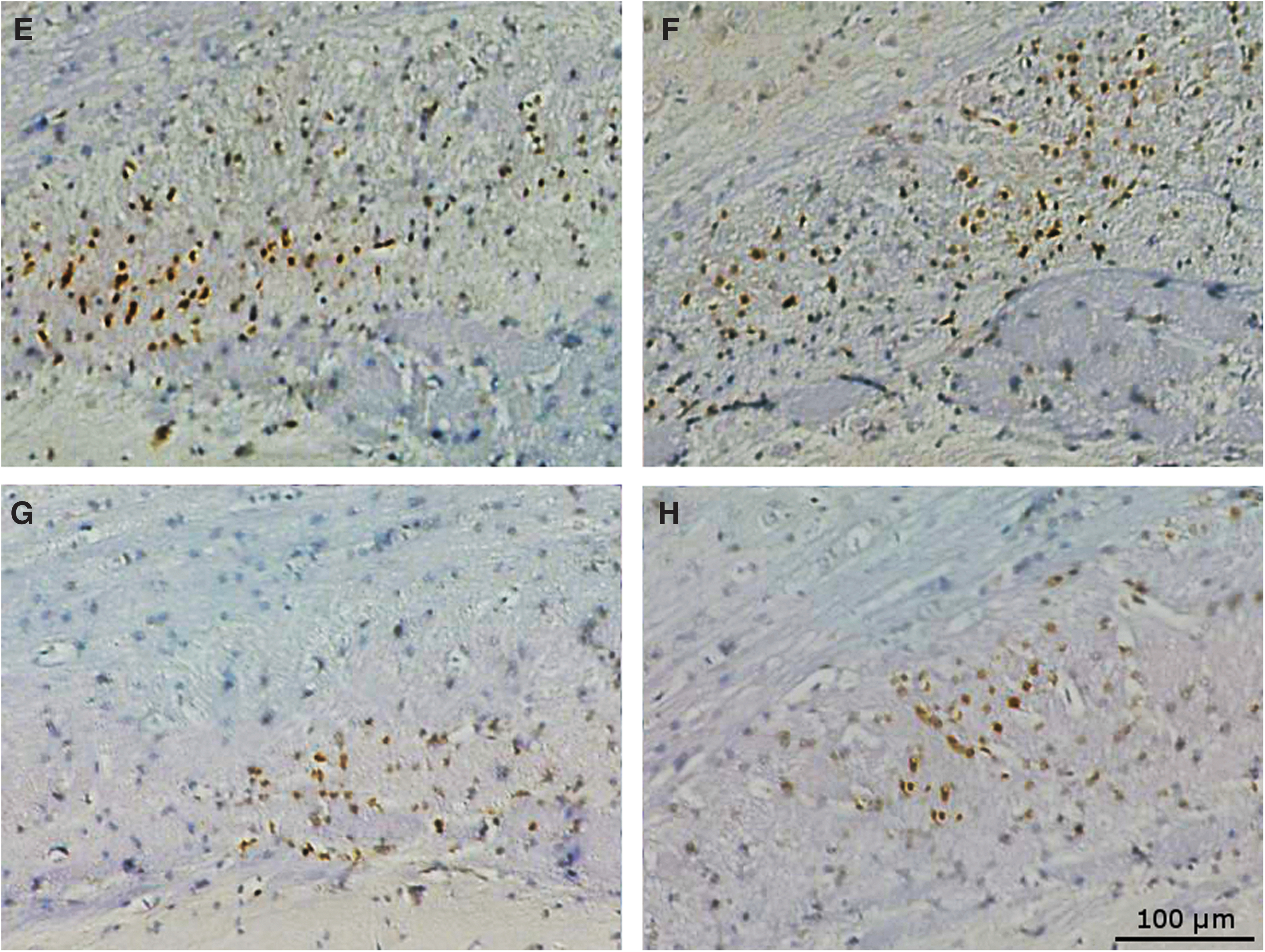
Corpus callosum ionized calcium-binding adapter molecule 1 (IBA1). (

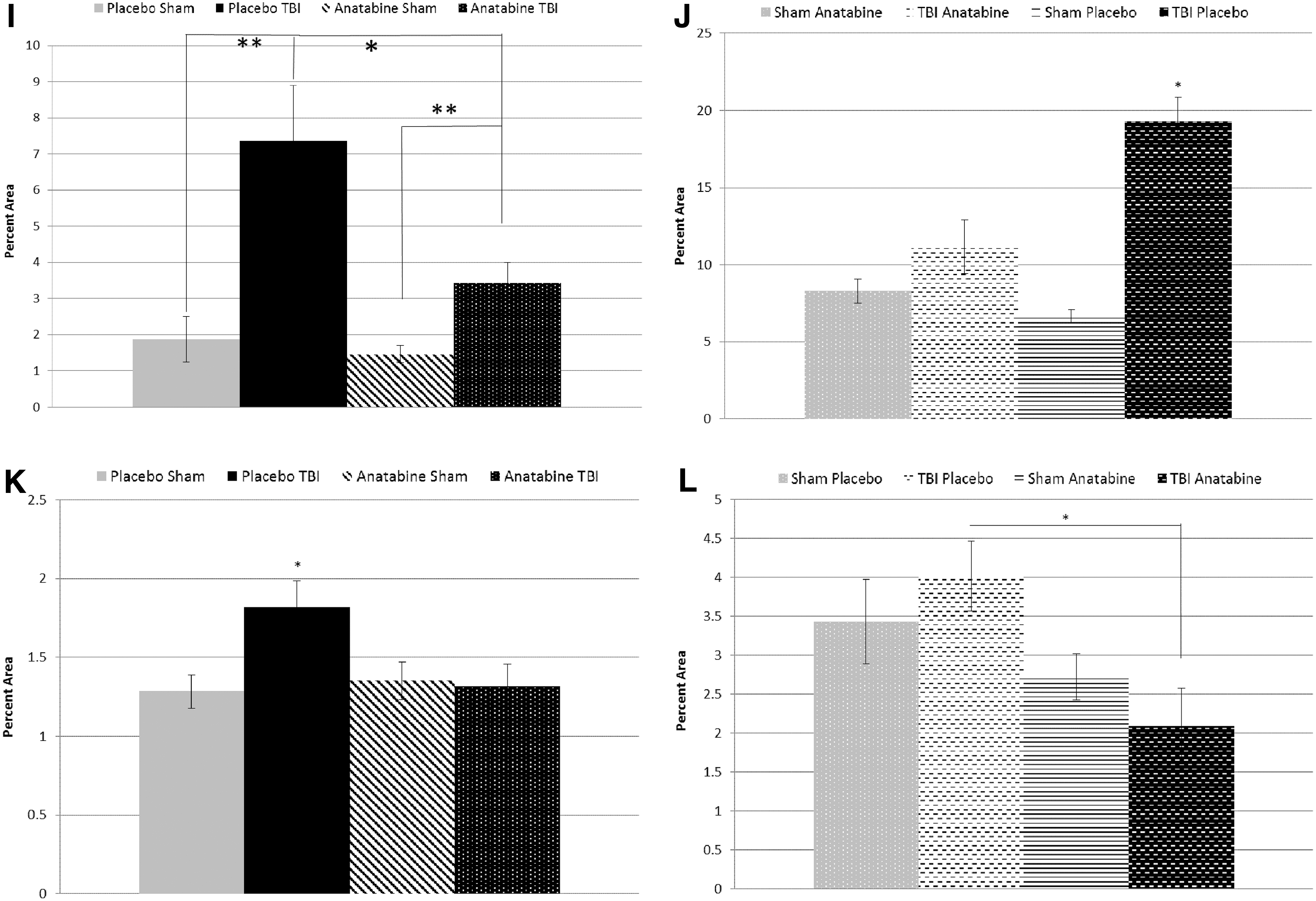
Eighteen months after TBI, post-crossover placebo-treated TBI animals had a higher level of microglial staining in the corpus callosum than post-crossover placebo-treated shams (19.3% vs. 6.7% area, p < 0.05) despite the improvement seen in early anatabine-treated animals at the 9-month time-point. Injured mice treated late with anatabine during the post-crossover phase showed no difference compared with their corresponding shams (p > 0.05). Injured mice treated late with anatabine showed less microglial staining in the corpus callosum than their placebo-treated counterparts (p < 0.01).
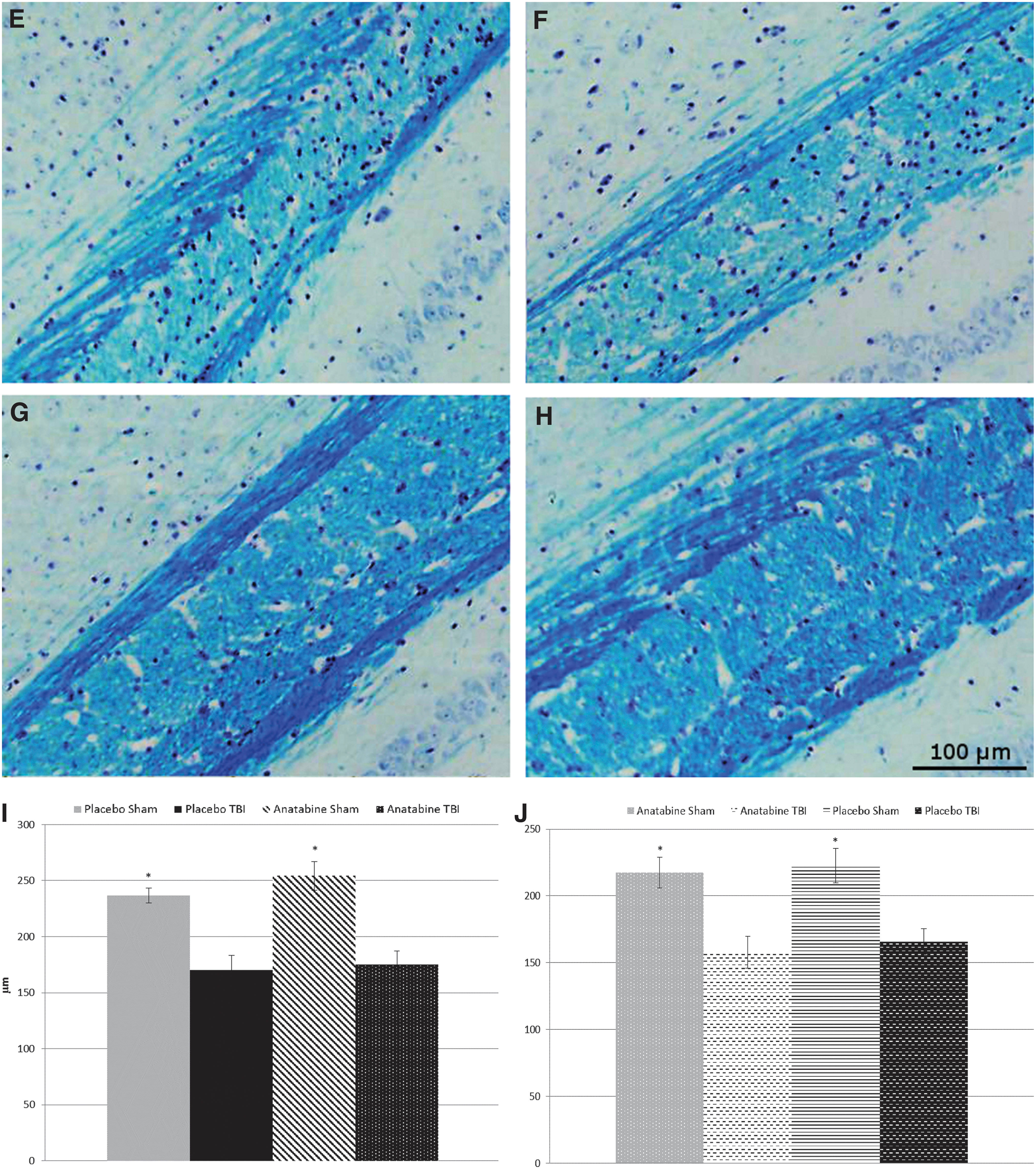
White matter degeneration
At 9 months post-TBI, LFB staining appeared paler within the body of the corpus callosum in TBI mice compared with sham mice (p < 0.05) but treatment with anatabine did not affect this TBI-induced reduction. This difference was maintained post-crossover at the 18-month time-point with the corpus callosum thickness showing reductions in both late anatabine- and late placebo-treated groups compared with their respective shams (p < 0.05) but with no differences between the treatment groups (Fig. 6).

Luxol fast blue staining of the corpus callosum. (
At 9 months after TBI, APP staining showed an increase in the number of APP positive axons throughout the body and splenium of the corpus callosum in both the placebo-treated and anatabine-treated TBI groups compared with their shams, with no difference between the treated groups. Placebo-treated TBI mice had an average of 30 positive axons compared with an average of 2 for placebo-treated sham mice (p < 0.05). Anatabine-treated TBI mice had an average 38 positive axons compared with an average of 2 for anatabine-treated sham mice. There were no differences between placebo-treated and anatabine-treated sham animals (Fig. 7). Eighteen months after TBI, 9 months after crossover, this pattern was maintained with TBI mice showing a greater number of APP positive axons (p < 0.05) regardless of treatment, and no differences between anatabine and placebo mice.
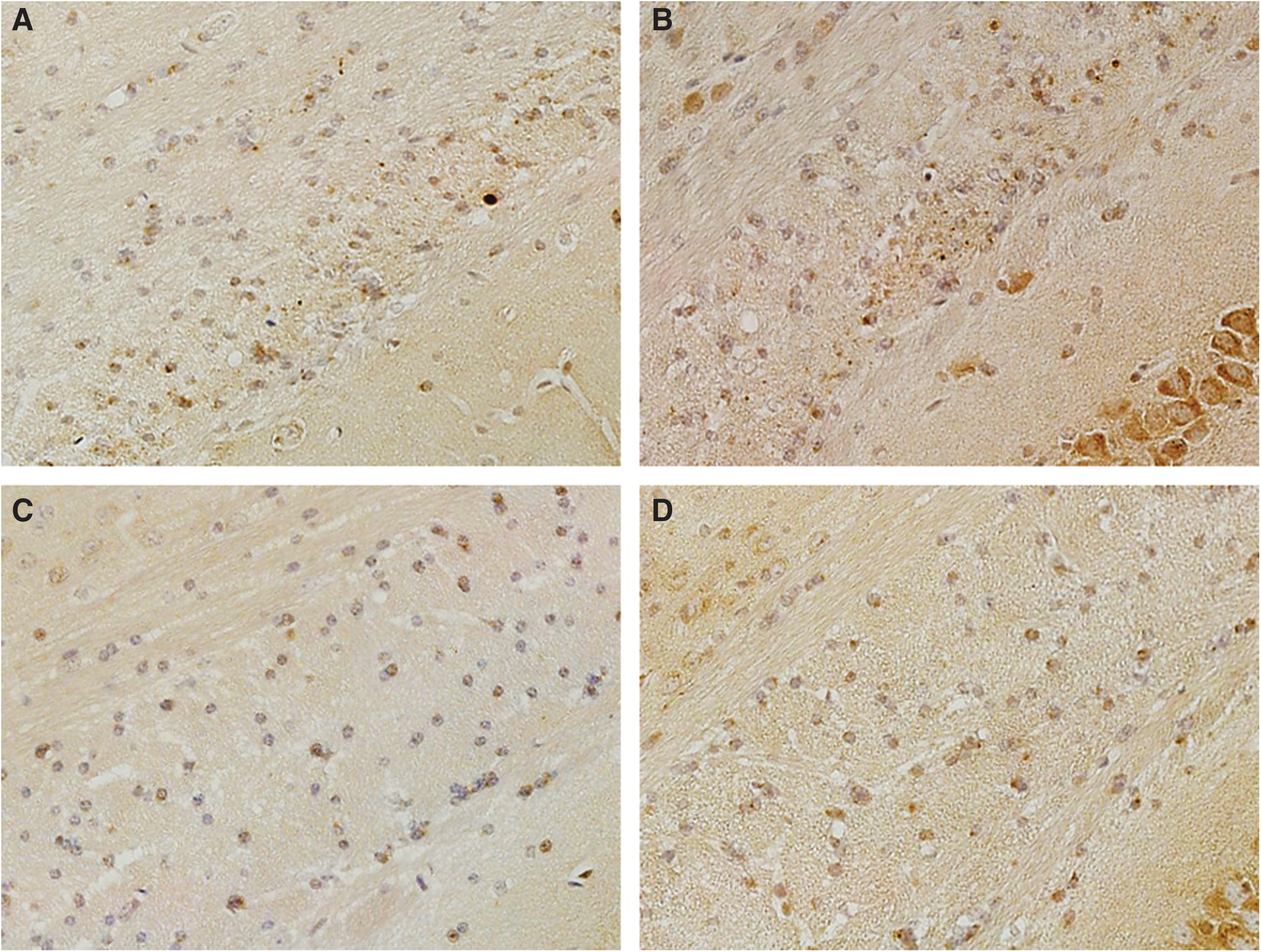
Amyloid precursor protein (APP) staining of the corpus callosum. (

STAT3 phosphorylation
At 9 months post-TBI we evaluated STAT3 phosphorylation to determine whether anatabine acts on STAT3 signaling to reduce inflammation in TBI. Staining for p-STAT3 revealed the corpus callosum was the only brain region to exhibit positively stained cells. Cell counts of positive cells throughout the corpus callosum revealed a higher number in placebo-treated TBI mice than in placebo-treated sham animals (64.2 on average vs. 6.3, p < 0.05). Anatabine-treated TBI mice also had an elevated number of positive cells compared with their corresponding shams (33.5 vs. 13) but fewer than in placebo-treated TBI mice (p < 0.05) (Fig. 8). At the 18-month time-point the p-STAT3 positive cell counts were elevated in all groups, with post-crossover placebo sham mice at 222.5 positive cells in the corpus callosum on average, post-crossover placebo TBI at 381.9, post-crossover anatabine-treated sham at 261.5, and post-crossover anatabine-treated TBI at 392.5 positive cells. Both post-crossover TBI groups showed elevated levels above shams regardless of treatment.

Phosphorylated signal transducer and activator of transcription 3 (p-STAT3) staining of the corpus callosum. (

At completion, our treatment study did not include any naïve (untreated) mice as all groups had at some point during the crossover design received anatabine, thus confounding our ability to separate treatment-dependent from age-dependent effects on STAT3 phosphorylation. However, we had tissue available from a previously published separate cohort of r-mTBI or r-sham mice (using the same paradigm) analyzed at chronic time-points. 13 Therefore, for comparison with the current study, we conducted p-STAT3 staining on slides from these r-sham or r-mTBI animals analyzed at 12 and 24 months post-TBI, time-points flanking the final time-point of our current study. Batches of samples from all four time-points from both studies combined were processed simultaneously for p-STAT3 to allow for direct comparison of data from the two studies. The treatment naïve r-sham animals had an average of 286 p-STAT3 positive cells in the corpus callosum at the 12-month time-point, and an average of 2725.8 at the 24-month time-point. Naïve r-mTBI mice had averages of 894.25 and 2616.4 at 12 and 24 months post-TBI respectively. Thus at 12 and 24 months post r-mTBI treatment naïve mice had higher numbers of p-STAT3 positive cells than either the early or late anatabine-treated r-mTBI animals at the 18-month time-point (Fig. 7).
Discussion
In this study we examined the effect of anatabine on neurobehavioral and pathological outcomes in a mouse model of r-mTBI. Our results showed no effect of treatment at acute time-points after injury, but that treatment with anatabine improved the spatial memory and reduced the neuroinflammation of mice at a later time-point after injury. A cross-over study of these mice showed that even with a 9-month delay, anatabine treatment can still reduce IBA1 staining and STAT3 phosphorylation, but a 9-month cessation of treatment results in a return of the IBA1 signal within the corpus callosum of the mice indicating that the suppression of the microglial response has been lost. Treatment with anatabine appears to reduce chronic inflammation in a regionally specific manner and improve the spatial memory in this model of TBI. Although previous human clinical studies targeting neuroinflammation have historically failed, this study reveals that later chronic time-points and treatments may be necessary to reduce sequelae of the disease.
Clinical trials of potential treatments targeting secondary injury after TBI have so far largely been unsuccessful. Neuroprotective compounds that have been administered to reduce secondary injury have all failed in Phase III clinical trials thus far. 24 –26 Anti-inflammatory treatments have had no success thus far either, and the Corticosteroid Randomisation After Significant Head Injury (CRASH) study showed increased risk of death in patients who received methylprednisolone for 48 h after TBI. 27 Even treatments with multifarious drugs, such as erythropoietin and progesterone, intended to target the pathology through multiple mechanisms of action have failed in Phase III clinical trials. Often these negative results are determined to have been due to difficulties arising from the heterogeneity of the patient population, delays in clinical trial enrollment before treatment, as well as uncertainties in the effective treatment window, dose, and bioavailability data from pre-clinical studies. The studies cited here were designed to treat patients with severe TBI, but with mTBI most human studies have looked at pharmacological compounds to treat the cognitive and emotional consequences without actually impacting the progression of pathological damage within the brain. These treatments have sought to alleviate the symptoms commonly reported in mTBI cases, but our study highlights the importance of the chronic phase after TBI and how a drug targeting neuroinflammation may not show acute improvement, and even chronic treatment may not provide sustained benefits if the treatment is halted for a prolonged period of time.
As in our previous studies, 12,13 we found that among the placebo-treated mice the r-mTBI mice exhibited worse motor coordination, spatial learning, and spatial memory in the rotarod and Barnes maze tests. Neuropathological analysis in the current study also revealed the same chronic, persistent neuroinflammation and white matter damage within the r-mTBI mice as we saw in our previous studies. At an acute time-point of 2 weeks there was no improvement of the anatabine-treated r-mTBI mice over placebo-treated r-mTBI mice, but at a chronic time-point 6 months after TBI we saw improvements in spatial memory during the probe trial of the Barnes maze that correlated with brain region specific improvements seen by immunohistochemical analysis. Hippocampal pathology, although not as prominent as white matter pathology in the corpus callosum, may be important to our neurobehavioral results. IBA1 was increased in the hippocampus and corpus callosum of placebo-treated r-mTBI mice at 9 months post-TBI, but anatabine-treated mice showed no elevation of IBA1 in the hippocampus at the 9-month time-point. It is important to note that the corpus callosum shows the largest amount of pathology of any brain region within our model of TBI. 12,13 We have also observed extensive optic nerve and retinal pathology in this model of TBI that may be affecting visual-based memory task results as well, 28 and future studies will examine the effect anatabine may have on the functional visual outcome of mice in this model.
GFAP staining showed an increase in the hippocampus of placebo-treated r-mTBI mice, but not in anatabine-treated mice. Although these differences in the hippocampal pathology are small, they may also contribute to the spatial memory differences that were seen in the Barnes maze 6 months after TBI. Interestingly, GFAP staining remained low in the hippocampus of post-crossover placebo r-mTBI mice, but became higher in post-crossover placebo sham animals at this time-point, whereas post-crossover anatabine-treated r-mTBI mice continued to show higher levels of GFAP staining. Within the r-mTBI group, early treatment appears to be necessary to prevent injury-dependent increases in GFAP staining, but the reason for the increase in post-crossover placebo-treated sham animals is unclear. Prevention of chronic inflammation within the hippcampal region appears to be important in rescuing spatial memory, even though the hippocampus exhibits far less inflammation at chronic time-points than the corpus callosum. More work needs to be done to characterize the region specific changes after TBI as well as following experimental treatment to determine which pathological hallmarks are the most important in determining outcome and therapeutic benefits.
Despite the reduction of IBA1 staining, neither GFAP nor APP staining were reduced in the corpus callosum of anatabine-treated r-mTBI mice at 9 months post-TBI. Additionally, staining with LFB showed a reduction in the thickness of the corpus callosum, but this was not abated by treatment with anatabine. This suggests that anatabine is not acting to reduce the structural changes and persistent white matter loss occurring after TBI. At the 18-month post-TBI time-point, 9 months after treatment crossover, IBA1 staining had increased in the post-treatment placebo animals compared with sham animals but had decreased to sham levels in post-crossover anatabine-treated r-mTBI mice. This suggests that the chronic inflammatory sequelae are only temporarily reduced by anatabine treatment. Axonal damage is not being prevented, and may be driving these prolonged inflammatory sequelae. The cessation of treatment then results in a return of the chronic inflammatory phenotype.
It has been suggested that previous animal studies failed in translation to human application often due to the failure to perform appropriate pre-clinical studies to establish the bioavailability and treatment window of their compounds. 29 In human mTBI cases, the initial injury may go undiagnosed for long periods of time until secondary clinical manifestations begin appearing at chronic time-points. 30 This is especially true in settings where r-mTBI is common such as in athletes. 31 To address the issue of delayed diagnosis and delayed treatment, our study utilized a cross-over design, allowing us to examine the effect of delayed treatment administration as well as the effects of treatment cessation.
Prior to the crossover, we witnessed an improvement in spatial memory during the probe trial of the Barnes maze that correlated with regionally specific reductions in IBA1 and GFAP staining. After the crossover, we saw an increase of IBA1 staining in the corpus callosum of post-crossover placebo-treated r-mTBI mice, whereas late-anatabine-treated r-mTBI saw a reduction to sham levels. Although we did not see TBI-dependent spatial memory impairments at the 18-month time-point, pathologically we begin to see a return of TBI-dependent neuroinflammation following the 9-month cessation period, suggesting that anatabine is not targeting the underlying causes of the inflammation, but is instead modulating the inflammatory response only for the duration of treatment.
Cessation of treatment did not result in reductions in spatial memory in the originally treated r-mTBI mice, but a non-significant trend of increasing latency to find the target hole was observed. In particular, the originally treated r-mTBI mice showed an increase in latency to find the target hole between the 3-month and 9-month post-crossover time-points (12 and 18 months post-TBI respectively). This combined with pathological data showing a resurgence of microglial staining in the corpus callosum suggests that sustained treatment may be required. A reduced n of 8 at the post-crossover time-points may have also obscured the statistical significance of the neurobehavioral trend toward a return of cognitive decline. It is interesting to note that there were some mice that performed nearly 2 standard deviations worse than the average. If we were to eliminate those mice from the data as outliers, there would be a very consistent pattern to the data showing both sham groups and the post-crossover treatment group all performing at the same level, whereas the post-crossover placebo group starts to show a return of the TBI effect. This shows that even delayed administration of anatabine may ameliorate TBI-dependent cognitive dysfunction at chronic time-points if it is administered for a chronic length of time, but prolonged cessation of treatment may result in the return of cognitive dysfunction.
To better understand the mechanism behind these changes, we examined STAT3 phosphorylation histologically as well. Anatabine has been shown to reduce phosphorylation of STAT3 and NF-κB, 20,21 which may be influencing microglial activation downstream from the axonal damage. Prior to treatment crossover, anatabine-treated r-mTBI had fewer p-STAT3 positive cells in the corpus callosum than placebo-treated r-mTBI mice, correlating well with their neuroinflammatory and spatial memory profiles. Following the treatment crossover there was no difference between p-STAT3 in the post-crossover treated and untreated r-mTBI animals, nor were there any differences compared with their shams. We noted a strong aging effect on STAT3 phosphorylation, which may have obscured the TBI-dependent signal, but inclusion of treatment naïve cohorts of mice was beyond the scope of the current study. Thus to place our findings in the context of aging-dependent STAT3 phosphorylation we analyzed tissue collected from treatment naïve mice in a previous study using the same r-mTBI/r-sham paradigm and euthanized at time-points flanking the current final time-point, namely 12 or 24 months post-injury/sham. By bracketing the 18-month time-point in this manner we were able to note a dramatic decrease in STAT3 phosphorylation in the anatabine-treated r-mTBI animals at 18 months post-injury compared with the treatment naïve r-mTBI animals at 12 or 24 months post-injury, regardless of whether anatabine was administered in the first 9 or last 9 months of the study. Both pre- and post-crossover treated r-mTBI mice had lower levels of STAT3 phosphorylation within the corpus callosum than untreated r-mTBI mice from the 12-month post-TBI time-point. Although this suggests that the lack of a TBI-dependent signal in STAT3 phosphorylation at the 18-month time-point is due to a treatment effect rather than aging, by the 24-month time-point in the untreated cohort the levels had increased to a point of saturation and obscured any TBI-dependent increase. This is consistent with previous reports of STAT3 activation's role in senescent macrophages. 32 Even if aging is obscuring a TBI-dependent signal in the post-crossover cohort, early or delayed treatment with anatabine appears to reduce the age-dependent increase observed in the sham animals as well when compared with the untreated cohort, supporting its potential utility regardless of its effect on injury-dependent inflammation.
Although we do not have any naïve tissue from an 18-month time-point for comparison, it is possible that anatabine treatment of the sham animals has also delayed the onset of the age-dependent increase seen in the naïve animals at the 24-month time-point. We hypothesize that anatabine treatment may have delayed the increase that was probably continuous and ongoing in the untreated animals between the 12- and 24-month time-points. This treatment-related reduction in STAT3 phosphorylation appears to be ongoing at chronic time-points after TBI and perseveres even after the cessation of anatabine administration. This may be important in reducing neurodegenerative chronic inflammation driving at least some of the neurobehavioral deficits seen in this model of mTBI.
Anatabine shows potential as a method of reducing chronic inflammation after TBI, although the reductions seem to be regionally specific and more complete in less severely affected regions. A wide variety of animal models as well as human studies have shown a common theme of chronic inflammation after TBI.
This study shows that chronic inflammation may be a key factor in mediating spatial memory loss after TBI, and may be amenable to treatment. Ideally, a potential therapeutic should be effective against the long-term consequences of TBI, and be able to reduce the chronic pathological and behavioral consequences. Previous problems in clinical trials have arisen due to difficulties in early recruitment as well as bioavailability and treatment dose and timing. Anatabine has a history of safe use as a dietary supplement, and as it is well-characterized with a low risk of adverse events, it shows potential as a compound for future clinical trials. It can be safely administered in a chronic treatment paradigm, which may be necessary to provide efficacy at long-term time-points after injury. Future studies will seek to quantify the minimum effective dose, examine the effect of delayed treatment, and examine time-points after the cessation of treatment in preparation for future human clinical trials.
Footnotes
Acknowledgments
This study was paid for by the Roskamp Foundation. Dr. Fiona Crawford holds a VA Career Scientist Award. Dr. William Stewart is supported by NIH grants NS038104 and NS094003, DOD grant PT110785, and by a NHS Research Scotland Career Researcher Fellowship.
Author Disclosure Statement
Dr. Michael Mullan was the former CEO of Rock Creek Pharmaceuticals. He was not involved in the design of the study or the interpretation of its results. Rock Creek Pharmaceuticals supplied the anatabine used in the study.