
Abstract
Resuscitation with polynitroxylated pegylated hemoglobin (PNPH), a pegylated bovine hemoglobin decorated with nitroxides, eliminated the need for fluid administration, reduced intracranial pressure (ICP) and brain edema, and produced neuroprotection in vitro and in vivo versus Lactated Ringer's solution (LR) in experimental traumatic brain injury (TBI) plus hemorrhagic shock (HS). We hypothesized that resuscitation with PNPH would improve acute physiology versus whole blood after TBI+HS and would be safe and effective across a wide dosage range. Anesthetized mice underwent controlled cortical impact and severe HS to mean arterial pressure (MAP) of 25–27 mm Hg for 35 min, then were resuscitated with PNPH, autologous whole blood, or LR. Markers of acute physiology, including mean arterial blood pressure (MAP), heart rate (HR), blood gases/chemistries, and brain oxygenation (PbtO2), were monitored for 90 min on room air followed by 15 min on 100% oxygen. In a second experiment, the protocol was repeated, except mice were resuscitated with PNPH with doses between 2 and 100 mL/kg. ICP and 24 h %-brain water were evaluated. PNPH-resuscitated mice had higher MAP and lower HR post-resuscitation versus blood or LR (p < 0.01). PNPH-resuscitated mice, versus those resuscitated with blood or LR, also had higher pH and lower serum potassium (p < 0.05). Blood-resuscitated mice, however, had higher PbtO2 versus those resuscitated with LR and PNPH, although PNPH had higher PbtO2 versus LR (p < 0.05). PNPH was well tolerated across the dosing range and dramatically reduced fluid requirements in all doses—even 2 or 5 mL/kg (p < 0.001). ICP was significantly lower in PNPH-treated mice for most doses tested versus in LR-treated mice, although %-brain water did not differ between groups. Resuscitation with PNPH, versus resuscitation with LR or blood, improved MAP, HR, and ICP, reduced acidosis and hyperkalemia, and was well tolerated and effective across a wide dosing range, supporting ongoing pre-clinical development of PNPH for TBI resuscitation.
Introduction
T
Traditional resuscitation solutions, such as crystalloids (i.e., saline, Lactated Ringer's [LR] solution), have many advantages, including their low cost of production, long shelf-life and stability, lack of need for refrigeration, long history of use, and proven safety profile. However, large volumes of these fluids are typically required for adequate resuscitation, which has been shown to contribute to increased brain edema—potentially exacerbating brain injury. 5 –7 Additionally, they have been shown to have other deleterious extracranial effects, including increased duration of mechanical ventilation and mortality. 8,9 In addition, in combat casualty cases, a resuscitation fluid of the smallest volume and weight would be highly desirable for the military medic. Colloids, such as albumin and starch-based solutions, have favorable resuscitative properties versus crystalloids, including reduced fluid requirements and improved hemodynamics; yet, surprisingly, recent evidence has shown their use in the presence of TBI may acutely worsen outcomes. 5 The gold standard for resuscitation, particularly in the setting of hemorrhage, remains blood products 10 ; however, the drawbacks of blood products are numerous, including need for cross-matching, short shelf-life, need for refrigeration, potential transmission of infection, and the potential contribution to systemic inflammation (e.g., including systemic inflammatory response syndrome and transfusion-related acute lung injury). 11 Numerous observational studies have demonstrated that blood transfusion in TBI patients is independently associated with worse neurologic outcome, suggesting blood transfusion in the brain-injured patient may have detrimental consequences. 12,13
Given these limitations, much interest has been on the development of new resuscitation fluids, including hemoglobin-based oxygen carriers (HBOCs). HBOCs are solutions of cell-free hemoglobin molecules that have been modified to prevent some of hemoglobin's deleterious effects in an extra-cellular environment, including nitric oxide (NO) scavenging and generation of free radicals contributing to oxidative stress. 14 –16 Polynitroxylated-pegylated hemoglobin (PNPH) is a novel HBOC currently being developed for use as a small-volume resuscitation solution and designed to mitigate drawbacks of its hemoglobin-based chemistry. PNPH is a pegylated bovine carboxyhemoglobin (CO-Hb) covalently labeled with nitroxide moieties. Polynitroxylation of the hemoglobin confers antioxidant effects and prevents NO scavenging, while the polyethylene glycol (PEG) side chains create a “hydrating shell” with a marked oncotic effect that is useful in resuscitation. 17 –19 PNPH, in our prior studies, dramatically reduced resuscitation fluid requirements, ICP, and brain edema, and attenuated neuronal death in vulnerable hippocampal regions. Unlike conventional free hemoglobins, PNPH is not toxic to neurons and has surprising neuroprotective effects in vitro. 17,19
In the ongoing pre-clinical development of PNPH, we carried out two additional studies in mice to further investigate its resuscitative properties and safety profile. First, we compared PNPH resuscitation versus equal volume whole blood in our established TBI+HS model. 20 Next, we performed a dose response study, comparing a wide range of PNPH doses for the resuscitation of TBI+HS. We hypothesized that PNPH would perform similarly to whole blood with regard to acute physiologic markers of resuscitation and that it would be safe and effective across a wide dosage range.
Methods
The Institutional Animal Care and Use Committee of the University Of Pittsburgh School of Medicine approved the experiments used in this study. C57/BL6 mice (n = 32; Jackson Laboratories, Bar Harbor, ME), 12–15 weeks of age were allowed ad libitum food and water and were housed in controlled environmental conditions until the study began.
Mice underwent a controlled cortical impact (CCI) and HS paradigm (Fig. 1) that has been used successfully in our laboratory for prior investigations, and its methods have been described in prior publications. 17,19,20 Our model is designed to mimic a clinically relevant scenario of a moderate-severe TBI sustained in the field followed by HS and subsequent resuscitation in both the pre-hospital and hospital phase.

Schematic of traumatic brain injury plus hemorrhagic shock model. In the “equal volume resuscitation” study, mice received only pre-determined resuscitation volumes without further fluid or blood administration. In the “dose response” study, mice received their randomly allocated resuscitation solution, then LR boluses for mean arterial pressure <70 mm Hg. They then received re-infusion of shed blood in the “Hospital” phase. CCI, controlled cortical impact; LR, Lactated Ringer's solution.
CCI and monitoring
Anesthesia was induced with 4% isoflurane in 2:1 N2O/oxygen delivered via nose cone and was then maintained with 1.5% isoflurane in a 2:1 N2O/oxygen mixture for surgical preparation. Under sterile conditions, an inguinal cut-down was performed and femoral venous and arterial catheters (modified PE-50 tubing) were placed. Mice were then placed in a stereotactic frame (Kopf, Tujunga, CA) and an incision was made over the left scalp. Using a dental drill, a 5 mm craniotomy was performed over the left parietal cortex and the bone flap was removed. A temperature probe (Physitemp, Clifton, NJ) was inserted into the left temporalis muscle to approximate brain temperature. Body temperature was monitored via rectal probe. Temperature was controlled throughout the study via heating lamp to maintain brain temperature at 37 ± 0.5°C. PbtO2 was monitored and recorded using a microelectrode (Unisense, 50 μm; Arhus, Denmark) stereotactically inserted via a burr hole through the cortex into the left dorsal hippocampus (ipsilateral to the CCI; bregma −2.5 AP, −2.0 ML; depth, 2.0 mm). After craniotomy, the isoflurane was decreased to 1% in room air for 10 min before introduction of CCI. A CCI was performed with a pneumatic impactor (Bimba, Monee, IL), using a flat 3-mm tip impounder on the left parietal cortex at 5 m/sec and to a depth of 1.0 mm.
CCI, pressure controlled HS mode: Comparison of resuscitation with PNPH, LR, and whole blood
After CCI, mice underwent a pressure-controlled HS model comprised of three phases: “Shock,” “Pre-hospital,” and “Hospital.” Shock was initiated 5 min after CCI by removal of 12 mL of blood per kg of body weight over 5 min via the venous canula. Blood was drawn into a syringe containing 0.07 mL of citrate anticoagulant (Cardian BCT, Lakewood, CO). An additional 12 mL of blood per kg of body weight was removed over the next 5 to 15 min for a total of ∼30% of the blood volume in a mouse. 21 Using this approach, a mean arterial pressure (MAP) of 25–27 mm Hg was achieved and this level was maintained by continued removal or re-infusion of autologous citrated blood from the venous catheter in 0.05 mL aliquots using the same syringe. The mice were maintained at this MAP for the duration of the 35 min Shock phase initiated at the time of blood removal. During the Shock phase, anesthesia was reduced from 1% isoflurane in room air to 0.5% isoflurane in room air. After the Shock phase, the mice entered the Pre-hospital phase and were resuscitated using one of three approaches: i) 4% PNPH solution in normal saline (SynZyme Technologies, Irvine, CA), 20 mL/kg (n = 10); ii) autologous whole blood, 20 mL/kg (n = 12); or iii) LR, 60 mL/kg (n = 10). A 3:1 ratio of crystalloid to blood/PNPH dosing was chosen based on a common clinical standard of crystalloid being equivalent to 1/3 the volume of whole blood infused. In this model, the amount of shed blood removed to achieve the desired MAPs of 25–27 mm Hg typically totals ∼25 mL/kg (∼30% total blood volume). Thus, administration of 20 mL/kg of autologous whole blood (a conventional clinical resuscitation volume) resulted in re-infusion of most of the shed blood after 35 min of HS in each case. All mice were resuscitated at the beginning of the Pre-Hospital phase with 20 mL/kg of their respective resuscitation fluid. Mice in the LR group then received additional 20 mL/kg boluses × 2 every 5 min to achieve a total of 60 mL/kg of total fluid resuscitation (once again to mimic clinical care, where a 3:1 ratio of crystalloid to blood is routinely administered). Unlike prior studies in our model, mice received no additional fluids for the duration of this experiment after their initial resuscitation. During the Pre-hospital phase, room air was used to mimic field resuscitation in combat casualty care, where oxygen is not readily available. During the model, standard physiologic outcomes were obtained at baseline and post-injury, including MAP, heart rate (HR), and blood chemistries/gases and brain tissue oxygen tension (PbtO2). After the 90-min Pre-Hospital phase, mice entered the 15-min Hospital phase. In the Hospital phase, in this study, mice received no additional blood or fluids. During that phase, 1% isoflurane in pure oxygen was administered, again mimicking clinical care in a combat support hospital or civilian emergency department, where 100% oxygen would be available. At the completion of the model, mice were sacrificed with an intravenous potassium chloride bolus.
Resuscitation of CCI+HS with PNPH: Assessment of dose response
To determine the relative effectiveness of PNPH resuscitation across a wide dosage range, as well as to determine its safety in higher doses, a 50-fold range was selected encompassing both the extreme low and high range of potential PNPH dosing. Thus, a total of six PNPH doses were chosen for investigation (2, 5, 10, 20, 50, or 100 mL/kg) and were compared with standard resuscitation with LR. Mice (n = 21, 3/group × 7 groups) underwent our standard CCI+HS model, as detailed above, although in this experiment PbtO2 was not measured. Instead, ICP was monitored continuously by an ICP transducer (1 Tr MIKRO-TIP) inserted through a burr hole into the right frontal lobe, contralateral to injury. After the normal 35-min HS period, mice were resuscitated with either LR or PNPH in one of the previously mentioned doses. Resuscitation was begun in all mice with 20 mL/kg total fluid, regardless of group to mimic the clinical standard of trauma care. Thus, mice in the 2, 5, or 10 mL/kg PNPH groups received additional LR (18, 15, and 10 mL/kg respectively) to achieve this goal. Given the high oncotic potential of PNPH, in an effort to prevent mortality or morbidity from acute volume overload, mice in the 50 and 100 mL/kg group started resuscitation with the same initial bolus volume of 20 mL/kg, but then received the remainder of their allocated fluid volume (30 and 80 mL/kg respectively) over the subsequent 60 min. This was felt to be a logical approach given that a 50 or 100 mL/kg bolus of PNPH would be substantially in excess of its projected standard of care use in clinical practice. After initiation of resuscitation in the Pre-Hospital phase, mice received ongoing fluid resuscitation with LR (10 mL/kg) for MAP <70 mm Hg. There are a total of 17 time-points in the Pre-Hospital phase in which mice could receive additional fluid, thus making the maximum total fluid resuscitation volume, including the initial 20 mL/kg bolus, to be 190 mL/kg. After 90-min of Pre-Hospital resuscitation, mice entered a Hospital phase, during which they had re-infusion of their previously shed blood and were placed on 100% oxygen. The primary outcomes of this study were similar to the previous experiment and included acute physiologic measures, including MAP, HR, and blood gas/chemistry analysis, and ICP. At completion of the model, mice were sacrificed via isoflurane overdose followed by decapitation, and their brains immediately removed for determination of brain edema by %-brain water via wet-weight/dry-weight method as previously described. 17
Statistical analysis
Physiologic parameters were analyzed by two-way analysis of variance for repeated measures; between-group comparisons for other outcomes were made using Student's t-test. Post hoc testing was corrected for multiple comparisons. All results are provided as mean ± standard error of the mean. Significance was determined by a p value ≤0.05.
Results
Comparison of resuscitation after TBI + HS with PNPH, whole blood, and LR
A total of 32 mice underwent the above paradigm (n = 10 PNPH, n = 12 blood, n = 10 LR). Overall acute survival to the completion of the model was 30/32 (93%); two mice in the LR group died near the end of the Pre-Hospital phase. Overall, MAPs were significantly different between all groups in both the Pre-Hospital and Hospital phase (p < 0.001). Phase average MAPs for PNPH, blood, and LR during the Pre-Hospital phase were 81.2 ± 0.4, 65.8 ± 0.6, and 42.8 ± 0.7 mm Hg respectively (Fig. 2A). PNPH-treated mice had stable MAP, after resuscitation with no significant difference in MAPs in the first 15 min after resuscitation versus the final 15 min of the model (78.0 ± 1.2 vs. 80.1 ± 1.1 mm Hg, p = 0.20). However, both groups resuscitated with LR and whole blood experienced a progressive decline in MAP after resuscitation and had significantly lower MAPs (p < 0.001) in the final 15 min of the model versus the immediate 15 min after resuscitation (blood: 77.2 ± 1.5 vs. 56.8 ± 1.8 mm Hg; LR: 47.7 ± 2.2 vs. 31.2 ± 1.4 mm Hg). The LR mice received three separate boluses every 5 min to complete resuscitation versus whole blood and PNPH receiving a single bolus at the initiation of the resuscitation phase. This comparison, therefore, represented the initial three recorded MAPs after full resuscitation in both groups.
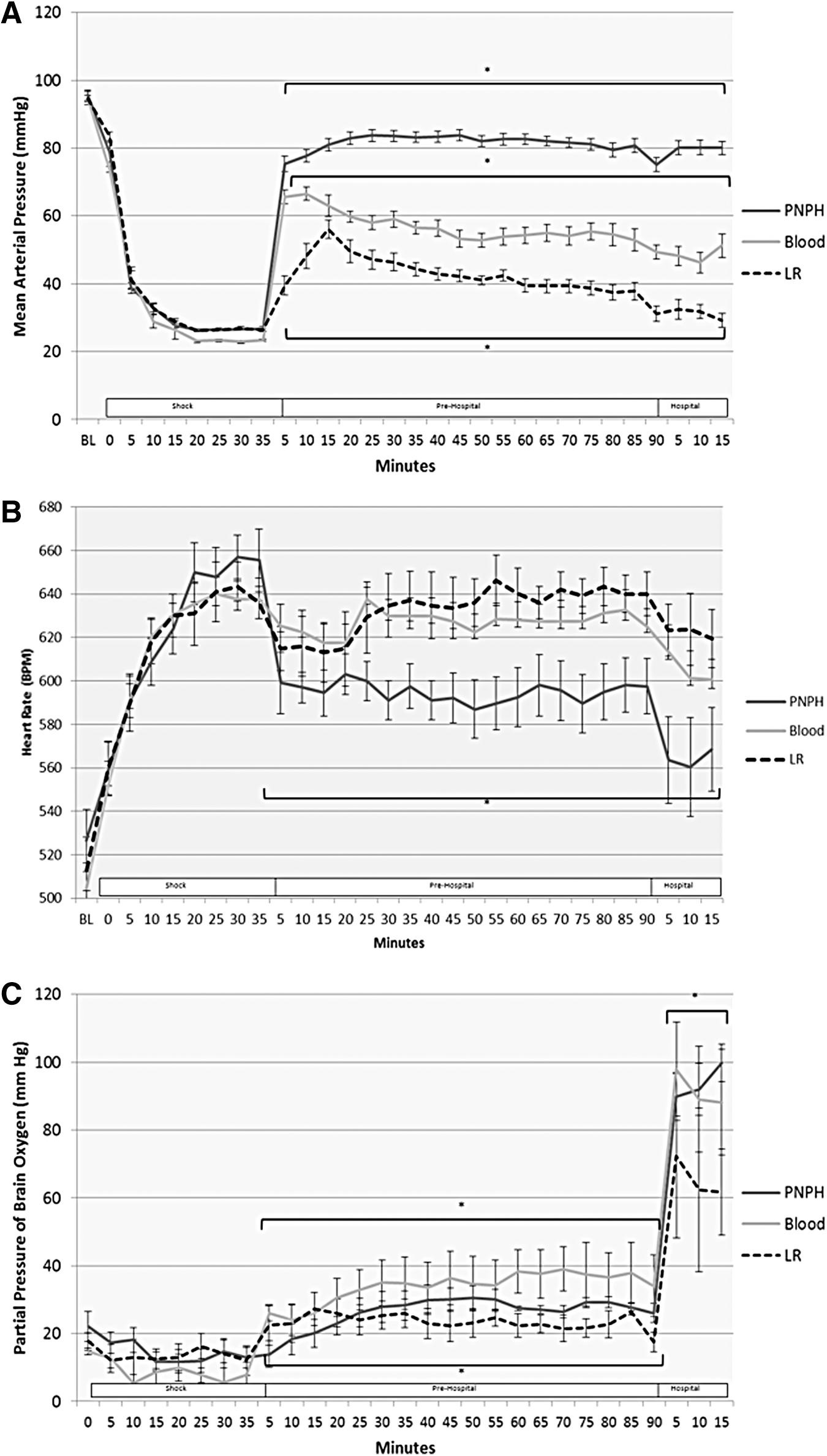
At baseline and during shock, there were no differences in HR between groups. After resuscitation, HRs in the PNPH- resuscitated mice were significantly lower than those of both the LR and blood groups in the Pre-hospital and Hospital phase (p < 0.05; Fig. 2B). There was no significant difference in HR between the LR and blood groups.
Brain tissue oxygenation (PbtO2) was similar between all groups during shock (13–15 mm Hg); however, during the Pre-hospital phase, all three groups' mean PbtO2 differed from each other, with the highest in the blood group (33.8 ± 1.6 mm Hg) followed by the PNPH group (26.2 ± 0.8 mm Hg), then the LR group (23.5 ± 0.9 mm Hg; Fig. 2C). PbtO2 in both the PNPH and blood groups was significantly higher than in the LR group during the Hospital phase, though the blood and PNPH groups did not differ from each other. During the Hospital phase, mice were placed on pure oxygen with 1% isoflurane, representing arrival at a receiving hospital, thus explaining the significant rise in PbtO2 in all groups during the Hospital phase. Also, the temporal trend of PbtO2 in the PNPH- resuscitated mice is noteworthy. Both the LR- and blood-resuscitated mice had a small but rapid initial improvement in their PbtO2 after the initial resuscitation bolus; however, the response in PNPH-resuscitated mice was much slower. These mice had a gradual rise in PbtO2 after resuscitation, and did not surpass baseline PbtO2 values until on average 20 min into the Pre-Hospital phase. In contrast, both LR- and blood-resuscitated mice had restoration of their PbtO2 to baseline values at the first time-point in the Pre-Hospital phase. While in PNPH-resuscitated mice PbtO2 was initially lower than that in LR-treated mice, this trend reversed as PNPH-treated mice had gradual improvement in their PbtO2 and while LR-treated mice had a gradual deterioration in PbtO2.
Blood gas analyses showed similar findings between all groups in baseline values and during the Shock phase (Fig. 3; Table 1); however, before resuscitation, oxygen saturations were slightly lower in the blood group during the Shock phase versus PNPH (p < 0.05). As expected in our TBI+HS model, all groups developed a significant lactic acidosis after Shock, with peak lactates between 8 and 9 mmol/L. During the Pre-Hospital and Hospital phases, significant differences developed between groups. The PNPH-treated mice had significantly higher pH versus both the LR- and blood-treated groups in the Pre-Hospital phase and versus the LR group in the Hospital phase (p < 0.05). Also, there was reduced hyperkalemia in PNPH-treated mice, with PNPH-treated mice having lower potassium levels versus LR- and blood-treated mice in both the Pre-Hospital and Hospital phase (p < 0.01). As expected, mice treated with blood had higher hemoglobin and hematocrit values versus both LR- and PNPH-treated mice in the Pre-Hospital and Hospital phase (p < 0.01). Surprisingly, blood oxygen saturations were significantly lower in the blood-treated mice during the Pre-hospital phase versus both the PNPH- and LR-treated mice (p < 0.05). However, with the higher hemoglobins in the blood-treated mice, oxygen content was significantly higher than both the PNPH and LR groups in the Pre-Hospital and Hospital phase (p < 0.01). During the Hospital phase after being placed on 100% oxygen, there were significant differences in PaO2 between all groups in the Hospital phase, with PNPH > blood > LR (p < 0.01). There were no differences in acid/base status in either the Pre-Hospital or Hospital phase, with similar lactates and base deficits between groups.
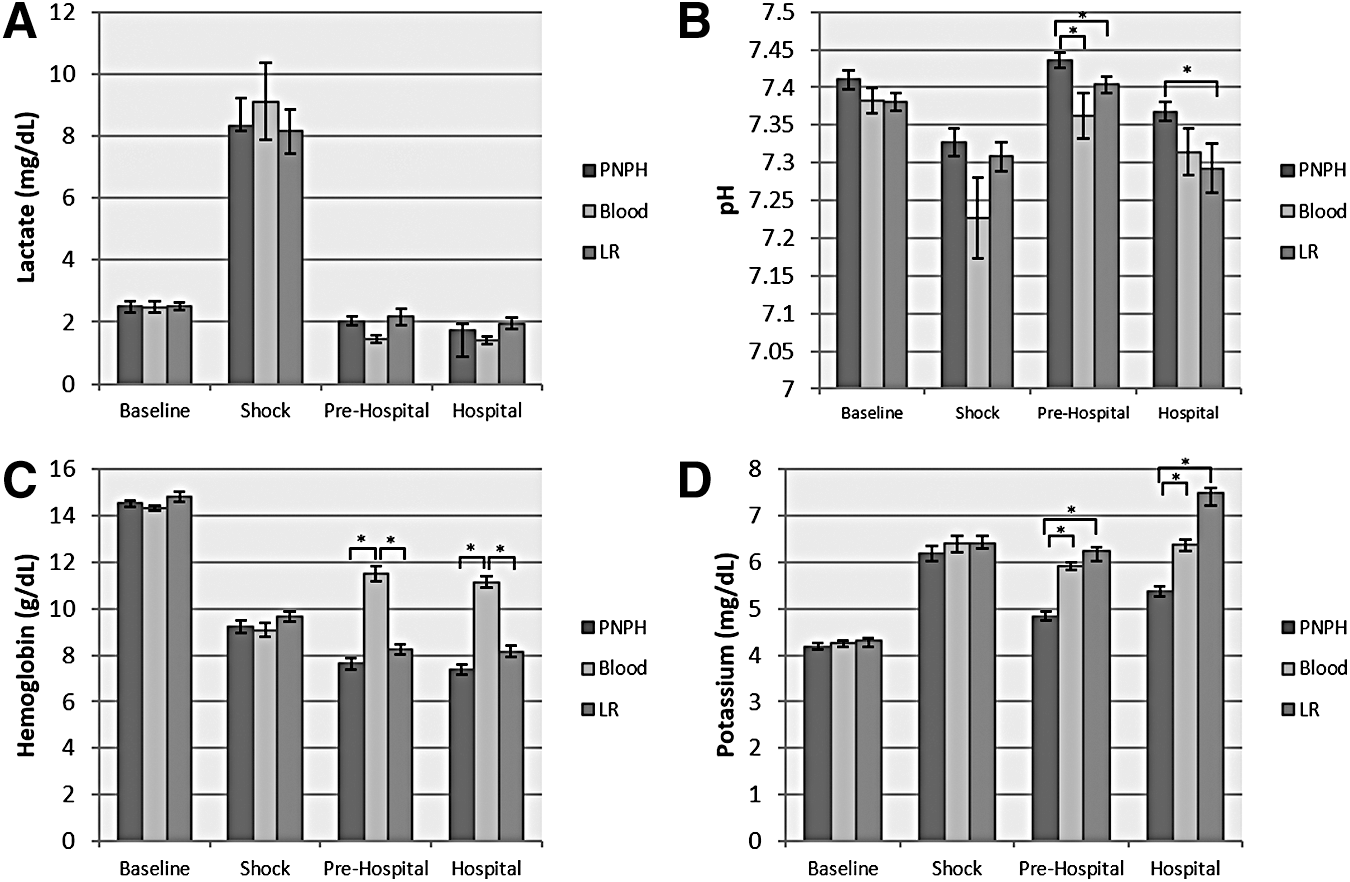
Blood gas and chemistry results during “equal volume resuscitation” study. There were no significant differences between groups in lactate
p < 0.05 PNPH vs. whole blood; ** p < 0.01 PNPH vs. whole blood; †p < 0.05 PNPH vs. LR; ‡p < 0.01 PNPH vs. LR; #p < 0.05 whole blood vs. LR; ##p < 0.01 whole blood vs. LR.
PNPH, polynitroxylated pegylated hemoglobin; LR, Lactated Ringer's solution; PaCO2 (mm Hg), PaO2 (mm Hg), SaO2, oxygen saturation (%); HCT, hematocrit (%); Hgb, hemoglobin (g/dL); O2 content, Oxygen content (g/dL); BE, base excess (mEq/L); HCO3, bicarbonate (mmol/L); Na, sodium (mmol/L); K, potassium (mmol/L); Cl, Chloride (mmol/L); Ca, calcium (mmol/L); Gluc, glucose (mg/dL); Lac, lactate (mmol/L); MetHb, methemoglobin (%); CoHb, carboxyhemoglobin (%); Osm, osmolality (mmol/kg).
Resuscitation after TBI + HS with PNPH: Dose response study
A total of 21 mice underwent the above paradigm (n = 3/group). One mouse in the 50mL/kg group died before initiation of resuscitation, and was therefore excluded from analysis. Thus, a total of 20 mice were included in the final analysis. All mice tolerated their assigned resuscitation volume well, including both the 50 and 100 mL/kg groups. No mice in the 50 or 100 mL/kg group developed hypertension, hemodynamic instability, or other evidence suggestive of acute volume overload. All groups resuscitated with PNPH showed significantly improved MAPs during the Pre-hospital phase versus resuscitation with LR (p < 0.01; Fig. 4A). As seen in previous studies in our model, 17,20 LR-resuscitated mice failed to achieve the MAP goal of 70 mm Hg until re-infusion of their shed blood during the Hospital phase. Notably, even mice receiving the lowest dosages of PNPH showed significant improvement in MAP for the duration of the model and remained significantly improved versus LR-treated mice (p < 0.01). There were no significant differences in HR between groups (Fig. 4B), though LR mice did have the numerically highest HR in the Pre-hospital and Hospital phase.
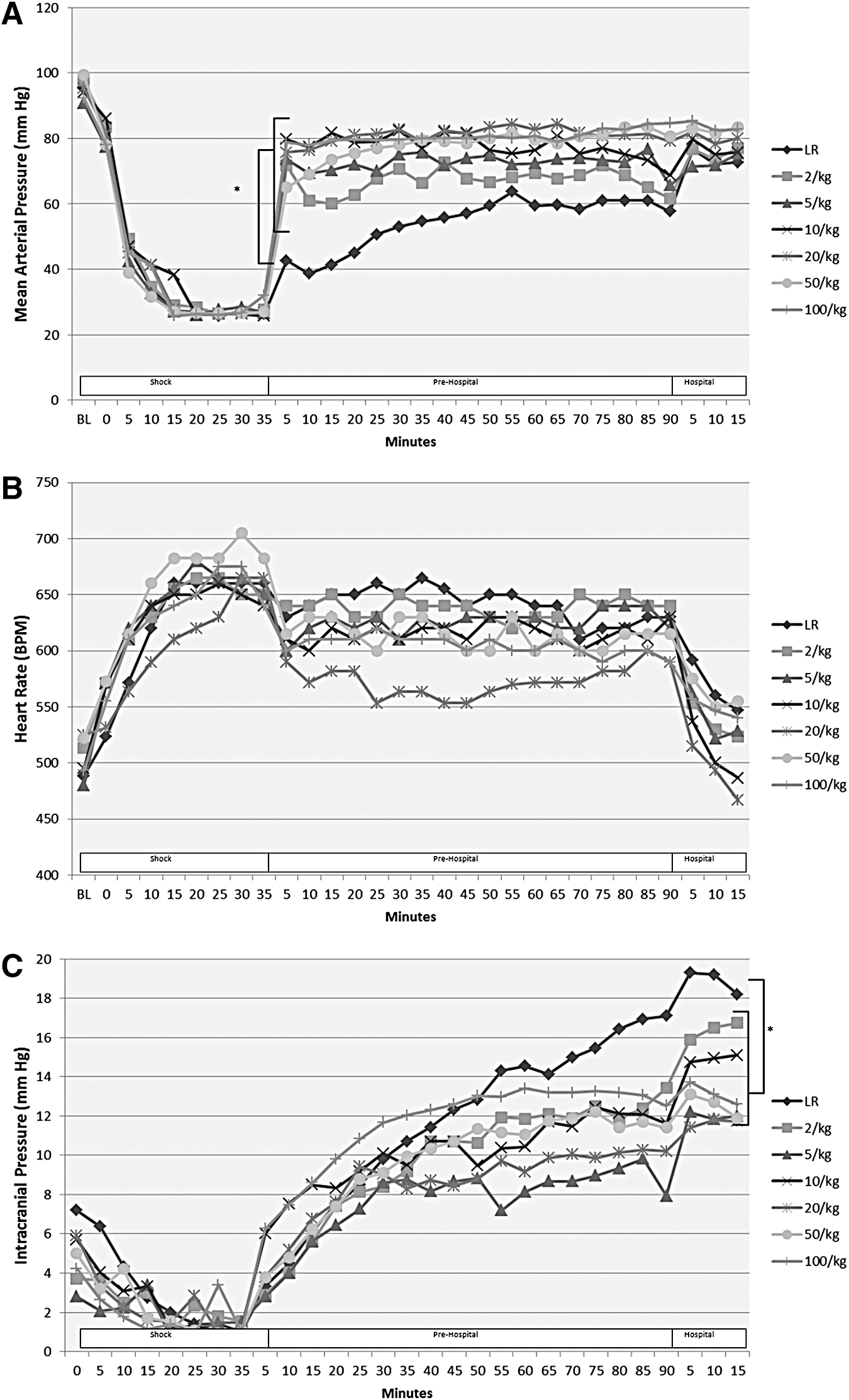
Panel
ICP in all groups was similar at baseline and during the Shock phase. LR-treated mice, however, developed significantly higher ICPs during the Pre-Hospital phase versus all groups (p < 0.05) except the 20 and 100 mL/kg PNPH groups. (Fig. 4C). Also, ICP in the LR group was highest in the Hospital phase versus all groups (p < 0.05) except 2 mL/kg PNPH.
Fluid requirements were greatest in the LR group and treatment with PNPH at all doses led to a significant reduction in total fluid requirements and LR resuscitation volume (p < 0.01) (Fig. 5). The PNPH dose showed a strong inverse logarithmic correlation to LR resuscitation volume requirements (R2 = 0.8312). Brain edema, as measured by %-brain water, did not differ significantly between groups in either the ipsilateral (injured) or contralateral (uninjured) hemisphere (Fig. 6); however, PNPH dose and %-brain water again exhibited a strong inverse logarithmic correlation in the ipsilateral hemisphere (R2 = 0.6363); in the contralateral hemisphere this correlation was much weaker (R2 = 0.1503).
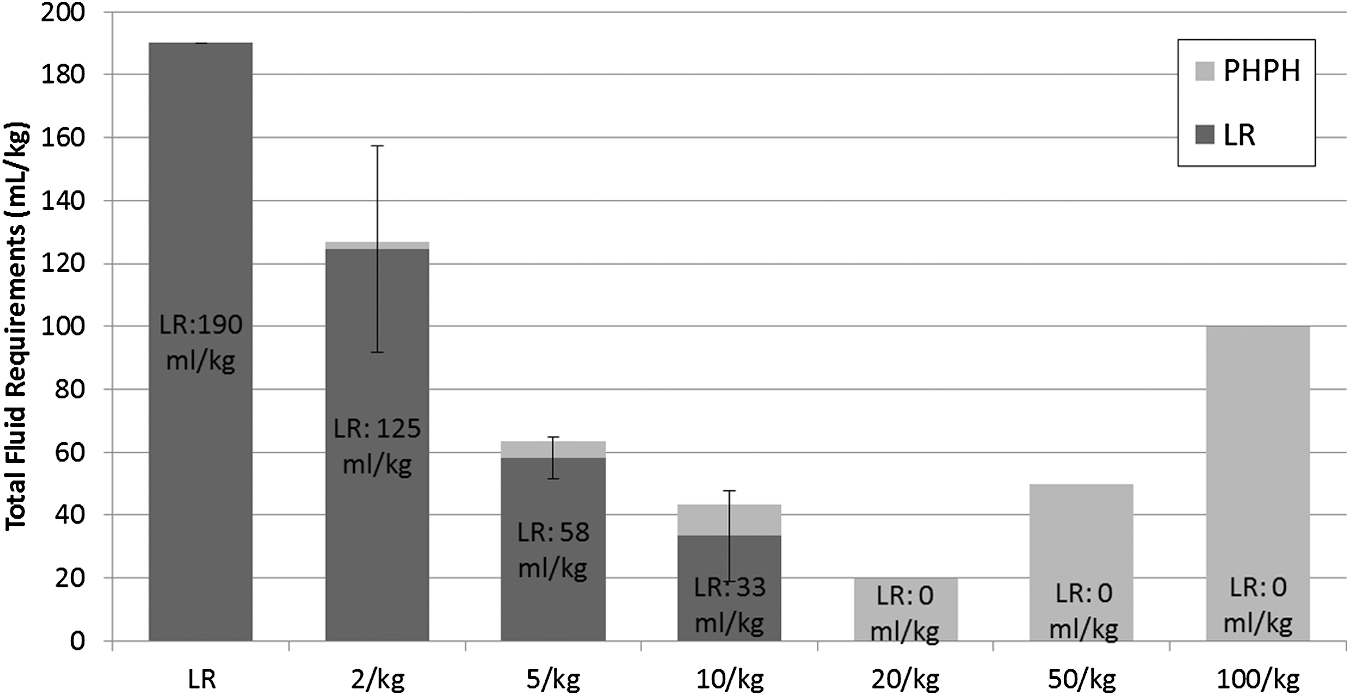
Fluid requirements during the polynitroxylated pegylated hemoglobin (PNPH) “dose response” study. Group designation (Lactated Ringer's solution [LR] or PNPH dose ranging from 2 mL/kg to 100 mL/kg) shown along the x-axis and amount of LR needed depicted within the vertical bars. Total fluid required depicted along the y-axis. Mice resuscitated with LR required the most fluid versus all PNPH dosing (across the range shown from 2 mL/kg to 100 mL/kg) groups (p < 0.01). Based on the protocol, which mandates administration of a 10 mL/kg LR fluid bolus every 5 min when MAP is <70 mm Hg during the “Pre-Hospital” resuscitation phase, PNPH resuscitation was associated with markedly reduced total fluid requirements versus LR at all dosages, as well as a significantly reduced LR resuscitation requirement versus the LR resuscitation group (p < 0.01 for all comparisons to LR). Even the low dose PNPH groups (both 2 mL/kg and 5 mL/kg) produced clinically relevant 1.5 to 3.2-fold reduction in total fluid requirements.
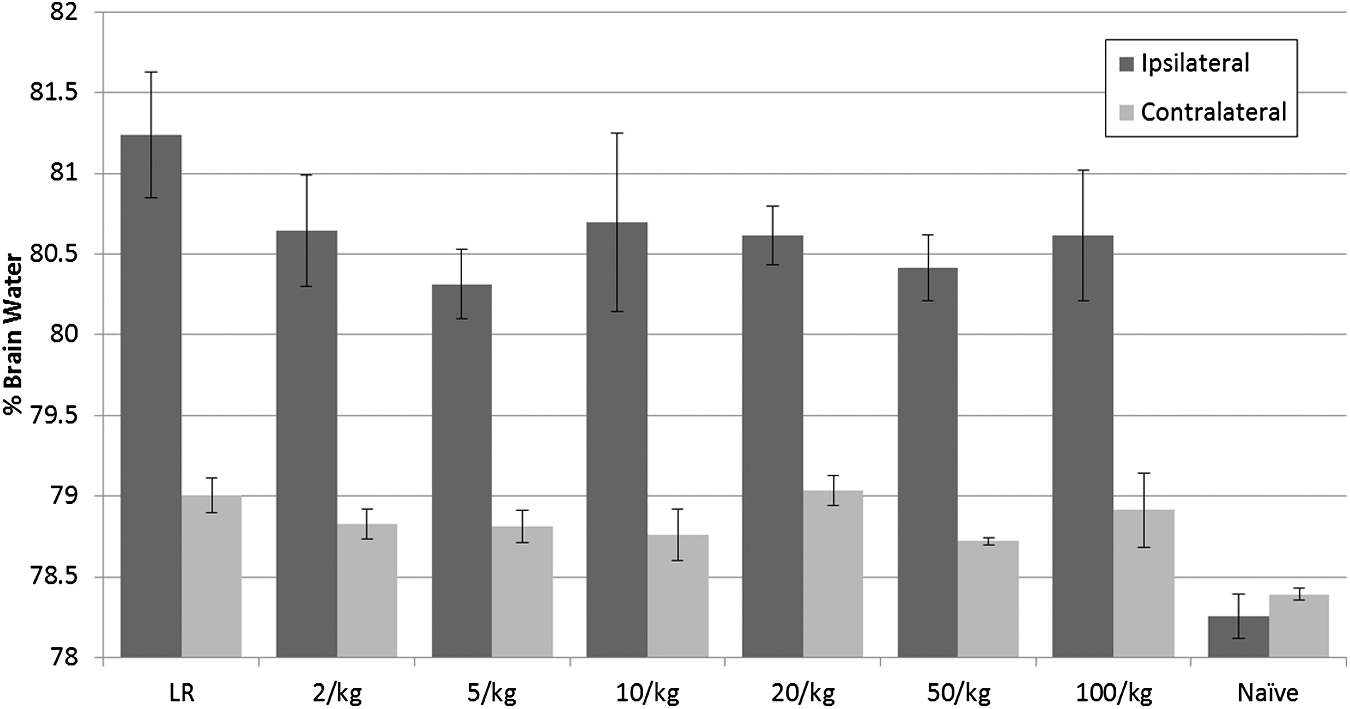
%-Brain water of mice in Lactated Ringer's solution (LR) or polynitroxylated pegylated hemoglobin (PNPH) groups (across the range shown from 2 mL/kg to 100 mL/kg) after resuscitation in the “dose response” study. There were no significant differences between groups in %-brain water at 24 h in the ipsilateral (injured) or contralateral (uninjured) brain hemispheres. %-Brain water was significantly greater in all injured groups than in naïve mice in the respective injured or contralateral hemisphere (*p < 0.05).
Blood gas and chemistry analysis showed significant differences between groups in both the Pre-Hospital and Hospital phase, with serum sodium, chloride, and osmolality all increasing significantly as PNPH dose increased (Fig. 7; Table 2). There was a direct linear correlation between PNPH dose and serum sodium (R2 = 0.6621), chloride (R2 = 0.5271), and osmolality (R2 = 0.4695). There also was a significant increase in CO-Hb and methemoglobin (Met-Hb) as PNPH dose increased with strong direct linear correlations (R2 = 0.8957 and 0.8953, respectively).

Blood gas and blood chemistries of mice during the polynitroxylated pegylated hemoglobin (PNPH) “dose response” study. Methemoglobin levels
p < 0.05; ** p < 0.05 vs. LR.
PNPH, polynitroxylated pegylated hemoglobin; LR, Lactated Ringer's solution; PaCO2 (mm Hg), PaO2 (mm Hg), SaO2, oxygen saturation (%); HCT, hematocrit (%); Hgb, hemoglobin (g/dL); BE, base excess (mEq/L); HCO3, bicarbonate (mmol/L); Na, sodium (mmol/L); K, potassium (mmol/L); Cl, chloride (mmol/L); Ca, calcium (mmol/L); Gluc, glucose (mg/dL); Lac, lactate (mmol/L); Osm, osmolality (mmol/kg); CoHb, carboxyhemoglobin (%); MetHb, methemoglobin (%).
Discussion
Resuscitation with PNPH led to many significantly improved acute physiologic outcomes versus equal volume resuscitation with autologous whole blood or three times the volume of LR. Mice resuscitated with PNPH had an immediate restoration of MAP to the target threshold of >70 mm Hg and remained stable the remainder of the experiment, with significantly improved HRs versus LR or whole blood. This robust recovery of MAP and reduction in HR is consistent with our prior work 17 –19 ; however, this is the first study comparing PNPH resuscitation to whole blood, which, when available, is the “gold-standard” for the resuscitation of a trauma patient with hemorrhage. 10 Initially, mice had comparable MAPs and HRs versus the blood and PNPH groups; however, early in the Pre-Hospital phase, the groups diverged. By the end of the study, blood-resuscitated mice had significant decline and overall greater lability of MAPs versus PNPH, but better than those of LR. However, their HRs remained significantly elevated for the duration of the model versus PNPH and never differed significantly versus the LR group.
The robust improvement and stability of MAP with PNPH resuscitation could be argued to represent a mild vasopressor effect 22 –24 ; however, evidence against this as a major mechanism is the improvement in HR of PNPH mice versus blood resuscitation and improved acid-base status. PNPH mice had significantly improved pH versus both the blood and LR groups and reduced hyperkalemia to a degree that could be considered clinically relevant. This reduced hyperkalemia may be, in part, secondary to intracellular potassium shifts given the higher pH in the PNPH mice. However, given the relatively small difference in pH between groups, this is unlikely the sole contributing factor for such large differences in potassium levels. It also may reflect improved tissue perfusion early in the model (leading to less tissue necrosis damage and potassium release); improved renal perfusion and clearance; and/or reduced potassium load from lysed red blood cells in from the whole blood–treated group.
Another potential explanation for the MAP differences seen between blood- and PNPH-treated mice could be in the citrate used for anti-coagulation during the HS phase. The blood, after being removed, blood is drawn into a syringe containing citrate to chelate calcium and prevent coagulation. At the end of the HS phase, when this blood was re-infused in the blood-resuscitated group, citrate may have had systemic effects, leading to depletion of available ionized calcium, negative cardiac inotropy and loss of vascular tone. 25 This is seen in clinical practice, although usually in massive transfusion, where calcium administration is necessary to prevent the systemic effects of citrate accumulation. 25,26 Citrate is typically rapidly metabolized into bicarbonate, primarily by the liver; however, in large quantities it may overwhelm the metabolic capacity, leading to prolonged systemic effects. 5,18 Refuting this theory is the persistence of differences in MAP between the blood and PNPH groups. Blood gases obtained at both the end of the Pre-Hospital and Hospital phase showed no differences in ionized calcium levels between any groups at either time-point. This would not be expected if citrate were the sole cause of the persistent hypotension and tachycardia in the whole blood–resuscitated group.
Our study is also the first to examine PbtO2 in PNPH- versus whole blood–resuscitated mice. Though not surprising, given the significantly higher hemoglobin and hematocrit (PNPH is a 4% Hb solution) and thus oxygen-carrying capacity, the blood-resuscitated mice had improved Pre-Hospital PbtO2 versus both PNPH- and LR-resuscitated mice, despite having lower oxygen saturations. However, there was no difference in PbtO2 between blood and PNPH in the Hospital phase. Also notable is a delayed and gradual rise in PbtO2 in the PNPH-resuscitated mice. This may reflect the initial CO-Hb state of PNPH and gradual dissociation of the CO group to form oxyhemoglobin. The timeframe in which this dissociation is expected to occur, as shown in a stroke model with a pegylated CO-Hb, coincides with the same time period of the gradual PbtO2 rise seen in the PNPH mice in our study. 27 Thus, we cannot exclude the possibility that this more rapid improvement in PbtO2 represents an advantage for whole blood versus PNPH. Only studies assessing acute and long-term behavior and neuropathology could answer that question.
We also showed that resuscitation with PNPH across a wide dosage range is safe in the acute phase and highly effective. PNPH at all doses tested, including doses as low as 2 ml/kg, produced robust improvement in MAP immediately after resuscitation. Even the smallest PNPH doses tested (2 mL/kg and 5 mL/kg) led to a very clinically relevant 1.5 to 3.2-fold reduction in total fluid requirements. That represents a potentially exciting finding for TBI resuscitation. Our prior work with PNPH investigated doses of 20 mL/kg 17,19 ; thus, demonstration of efficacy at doses 10 × lower is an important finding. Although not shown in the current study (possibly due to the small number of mice per group), we previously reported a direct correlation between fluid resuscitation volume and %-brain water. 17 Large resuscitation volumes exacerbate brain edema and may blunt restoration of oxygen delivery. Volume-limiting approaches have been shown to be neuroprotective with reduced brain edema. 6,7,28 Also, ICP was highest in the LR- treated group (also consistent with our prior work); however, in this study we showed a strong correlation of lower %-brain water in the injured hemisphere to higher PNPH doses. ICP may be greatly underestimated in our model given the presence of a craniotomy. 29,30 Although sealed in place after CCI, this may still provide some decompression. The acute nature of the study also represents the earliest point one would anticipate raised ICP from brain edema. Peak ICP in TBI patients is typically days rather than hours after injury. 31 Patients with early intracranial hypertension are at risk for poor outcome. 32,33 Taken together, our findings are of a magnitude that would be clinically relevant, particularly in the setting of TBI resuscitation.
The increased levels of Met- and CO-Hb are expected at the extreme high ends of PNPH dosing. We previously reported increased Met-Hb levels after PNPH administration 17 and concluded that it likely reflects direct measurement of the PNPH molecule itself rather than a consequence of therapy on residual native hemoglobin. There is no evidence in this or prior studies of impaired oxygen delivery as a consequence of this low level of Met-Hb. At PNPH doses of 2, 5, 10, or 20 mL/kg, the level of Met-Hb observed would in general not be a concern. However, we recognize that for therapies associated with an increase in Met-Hb, they are generally weaned when levels of ∼2% are reached. Also, the finding of CO-Hb is likely a direct measurement of PNPH, which has not yet dissociated with CO from its stored state. Significantly elevated CO-Hb levels were seen only in the 50 and 100 mL/kg group. These groups received prolonged infusion of PNPH over 60 min to prevent acute volume overload. Thus, in these groups, the blood gas drawn at the end of the Pre-hospital phase represented a time-point only 30 min after completion of the PNPH infusion, versus 90 min in the 2–20 mL/kg groups. By the end of the Hospital phase, only 15 min after the previous blood gas measurement, CO-Hb levels had markedly decreased in both groups. Again, this is consistent with the reported dissociation time of CO from PNPH 27 and supports our earlier finding on the delayed rise in PbtO2 in the PNPH-treated mice. Finally, the 50 and 100 mL/kg doses were chosen solely to study the extremes of PNPH dosing and would not likely be considered for clinical use.
Limitations
This study has a number of limitations, some of which were already mentioned. An additional limitation is in the difficulty modeling such a complex pathology of clinical TBI and hemorrhage, which is often associated with polytrauma. 1,34 These patients warrant close monitoring and prolonged ICU care (e.g., careful fluid titration, medication administration, mechanical ventilation, etc.), and extrapolation of acute physiologic data to longer-term outcomes may not be fully valid. Longer-term outcomes would be desirable and informative; however, it is unfeasible to provide intensive care unit care to these mice outside of the acute period. Thus, we investigated immediate and 24-h outcomes since they can at least reasonably approximate clinical care. We have in prior studies reported 7-day outcomes in a similar mode of TBI+HS and observed reduced neuronal death in the hippocampus with PNPH resuscitation versus either LR or hetastarch. 19 However, those studies were carried out with a more mild level of HS than in the current investigations.
In developing this TBI+HS model, a MAP of 70 mm Hg was selected to provide a cerebral perfusion pressure (CPP) of ∼60 mm Hg (MAP - ICP = CPP), which is the recommended CPP goal for adult TBI patients. 35 However, the optimum MAP for TBI patients has not been established, and the selection of a lower MAP target would likely have reduced resuscitation volumes and might produce different results from those observed. While the optimum MAP has not been established for TBI patients, in animal models and in patients, important deleterious effects of hypotension in TBI patients are established. 36 –38 Rapid resuscitation is thus paramount. It is likely that there is no true optimum MAP that can be applied to all patients. MAP is a reflection of vascular tone and blood volume and does not directly inform on blood flow. 39 Resuscitation goals should be targeted towards re-establishment of tissue perfusion; however, in the absence of available information such as tissue oxygenation and blood flow, surrogate clinical markers like MAP must be used. Also, other available monitoring techniques such as PbtO2 monitors, microdialysis, laser and transcranial Doppler, and near-infrared spectroscopy may underestimate the degree of blood flow derangement in the vulnerable pericontusional regions, which are known to exhibit compromised blood flow. 39 –43 Also, traditional blood pressure goals may have a reduced relevance in the setting of novel resuscitation solutions, such as cell-free hemoglobins or other novel agents. 39,44 –46
To target the specific milestones set forth in our U44 drug development plan, we focused on physiological and dose response effects on MAP, ICP, CPP, and brain edema rather than on mechanistic end-points, such as NO biology or markers of oxidative stress. Future definitive long-term outcomes studies on PNPH in TBI resuscitation should assess those endpoints in addition to traditional behavioral and histological TBI outcomes.
In conclusion, we demonstrated robust resuscitative properties of a broad range of PNPH doses versus both LR and surprisingly autologous whole blood in our TBI+HS model. Specifically, resuscitation with PNPH, versus LR or blood, improved MAP, HR, and ICP, reduced acidosis and hyperkalemia, although whole blood showed the greatest improvement in PbtO2 among the three therapies. Taken together, these studies add substantial evidence to the potential efficacy of PNPH in TBI resuscitation and support ongoing development of this small volume resuscitation agent.
Footnotes
Acknowledgments
This study was supported in part by U44 NS070324 (CH, LM, PMK) from the National Institute of Neurological Disorders and Stroke. Drs. Kochanek and Dixon were also supported by grants WH81XWH-10-1-0623 and WH81XWH-14-2-0018 from the U.S. Department of Defense. Dr. Brockman was supported by T32 HD-040686 from NICHD. Catherine Byrd was supported by the Career Education and Enhancement for Health Care Research Diversity (CEED) Program at the University of Pittsburgh School of Medicine.
Author Disclosure Statement
Dr. Hsia is a shareholder in SynZyme Technologies, LLC. For the remaining authors, no competing financial interests exist.