
Abstract
Closed-head concussive injury is one of the most common causes of traumatic brain injury (TBI). Isolated concussions frequently produce acute neurological impairments, and individuals typically recover spontaneously within a short time frame. In contrast, brain injuries resulting from multiple concussions can result in cumulative damage and elevated risk of developing chronic brain pathologies. Increased attention has focused on identification of diagnostic markers that can prognostically serve as indices of brain health after injury, revealing the temporal profile of vulnerability to a second insult. Such markers may demarcate adequate recovery periods before concussed patients can return to required activities. We developed a noninvasive closed-head impact model that captures the hallmark symptoms of concussion in the absence of gross tissue damage. Animals were subjected to single or repeated concussive impact and examined using a battery of neurological, vestibular, sensorimotor, and molecular metrics. A single concussion induced transient, but marked, acute neurological impairment, gait alterations, neuronal death, and increased glial fibrillary acidic protein (GFAP) expression in brain tissue. As expected, repeated concussions exacerbated sensorimotor dysfunction, prolonged gait abnormalities, induced neuroinflammation, and upregulated GFAP and tau. These animals also exhibited chronic functional neurological impairments with sustained astrogliosis and white matter thinning. Acute changes in molecular signatures correlated with behavioral impairments, whereas increased times to regaining consciousness and balance impairments were associated with higher GFAP and neuroinflammation. Overall, behavioral consequences of either single or repeated concussive impact injuries appeared to resolve more quickly than the underlying molecular, metabolic, and neuropathological abnormalities. This observation, which is supported by similar studies in other mTBI models, underscores the critical need to develop more objective prognostic measures for guiding return-to-play decisions.
Introduction
T
mTBI is the signature injury of military conflicts in Iraq and Afghanistan; >230,000 deployed United States service members have sustained an mTBI over the last 14 years. 8 In addition to combat-related injuries, >84% of service member TBIs are diagnosed in a nondeployed setting, often resulting from vehicle crashes, falls, sports and recreation, and military training. Similar statistics have been cited for sport-related concussions; estimates of affected individuals range from 300,000 to 3,800,000 cases per year in the United States, with the greatest incidence reported from injuries sustained during football. 6,9,10 As a result, considerable attention has focused on amateur and collegiate athletes playing popular contact and inadvertent contact sports including football, rugby, soccer, wrestling, ice hockey, baseball, and softball. 10 –14 To promote concussion awareness, legislative efforts, combined with campaigns by the Centers for Disease Control (CDC), the National Football League (NFL), and the National Collegiate Athletic Association (NCAA) have launched initiatives directed at the prevention, diagnosis, and treatment of sports-related brain injury. 15 The vast majority of TBIs (70–90%) are categorized as mTBI, and return to play or duty with a progressive return to activities usually occurs within 7–10 days. 1,16 Nonetheless, TBI is a highly heterogeneous injury, and additional factors such as age, sex, and history of prior concussions further complicate return-to-duty and return-to-play guidelines. 17 Importantly, in a subset of head impacts that occur during contact or collision sports, symptoms and visible signs of neurological dysfunction may not outwardly manifest and are often not clinically recognized or identified as a concussion. Such individuals, especially those with repeat injuries, may develop progressive multiple symptoms that may appear months or even years after initial trauma and persist for extended periods of time. 18 –21
Studies have suggested that repetitive mTBI (rmTBI), which occur within a period of increased cerebral vulnerability, may result in disproportionally severe acute outcomes. 16,17,22,23 Athletes who sustain multiple repeated concussions often perform more poorly on neurocognitive testing and show slower processing speed. 18 The duration between brain injuries further confounds studies of multiple concussions. Both clinical and experimental studies have shown that repetitive injuries with shorter durations between insults produce greater cognitive, histological, and behavioral impairments than do injuries separated by a longer period. 24 –26 The cumulative effects of mTBI may result in chronic neuropsychological disorders including physical, mental, emotional, and cognitive impairments, as well as contributing to the development of late onset neurodegenerative chronic traumatic encephalopathy (CTE). 27 –30 Individuals subjected to repetitive injury may, therefore, represent a high-risk population with lowered concussion threshold. Nevertheless, the degree to which a single concussive event increases susceptibility to damage from subsequent concussions remains unclear. Given the high prevalence of mTBI and its link with comorbidities, it has become increasingly important to elucidate the underlying secondary injury mechanisms that sensitize the brain to subsequent mild head traumas, contributing to a period of increased vulnerability, and resulting in more severe cumulative chronic effects.
Animal models of clinically-relevant mTBI often serve as functional and physiological surrogates used to identify underlying pathology and putative prognostic indicators. Such indicators can: 1) reflect the temporal resolution of concussion symptoms and 2) help identify patients with increased risk of developing residual clinical sequelae. To better understand the changes in the brain following a concussion/mTBI, there has been a surge of interest in developing animal models of concussion. Given the complexity and heterogeneous nature of brain injury, various research groups have endeavored to achieve this goal by adjusting the injury parameters of existing TBI models, including a modified controlled cortical impact (CCI) model, 31 –37 the Marmarou weight drop models, 38 –41 the lateral fluid percussion injury (LFPI), 42 and developing primary blast injury TBI models. 43,44 Although researchers have successfully demonstrated the ability to achieve mild to moderate injury severity levels using these TBI models, there are some caveats with regards to mimicking true closed-head concussive impact injury. More specifically, primary blast injury is not concussive, and surgical procedures used in the direct impact models often include scalp incision and/or cranioectomy. These procedures can trigger proinflammatory responses and related behavioral changes that may complicate data interpretation. 45 In addition to the number of concussive impacts, the duration of inter-injury intervals can be modified, ranging from hours 38,46 to days between injuries, 47,48 resulting in varying degrees of neurofunctional impairment. 49 –51
We recently reported on the development of a rodent projectile concussive impact (PCI) injury model that produces a true closed-head concussive event resulting in acute cellular, molecular, and sensorimotor changes without gross contusional injury. 52,53 An important feature of this model is that the anesthetized animal is placed in a supine position and lightly restrained such that the direct impact from a high-speed projectile produces linear and rotational motions on the rat's head. Custom-designed protective helmets, lined with pressure-sensor film on the inner and outer surfaces, are used to prevent skull fractures and subdural hematomas, while allowing for the translation of sufficient impact force to create mTBI. The present study was designed to comprehensively characterize the PCI model from the acute through the chronic phases of injury by comparing the functional alterations following a single and repeated closed-head concussive impact injury and identifying potential TBI-related neurological and molecular signatures that are poised to have prognostic or theranostic value.
Methods
Subjects and experimental groups
Male Sprague–Dawley rats (270–330 g, Charles River Labs, Raleigh, VA) were used in all experiments. All procedures were approved by the Walter Reed Army Institute of Research Institutional Animal Care and Use Committee. All research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals, and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council (NRC) Publication, 1996 edition. Animals were housed individually under a 12 h light/dark cycle (lights on at 0600) in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and allowed 7 days for acclimation to the housing facility before procedures were performed.
PCI procedure
Animals were randomly assigned to four groups: Sham (SHAM), single PCI (mTBI), repeated PCI (rmTBI; four injuries spaced 1 h apart), and repeated sham (rSHAM). The repetitive injury paradigm was chosen in order to model the multidimensional symptoms of repetitive concussive and subconcussive blows to the head, and serve as a comparison for a single mTBI event. All rats were initially anesthetized with 3.5% isoflurane for 4 min in an induction box, and then placed on an elevated platform in the right, lateral recumbent position. The platform was housed inside a Plexiglas chamber filled with 2% isofluorane. In order to prevent skull fractures, a custom-designed carbon-fiber helmet (U.S Army Research Laboratory, Aberdeen Proving Ground, MD) was placed on the animal's head and shielded the direct impact as previously described. 52 Pressure-sensor (Sensor Products Inc., Madison, NJ) film was adhered to the inner and outer surfaces of the helmet surfaces to record the distribution and magnitude of impact force from the projectile. The anesthetized rat was placed on the elevated platform with its head positioned above an oval opening in the elevated platform such that the right hemisphere of the helmet-protected head was exposed to the projectile angled 45 degrees from the sagittal plane. A computer-controlled program was used to trigger the pressurized, rapid-release release of a steel ball (3.52 g) aimed at the right, dorsal–frontal quadrant of the rat brain. After impact, the helmet was removed and the animal was returned to its home cage. Sham animals received identical experimental procedures as PCI-injured rats (anesthesia level and time, helmet and film alignment, placement on computer-controlled pressure release system) except for the projectile impact. Following PCI or sham procedure, animals were immediately transferred to the home cage for quantification of righting reflex. Measurements of Pressure, Contact Area, and Force were calculated from sensor films using Topaq Pressure Analysis System (Sensor Products Inc., Madison, NJ). All subsequent assessment metrics were conducted following PCI or sham procedure. An experimental design schematic of TBI and assessment metrics is shown in Figure 1.

Experimental design. Schematic for cohorts in which rats were subjected to single and repeated mild traumatic brain injury (mTBI) (day 0) and assessed using a variety of behavioral and physiological measurements. B, baseline; D, day post-injury; PCI, projectile concussive impact; CW, CatWalk; RR, rotarod; BBB, Basso, Beattie, Bresnahan locomotor testing; NSSR, Neurobehavioral Severity Scale Revised; INFL, inflammatory measurement; IHC, immunohistochemistry; EPM, Elevated Plus Maze; MWM, Morris Water Maze.
Behavioral assessments
Righting reflex (RR)
Immediately following the sham procedure or PCI injury, animals were placed on their backs (supine position) in their home cage. RR was defined as the length of time needed for an animal to regain its normal posture (prone) after each individual impact or sham procedure. RR time was recorded using a stopwatch, and calculated from the time of impact to the time posture returned to normal.
Neurobehavioral Severity Scale Revised (NSS-R)
Neurological deficits were scored using the NSS-R at 1 h, 4 h, 24 h, 72 h, and 3 months post-injury in separate cohorts to reduce the effects of repeated testing (n = 8–10 rats/group/time point). The revised NSS-R consists of 10 tests designed to assay motor, sensory, and reflex skills and was originally designed to model a human clinical neurological examination. 54 The NSS-R uses a three-point Likert scale, in which a normal, healthy response is assigned a “0,” a partial or compromised response is assigned a “1,” and the absence of a response is assigned a “2.” This three point scale reliably allows for greater discrimination based on sensory-motor responses. A higher score indicates greater impairment, with 20 points representing the maximum (worst) outcome. All assessments were conducted by experimenters blind to groups.
Open-field locomotor assessment (Basso, Beattie, Bresnahan [BBB] score)
Functional behavioral following single mTB, rmTBI, and respective sham was assessed using the BBB Locomotor Rating Scale at 1 h post-injury in an open field by investigators blind to injury groups (n = 8–10 rats/group). The locomotion test is a 21 point locomotion scoring system, which categorically documents limb movements and walking characteristics in an open-field environment (a score of 21 indicates normal walking behavior and a score of 0 designates complete hindlimb paralysis). The scale assesses metrics of paw placement, coordination, toe clearance, and trunk instability as a means to document “global” attributes of locomotor ability, and is a valid and predictive measure of locomotor recovery following central nervous system (CNS) injury. 55
Vestibular/balance assessment
The Rotemex-5 rotarod apparatus (Columbus Instruments, Columbus, OH) was used to measure motor coordination and balance. Two different assessment paradigms were used for their ability to detect injury-induced vestibular deficits. Separate cohorts were used for each paradigm (n = 8–10 rats/group).
Acute–subacute rotarod assessment
Prior to PCI, rats were trained to criteria on the rotarod task: ability to maintain their balance on a rotating rod for a minimum of 60 sec at constant speed of 5 rpm for three trials, 2 min inter-trial interval, for 2 consecutive days immediately prior to injury. At specified time points following PCI (15 min, 1 h, 24 h, 7 days, and 14 days, n = 8 rats/group) or at 24 h only (n = 8–10), animals were tested using an accelerated rotating rod testing paradigm (0.1 rpm/sec increment) for a maximum of 150 sec per trial, two trials, 2 min inter-trial intervals. The primary outcome measure for the rotarod test was mean latency (two trials, averaged) to fall off the rotarod.
Chronic rotarod assessment
To determine the effects of mTBI on chronic vestibular dysfunction, rodents were challenged by a more vigorous “graded exercise testing” on rotarod assessment. No pre- or post-injury training was conducted. At 3 months post-injury, rats were tested using an accelerated rod rotating testing paradigm (0.1 rpm/sec), first set: five trials, 2 min inter-trial intervals. Rats were then given a 2 h rest period followed by a second set (five trials, 2 min inter-trial intervals). Rats were then given a 1 week rest period and then challenged by a third set: five trials, 2 min inter-trial intervals. Raw latency to fall off the rotating rod values for individual trials were averaged within groups and plotted across trials. Average latency to fall for each five trial set was compared between injured and respective sham groups.
Gait analysis
Gait analysis was conducted 3 days prior to mTBI (baseline measures) and at 2 h, 24 h, 72 h, 9 days, 28 days, and 3 months post-injury using the CatWalk automated gait analysis system (Noldus Information Technology Inc., Leesburg, VA) as previously described.
53,56
Individual footprints were identified, and data pertaining to static and dynamic gait parameters were generated for each trial (Table S1) (see online supplementary material at
Quantitative electroencephalogram (qEEG) recording and spectral analysis
Separate cohorts of animals were used for qEEG recordings (n = 12/group). EEG implantations and recordings were conducted as described. 57 Immediately after the last PCI or respective sham procedure, each anesthetized rat was prepared with four skull EEG electrodes that were symmetrically implanted over the bilateral frontal and parietal regions of the brain (anteroposterior [AP]: +1 and −4 mm; mediolateral [ML]: ±3.5 mm to bregma) and secured by dental acrylate. A reference electrode was implanted posterior to lambda over the transverse sinus. The free end of each electrode wire was soldered to a Dale multi-pin connector (March Electronics, West Hempstead, NY). Following surgery, animals were immediately placed in custom-designed Plexiglas EEG recording chambers equipped with multi-channel gold contact swivel communicators (Dragonfly Inc., Ridgeley, WV). EEG signals were collected from the ipsilateral (right) and contralateral (left) hemispheres independently using the Stellate amplifier and Harmonie software (Natus Medical Inc. San Carlos, CA). EEG and synchronized video recordings were initiated immediately after the surgery, and continued for 14 days in unanesthetized freely moving animals.
qEEG power spectral analysis was performed off-line on each animal based on a 60 sec wake (verified by video recording) EEG epoch selected from each animal at 12 h, 24 h, 48 h, 72 h, and on the 7th and 14th days post-injury. The EEG global frequency band (0.5–30 Hz) of each epoch was divided into four frequency bands: delta band (0.5–4 Hz), theta band (4–8 Hz), alpha band (8–12 Hz), and beta band (12.5–30 Hz). The EEG relative power was defined as the mean power of each frequency band calculated as the percentage of the total EEG power of the global band, and was analyzed using Harmonie software (Natus Medical Inc., San Carlos, CA). The ipsilateral and contralateral EEG relative power of each frequency band was performed separately for each individual animal.
Morris Water Maze (MWM)
At 6 months post-injury, spatial learning was assessed in the MWM task equipped with a video-tracking system (EthoVision XT; Noldus Information Technology, Wageningen, The Netherlands). 58 The animals were placed in a pool (75 cm deep and 175 cm in diameter) at one of four pseudo-randomly selected starting points (north, south, east, or west), and given 60 sec to locate the submerged Plexiglas platform. Animals that failed to find the platform within the allotted time were gently guided to the platform and allowed to rest there for 10 sec. All rats were given two 60 sec trials (5 min inter-trial interval). Primary outcome metrics included latency to find the platform, swimming distance, and swim velocity (spatial learning).
Molecular analyses
Descriptions of sample collection, processing, and detection methods for subsequent molecular analyses are listed subsequently. The appropriate positive and negative controls for antigen detection in Western blots, arrays, quantitative polymerase chain reaction (qPCR), and immunohistochemistry (IHC) were conducted.
Biofluid sample collection
For protein biomarker measurements, cerebrospinal fluid (CSF) and serum were collected at terminal end-points (1 h, 24 h, 72 h, 7 days, and 3 months post-injury). CSF was collected by puncture of the cisterna magna into heparin-coated tubes, and centrifuged to remove cells and/or debris. Whole blood was collected via cardiac puncture, and aliquots of blood were allowed to clot for 30 min at room temperature in 1.3/Z clotting tubes (Sarstedt, Newton, NC). Each sample was centrifuged at 1.2–1.3kg, for 10 min at 4°C to remove cells and/or debris. The resulting clarified CSF and serum were collected into Eppendorf tubes supplemented with Halt Protease/Phosphates Inhibitor mix (1:100 dilution) and stored at −80°C until use. For corticosterone (CORT) levels, samples were collected during the light cycle when levels are at their lowest concentration, and while animals were at rest. Effort was made to minimize stress to the animals during the collection process, and to avoid startle-related CORT spiking. Collection timing was designed to avoid masking the effects of mTBI by potential rise in the circadian rhythm of CORT levels during the dark cycle.
Calibration standard curve and quality controls (QCs) preparation
CORT, dimethyl sulfoxide (DMSO), and formic acid were purchased from Sigma-Aldrich (St. Louis, MO). Norit-A neutral decolorizing carbon and high performance liquid chromatography (HPLC)-grade acetonitrile were purchased from Fisher Scientific (Hanover Park, IL). Deionized water was obtained from a Solution 2000™ water purification system (Montreal, Canada). Serum standard curve and QCs were prepared from blank rat serum, which had been stripped of endogenous CORT using Norit-A neutral decolorizing carbon. Charcoal was added to blank rat serum at a concentration of 1 g/25 mL. The suspension was stirred at room temperature for 7 h and centrifuged overnight at 16,000g in a bench-top PrismR centrifuge (Labnet International, Inc., Edison, NJ) at 4°C. The supernatant was filtered using 0.45 μm sterile Nalgene filters (Nalgene Nunc International, Penfield, NY) and stored at −80°C until use. A standard CORT stock solution of 1 mg/mL was prepared in DMSO and used to make a 10 μg/mL stock dilution in acetonitrile. The calibration standard curve and QCs were prepared by spiking the stripped rat serum with the 10 μg/mL corticosterone stock solution. The calibration standard curve ranged from 1.0 to 1000 ng/mL. QC samples were prepared to cover the low (1.0 ng/mL, 2.5 ng/mL, and 5.0 ng/mL), medium (300 ng/mL), and high (750 ng/mL) concentration ranges of the standard curve. Standard curve and QC samples were then extracted with two volumes of acetonitrile with an internal standard (mefloquine in acetonitrile) for each volume of sample. A zero sample was prepared in the same manner. Each sample was vortexed and centrifuged at 16,000g for 10 min at 4°C. The supernatant was transferred to a 96 well plate for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. The CORT standard curve was constructed from peak area values obtained from LC-MS/MS analysis. Charcoal serum preparation was conducted as described.
LC-MS/MS analysis of CORT
A Waters ACQUITY separation module UPLC system was coupled with an AB Sciex QTrap 4000 triple quadruple mass spectrometer equipped with a Turbo-V source (Waters Corporation, Milford, MA). The analysis of CORT was performed using an Xterra® (2.1 × 50 mm, 3.5 μm) column, maintained at room temperature. The auto-sampler was maintained at 4°C to minimize evaporation, and 10 μL of sample was injected onto the column. Samples were eluted using a linear gradient from 5% to 98% (acetonitrile/0.1% formic acid in water) over the course of 2.0 min at a flow rate of 0.400 mL/min. The analysis was performed in multiple reaction monitoring (MRM) in positive electrospray ionization mode by monitoring the ion transitions from m/z 347.10 → 121.20 (corticosterone) and m/z 379.10 → 361.10 (mefloquine). Compound parameters and source parameters were optimized to obtain the highest intensity of the analyte. The collision energy (CE), collision cell exit potential (CXP), declustering potential (DP), and entrance potential (EP) for CORT were 39.00 V, 10.00 V, 116.00 V, and 10.00 V, respectively. The CE, CXP, DP, and EP for mefloquine were 35.00 V, 10.00 V, 71.00 V, and 10.00 V, respectively. The source parameters were optimized as follows: curtain gas (CUR), 20.00; collision activated dissociation (CAD), high; ionspray voltage (IS), 5500.00 V; nebulizer gas (GS1), 30.00; heater gas (GS2), 30.00; and source heater (TEM), 300.00. The instrument was controlled and data were collected using Analyst® software.
Inflammatory cytokine and chemokine detection and quantification
Serum and CSF inflammatory cytokine and chemokine levels were evaluated at specified time points (1 h, 24 h, 72 h, and 7 days post-injury) using a Proteome Profiler Rat Cytokine Array Panel A (R&D Systems, Minneapolis, MN) according to the manufacturer's specifications. Briefly, nitrocellulose membranes prepared to detect 29 different cytokines were blocked with blocking buffer on a rocking platform shaker. Concurrently, biofluid samples (200 μL serum and 100 μL CSF) were incubated in Detection Antibody Cocktail for 1 h at room temperature. Samples were then incubated on membranes at 4°C overnight. Following washes, membranes were incubated for 1 h at room temperature in Streptavidin-horseradish peroxidase (HRP) solution and subsequently washed. Levels of cytokines and chemokines were detected by chemiluminescence with automated background subtraction using an ImageQuant™ LAS 4000 Imager (GE Healthcare, Pittsburgh, PA). Pixel intensities were averaged between duplicate spots for each marker, and normalized to internal controls on each blot. Data derived from individual animals were expressed as fold change over respective sham.
Enzyme-linked immunosorbent assays (ELISA)
Glial fibrillary acidic protein (GFAP) assays were developed in-house. CSF or serum samples (n = 8–10/group/time point) were diluted 1/3 in 1 × phosphate-buffered saline (PBS), pH 7.8 (Biorad, Hercules, CA) then incubated in plates manually coated with 25 ug/mL polyclonal anti-GFAP capture antibody (Banyan Biomarkers, Carlsbad, CA) in 1 × PBS, pH 7.8. Plates were then incubated with mixed monoclonal anti-GFAP detection antibodies (BD Biosciences, San Jose, CA) and anti-mouse sulfo-tag antibody (Meso Scale Discovery [MSD], Rockville, MD) at 0.5 ug/mL each in 0.5% Blocker B (MSD) prepared in 1 × PBS, pH 7.8. Tau assays were conducted according to the manufacturers' specifications (Meso Scale Discovery, Rockville, MD). Protein content was derived from standard curves using recombinant human GFAP protein (Banyan Biomarkers, Alachua, FL; standard range: 0.039–10 ng/mL; CSF LoD ∼0.04 ng/mL; CSF LoQ ∼0.08 ng/mL; Serum LoD ∼0.08 ng/mL; CSF LoQ ∼0.16 ng/mL) or tau protein (Meso Scale Discovery, Rockville, MD; standard range: 0.24–1000 ng/mL; CSF LoD/LoQ ∼0.24 ng/mL; serum LoD ∼0.24 ng/mL; serum LoQ ∼2.5 ng/mL). Values derived from PBS (for CSF) or neat serum (BioreclamationIVT, Baltimore, MD) were used as blanks (0 ng/mL) and subtracted from all samples and standards. Analyte quantitation was determined by electro-chemiluminescent signal with a Meso QuickPlex SQ 120 (MSD, Rockville, MD). Quantitative values (ng/mL) were converted into fold change values compared with respective SHAM or rSHAM controls.
Western blot
Tissue was homogenized on ice in 1 × radioimmunoprecipitation assay (RIPA) buffer with Pierce Halt Protease and Phosphatase Inhibitor Cocktail (ThermoFisher-Scientific, Waltham, MA). Total protein concentration was determined with the Pierce BCA Assay Kit (ThermoFisher-Scientific, Walham, MA). Blots were blocked in 5% milk and 1 × PBS, pH 7.4. Total tau was evaluated using primary anti-tau-1 antibody MAB3420, diluted 1:2000 (EMD Millipore, Billerica, MA) and phosphorylated tau was evaluated using primary Phospho-Tau (Ser202) antibody #11834 diluted 1:1000 (Cell Signaling Technology, Danvers, MA). Blots were washed three times with 1 × PBS containing 0.01% tween-20 and incubated with secondary antibody 1:20,000, anti-mouse IgG NA9310V or 1:5000, anti-rabbit IgG NA9340V (GE Healthcare, Wauwatosa, WI). Chemilluminescent signal was detected with Clarity Western ECL (BioRad, Hercules, CA). Blots were re-probed for β-actin. Protein levels for each sample were normalized to β-actin. Band intensity was analyzed using an LAS4000 with ImageQuant TL software (GE Healthcare, Pittsburgh, PA).
Quantification of microRNA levels
Total RNA was isolated from serum using the mirVana isolation kit (Ambion Inc.). Briefly, serum was placed in a denaturing solution followed by a phenol: chloroform extraction. Following centrifugation, the aqueous phase was washed with absolute ethanol, passed through a filter, and eluted using DNAse/RNAse free water (ICN Biochemicals, INC). Total RNA quantitation was done using a Nanodrop spectrophotometer 1000 (Thermo Scientific, Inc.). Samples were stored at −80°C until use. Reverse transcription was performed using the TaqMan miRNA reverse transcription kit and megaplex reverse transcription primers pools A & B (Applied Biosystems). Reactions were incubated at 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min. Pre-amplification was performed using reverse transcription product and megaplex preamp primers. Reactions were incubated at 95°C for 5 min, 55°C for 2 min, and 72°C for 2 min, and 12 cycles of at 95°C for 15 sec, 60°C for 4 min, and 99.9°C for 10 min. Samples were stored at −20 °C until use. Taqman rodent microarrays were used to measure levels of individual microRNAs. Each sample was measured as an individual singlet array and normalized to levels of MammU6 endogenous reference RNA. Samples were run using the Quantstudio 12flex PCR system (Applied Biosystems) and data were analyzed with expression suite software. Data were expressed as fold change over sham.
IHC
For IHC processing, animals were deeply anesthetized with ketamine/xylazine (70 and 6 mg/kg, i.p., respectively) and perfused via the ascending aorta with saline followed by phosphate-buffered 4% paraformaldehyde (FD Neurotechnologies, Columbia MD). Whole brains were extracted and post-fixed in the same fixative for 6 h at 4°C, and subsequently cryoprotected with 0.1M phosphate buffer (PB) (pH 7.4), containing 20% sucrose for 72 h and then rapidly frozen in isopentane pre-cooled to −70°C with dry ice. All brains were stored in a freezer at −80°C prior to sectioning. Serial sections (40 μm) were cut coronally using a cryostat through the cerebrum (∼ 3.72 mm to −6.84 mm from bregma) and the brainstem (∼-10.20 mm to −11.16 mm from bregma). Twelve sets of serial sections were collected for use with known markers of cellular responses/injury. For GFAP staining, endogenous peroxidase activity was first inactivated with hydrogen peroxidase, and, subsequently, sections were incubated free floating in 0.01M PBS (pH 7.4) containing 1% normal blocking serum, 0.3% Triton X-100 (Sigma-Aldrich, St. Louis, MO) and then incubated with rabbit anti-GFAP (1:8000, DAKO, Carpinteria, CA) for 24 h at 4°C. Tissue slices were then incubated in PBS containing Triton-X, normal blocking serum with biotinylated secondary antibody for 1 h at room temperature, and then in PBS containing avidin-biotinylated HRP complex for 1 h using the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA). Sections were then incubated for 5 min in 0.05M Tris buffer (pH 7.2) containing 0.03% 3′,3′-diaminobenzidine (Sigma-Aldrich, St. Louis, MO) and 0.0075% H2O2. Sections were then rinsed in distilled water, mounted on slides, dehydrated in ethanol, cleared in xylene, and cover-slipped in Permount® Mounting Medium (Fisher Scientific, Fair Lawn, NJ). For animals euthanized at 3 months post-injury, one set was stained with hematoxylin and eosin (H&E) for gross morphological assessment of injury. To serve as a negative control, one series of sections received all staining conditions except incubation with primary antibodies.
Fluoro-Jade B (FJB) staining
For FJB staining, brain tissue sections were mounted on gelatin-coated microscope slides and stained with FJB (Histochem, Jefferson, AR). Briefly, after dehydration in 100%, 75% ethanol and rinses in distilled water, sections were immersed in 0.06% of potassium permanganate (Sigma, St. Louis, MO) for 15 min, followed by incubation in 0.1% acetic acid containing 0.0001% FJB for 1 h at 4°C. After thorough washes in distilled water, sections were dehydrated in ethanol, cleared in xylene, and cover-slipped with Permount® Mounting Medium (Fisher Scientific, Fair Lawn, NJ)
Image analysis
FJB-postive cells were visualized using an Olympus IX83 equipped with an Olympus DP26 camera (Olympus America Inc., Center Valley, PA). Full slice images of 12 sections were acquired at 4 × magnification and stitched using Olympus CellSens Dimension v. 1.11. For FJB quantification, images of FJB-positive brain regions were taken at a 10 × objective, and individual cells were quantified using Image-Pro Plus v. 7.0. High-resolution images were taken as a z-stack using an Olympus Flouview FV1200 confocal microscope (Olympus America Inc., Center Valley, PA). For GFAP quantification, all images were acquired under identical acquisition settings using an Olympus VS120 light microscope slide scanner, and regions of interest (ROI) of the hippocampus and cortex were traced in three separate sections using Olympus CellSense Dimension (Olympus America Inc., Center Valley, PA). GFAP reactivity was quantified by threshold analysis and expressed as a percentage of the ROI area. The corpus callosum isthmus thickness was measured at the hemispheric junction and averaged across four adjacent brain H&E-stained tissue sections. Ipsilateral and contralateral ventricular volumes were defined by tracing the ventricular area, and summing across six serial sections. All image acquisition and quantification was conducted by an experimenter blind to cohort groups.
Statistical analysis
For group-wise comparisons, the data were first assessed for normality using the Shapiro–Wilk (SW) test for normality. Normally distributed data were analyzed using a two way or two way repeat measures analysis of variance (ANOVA), or one way ANOVA followed by Tukey post-hoc multiple comparisons when appropriate. Descriptive statistics were expressed as mean ± SEM. Non-normal data were first analyzed by the Kruskal–Wallis test with Dunn's post-hoc if significant. Data were presented as median with interquartile range. For analysis of gait alterations, statistical tests were performed with Systat 13 software (Systat Software Inc., San Jose, CA). Statistical significance was evaluated using two way repeated ANOVA followed by post- hoc pairwise multiple comparisons using the Fisher least significant difference (LSD) method when appropriate. The F values and H statistic for statistical comparisons are shown in Tables S2–S5(see online supplementary material at
Results
Acute neurological deficiencies
Animals were subjected to either single or multiple PCI-induced blunt head traumas. Pressure sensor films located on the helmet surfaces confirmed injury location and uniformity of force, pressure, and impact area in all rats, for which representative images and data are shown (Fig. 2A). Immediately following injury, all mTBI rats demonstrated a significantly prolonged period of unresponsiveness evidenced by longer RR recovery latencies (range 3–20 min) when compared with uninjured anesthesia-only controls (range 1–6 min, one way ANOVA, F (3.190) = 22.96, p < 0.0001; mTBI vs. Sham, p < 0.0001; rmTBI vs. rSHAM, p < 0.0001). In rmTBI cohorts, RR remained consistently longer following each successive impact when compared with sham controls (rSHAM). Within subjects, comparisons of RR following sequential impacts showed significant increases between the first and second impact but no cumulative effect was detected between the second and subsequent impacts on this measure (Fig. 2B, C).
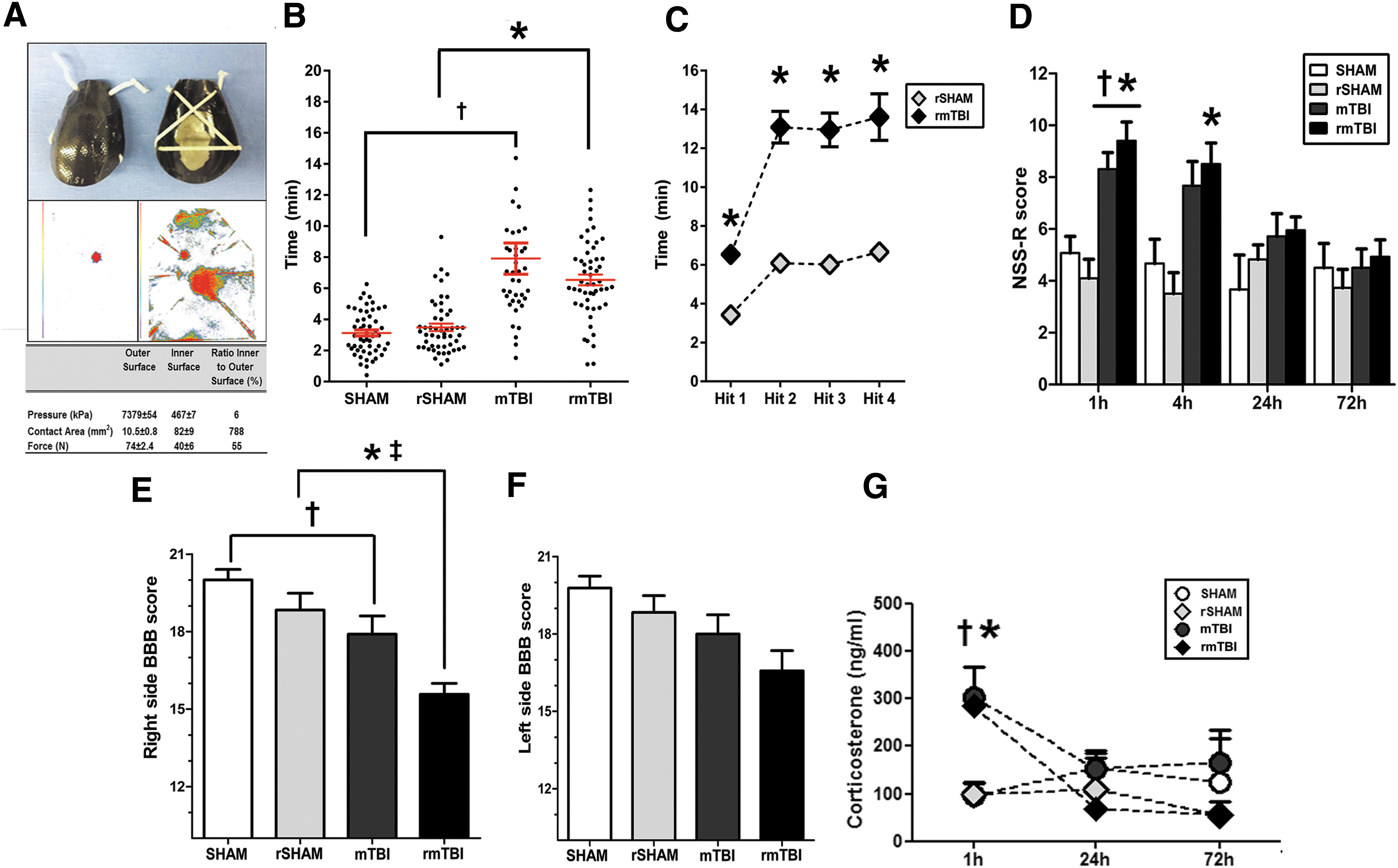
Single and repeated mild traumatic brain injury (mTBI) induces acute neurological dysfunction.
NSS-R neurobehavioral testing revealed significant dysfunction in both mTBI and rmTBI cohorts compared with uninjured counterparts (SHAM and rSHAM, respectively). Deficits were acute and transient; impairments manifested within 1 h post-injury in both single repeated concussion. NSS-R scores for rmTBI rats remained elevated at 4 h post-injury, but resolved by 24 h post-injury (Fig. 2D). In addition, the BBB test was used to assess potential alterations in CNS-controlled coordination and hindlimb function following injury. At 1 h post-injury, both mTBI and rmTBI rats showed asymmetric hindlimb locomotor deficits, evidenced by reduced BBB scores compared with respective controls (Fig. 2E, F). Although both right (ipsilateral to the injured hemisphere) and left (contralateral) hindlimb paws showed reduced BBB scores, significant differences were only detected on the right side following injury (p < 0.05, rmTBI vs rSHAM; p < 0.05 mTBI vs. SHAM). Injured rats failed to perform proper toe clearance during voluntary movement and displayed external paw rotation on the right side. The deficits appeared to be impact dependent; repeated concussions resulted in worsened performance compared with that from a single concussion alone (p < 0.05 mTBI vs rmTBI). Interestingly, the use of the forelimb asymmetry tests failed to detect any neurological changes in the forelimbs (data not shown). These early time points of neurological impairment corresponded with an increased release of the neuroendocrine stress-related hormone, CORT. CORT levels were robustly elevated following both single and repeated head trauma (1 h post-injury) and subsided by 24 h (Fig. 2G). The rise in CORT levels was not dependent on the number of impacts, because there were no significant differences noted in CORT levels between single and repeated injury groups (p > 0.05, mTBI vs rmTBI).
Single and repetitive mTBI impaired vestibular function and balance
Changes in balance and vestibular function were assessed on the rotarod initially at 15 min following the single/last impact with anesthesia, and then repeatedly over 14 days. Overall, all mTBI-injured rats exhibited an acute reduction in rotarod performance during the initial hour post-injury when compared with their respective sham control groups. A significant reduction in latency to fall on the rotarod task was detected in rats sustaining multiple impacts at both 15 min and 1 h post-injury, (p < 0.05, rmTBI vs. rSHAM), but this effect was resolved by 24 h (Fig. 3A). A similar, albeit not significant, trend was seen following a single impact. Because the observed recovery at 24 h may have been aided by the potential confound of repeated exposure to testing, we assessed a separate cohort of animals at 24 h post-injury only (Fig. 3B). In this case, animals that had been exposed to repeated mTBI displayed significant impairment in their ability to maintain balance on the rotarod at 24 h, compared with all other groups (Fig. 3B).
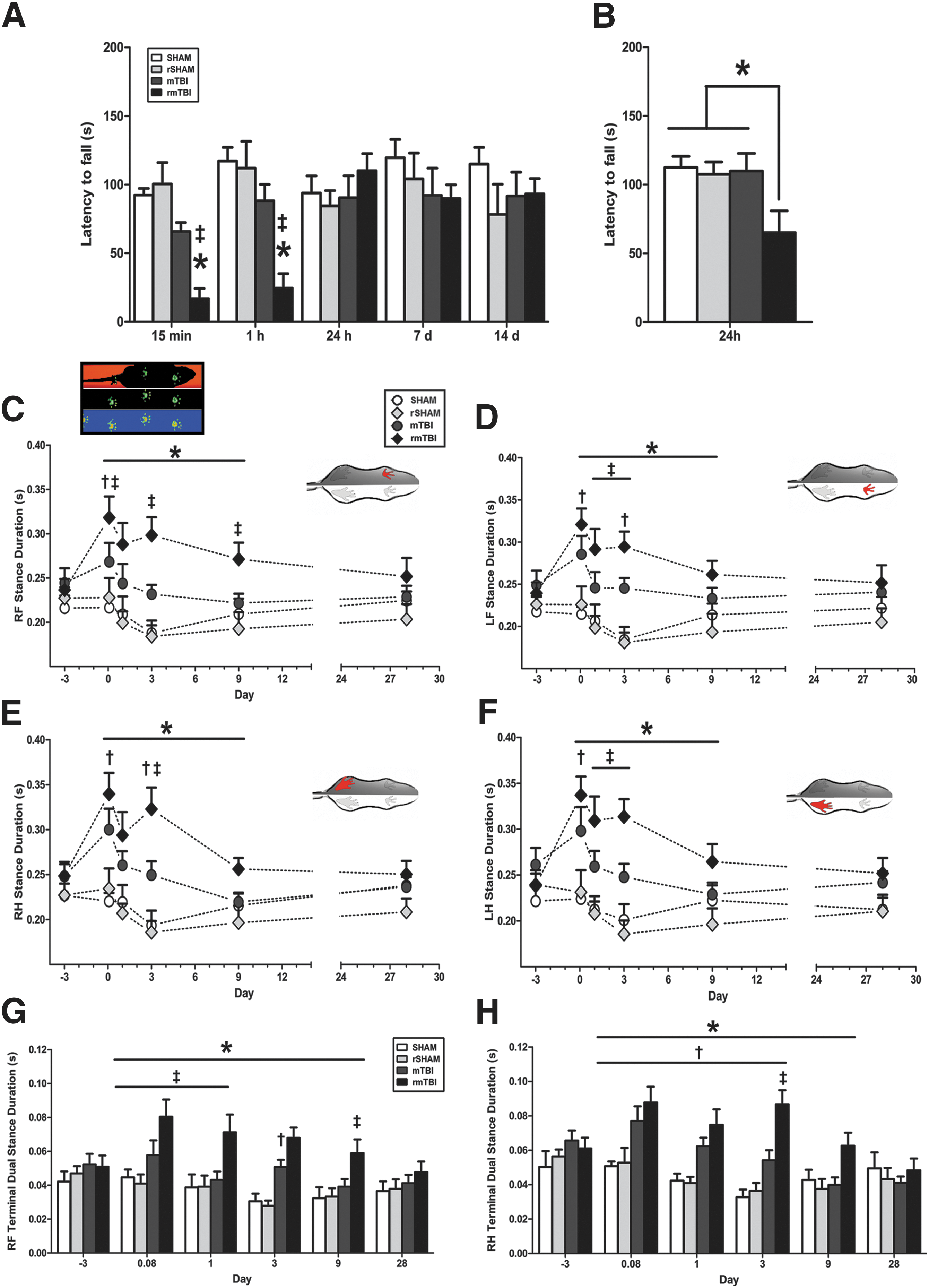
Single and repeated traumatic brain injury (TBI) alter acute sensorimotor performance
Single and repeated mTBI induced sensorimotor dysfunction
Because disruption of sensorimotor pathways can manifest as abnormal walking patterns, we measured changes in rodent gait following a single and repeated mTBI. Regardless of the injury cohort, no animals displayed gross behavioral or motor abnormalities. Importantly, a large proportion of the gait parameters on the CatWalk test remained unchanged in injured animals compared with respective controls over the course of the study. However, all injured rats did display a number of significant, yet subtle, sensorimotor abnormalities (Table S2). A single mTBI resulted in transient deficits on a limited number of dynamic gait parameters at 2 h post-injury, which resolved over the subsequent 9 days. The majority (80%) of the gait abnormalities had returned to sham levels by 1 month post-injury. In contrast, rats in the rmTBI cohort displayed significant deficits across 35 separate gait parameters, and ∼50% of these abnormalities were still present at 28 days post-injury. Notably, gait deficits were not restricted to one side of body, but instead appeared bilaterally and equally distributed on both front and hind paws (Figs. 3C–H and 4A–H). Injured rats displayed abnormal gait patterns with increased stance duration, swing phase duration, and step cycle as well as decreases in swing speed, stride length, and cadence. The stance phase was further subdivided into three segments, including 1) initial dual limb stance, 2) single limb stance, and 3) terminal dual limb stance. Notably, these three parameters were elevated over 9 days following repeated head impacts, indicating impairments in proprioception and potential increases in pain sensitivity. A comprehensive heat map shows the relative changes between mTBI and rmTBI against respective sham animals and corresponding statistical analyses (Fig. 4I). Repeated impacts also resulted in gait patterns employing significant increases in three point contact support and corresponding decreases in dual limb support. Reductions in base of support (BOS) between front and hind paws were noted in rmTBI rats through 9 days post-injury. Despite these significant gait abnormalities, step patterns and regularity index (major indices of overt coordination) showed no differences among groups.
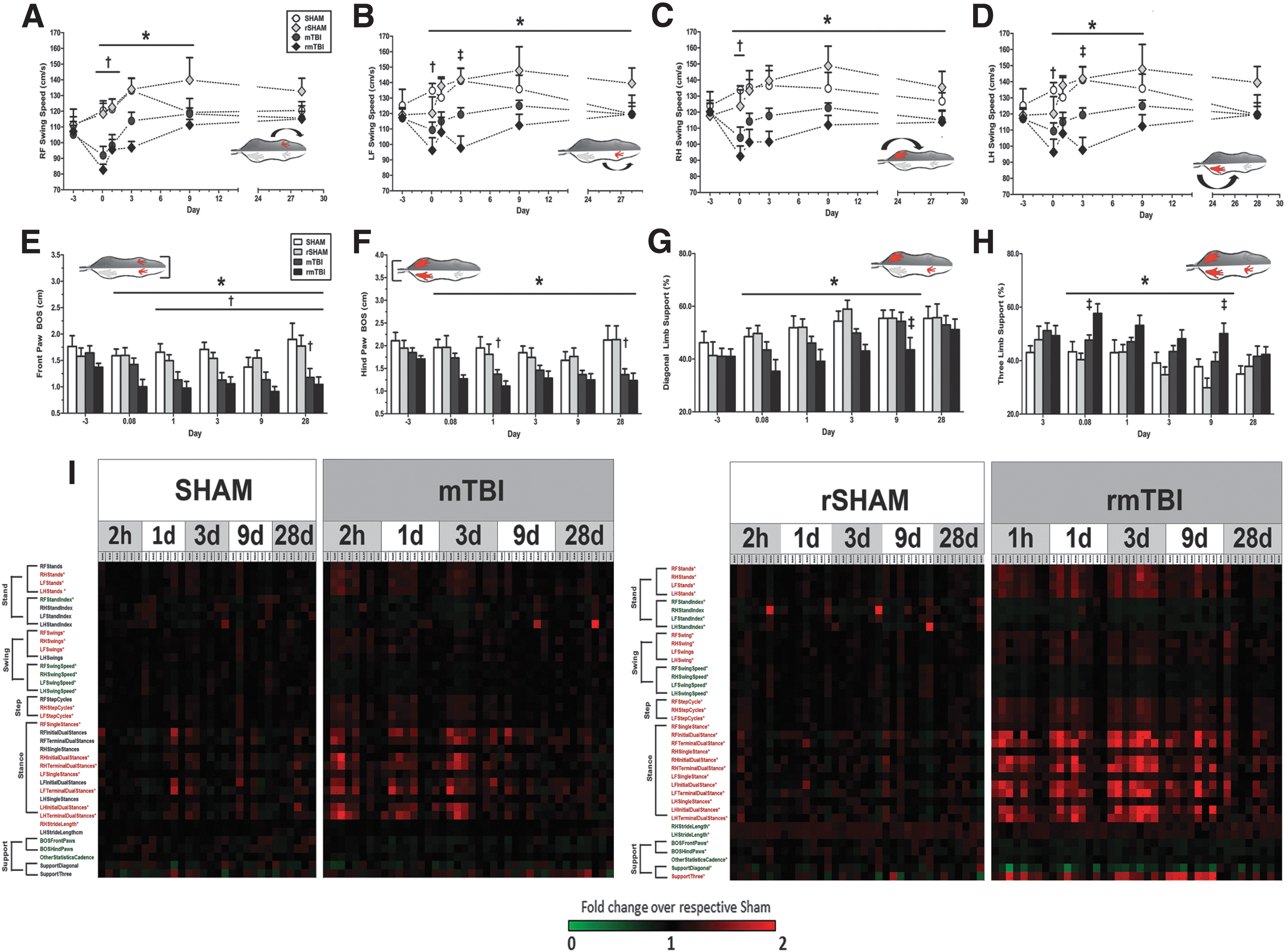
Single and repeated brain injury results in prolonged gait/balance ataxia. Gait patterns were altered following single and repeated brain injury. Single mild traumatic brain injury (mTBI) and repetitive mild TBI (rmTBI) rats showed reductions in swing speed
Single and repeated mTBI promoted systemic and local neuroinflammatory responses
To characterize both the systemic and local CNS–specific inflammatory response, we screened rodent serum and CSF for 29 distinct inflammatory cytokines and chemokines starting acutely (1 h) through 7 days post-injury (Table S6) (see online supplementary material at
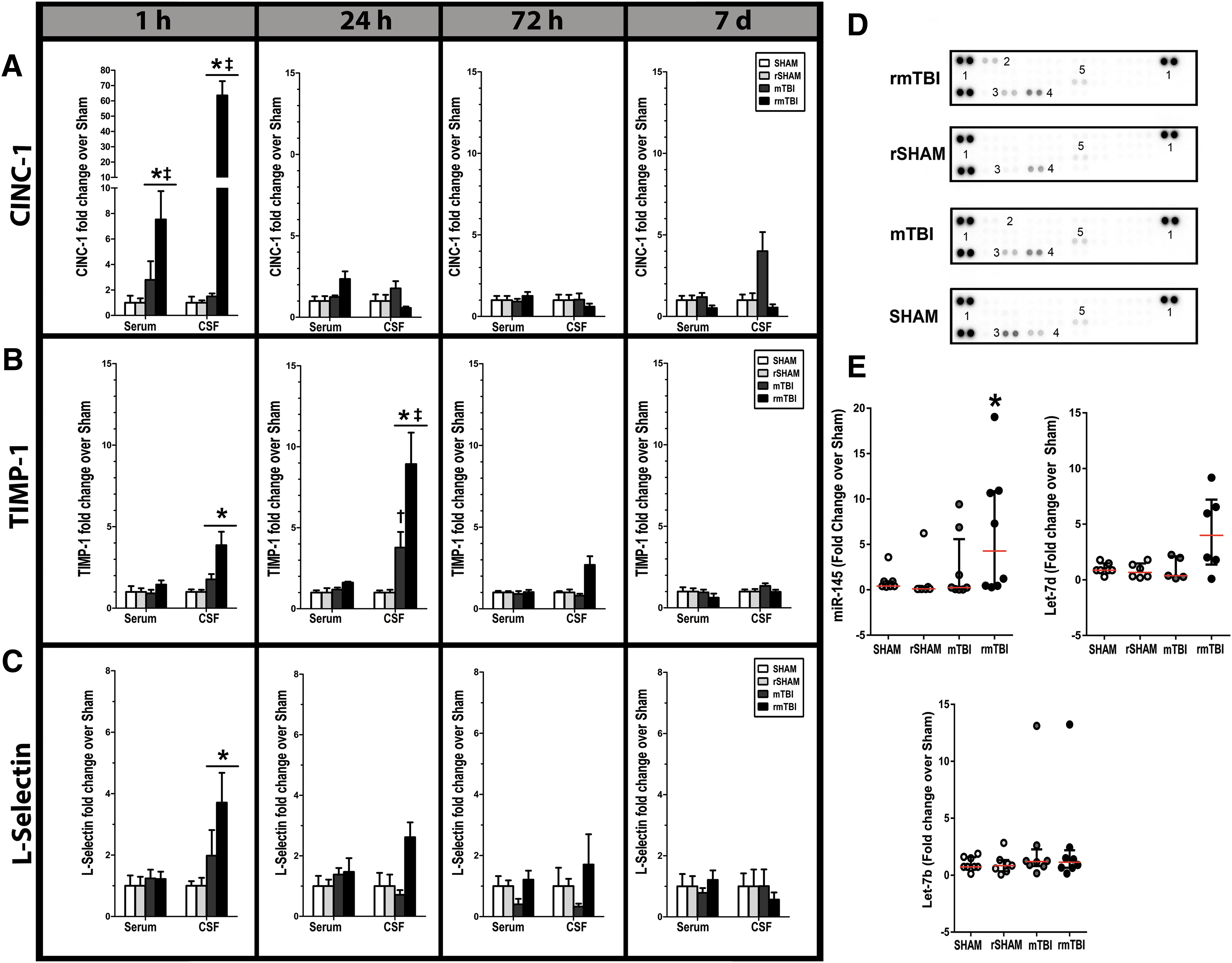
Single and repetitive mild traumatic brain injury (rmTBI) results in local and systemic upregulation of inflammatory cytokines and chemokines. Cytokine levels:
Single and repeated head impacts also significantly elevated TIMP-1 expression in the CSF (Fig. 5B). TIMP-1 levels were elevated at 1 h, peaked at 24 h post-injury in rmTBI rats (8.5-fold over rSHAM), and returned to control levels by 7 days (Fig. 5B). Interestingly, rats subjected to a single impact (mTBI) showed a similar, albeit dampened, TIMP-1 expression pattern with peak levels occurring at 24 h (4-fold over sham) post-injury. TIMP-1 levels were not significantly altered in the serum.
Levels of the leukocyte adhesion molecule, L-selectin, in CSF were significantly increased during the first 1 h following repeated impact (rmTBI), and trended toward higher levels at 24 h post-injury (Fig. 5C). No other changes were detected in serum or CSF neuroinflammatory markers including interleukin (IL)-6, IL-4, IL-10, and tumor necrosis factor (TNF)-α at any other time point. Given the profile of elevated cytokine levels following head impacts, serum from all injured and uninjured cohorts was screened for additional changes in microRNAs known to be involved in inflammation. rmTBI significantly increased systemic miR145 levels compared with the rSHAM group (6-fold, Kruskal–Wallis test, p < 0.05, Fig. 5E) at 24 h post-injury. Animals subjected to a single mTBI showed similar miR145 increases; however, these changes were not significant. Likewise, similar trends were seen for Let-7b and Let-7d microRNA levels for injured animals.
Single and repeated mTBI induced transient changes in qEEG spectra
A separate cohort of animals was assessed for injury-induced changes in EEG brain patterns. Continuous qEEG recordings were collected for all animals, and power spectral analysis was performed at 12 h, 24 h, 48 h, 72 h, and 14 days post-injury or sham procedure. Injured animals showed acute and transient alterations in qEEG power spectra and bilateral EEG slowing at 12 h and 24 h post-injury; EEG spectra returned to normal levels by 72 h (Fig. 6A, B). mTBI and rmTBI animals showed similar qEEG changes; significant increases in delta activity (0.05–4 Hz) were compensated for by a decrease in power in all other EEG bands. The most significant compensation was measured in the theta band (5–8 Hz).
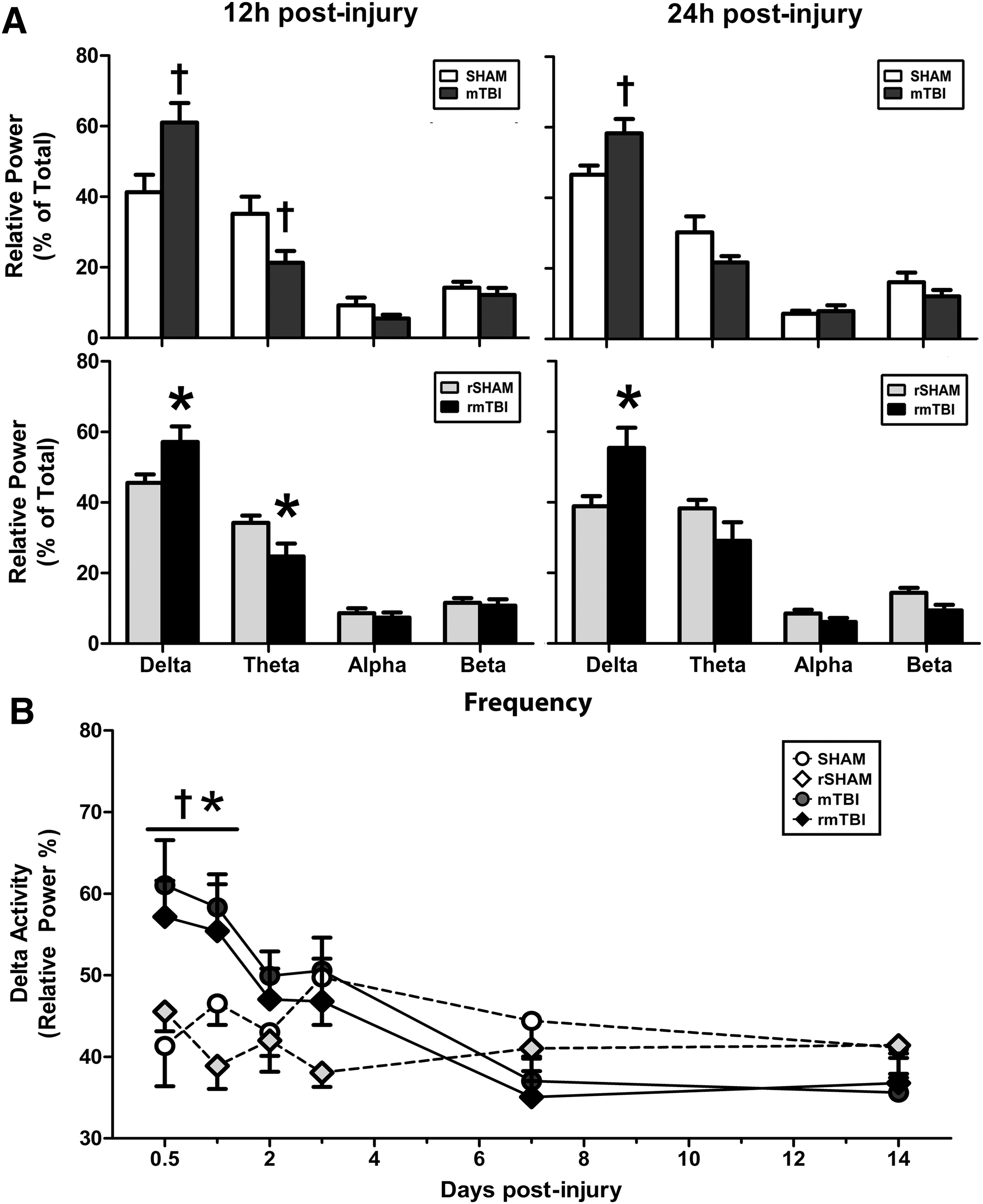
Single and repeated mild traumatic brain injury (mTBI) induces transient changes in quantitative electroencephalogram (qEEG) spectra. Time-dependent changes of delta, theta, alpha, and beta spectral powers presented as % of total power in freely moving rats at 12 h
Neuronal damage was exacerbated by repetitive head trauma
Macroscopic examination of mTBI and rmTBI rats did not reveal evidence of skull fractures, subarachnoid hemorrhage, or tissue damage. Additionally, initial microscopic examinations did not show lesioned brain tissue or an overt influx of inflammatory cells in mTBI-injured rats. However, we did detect significant neuronal death, indicated by FJPpositive cells, which occurred within 24 h post-injury in both injured cohorts (mTBI, 6405 ± 4036; rmTBI, 14,264 ± 3609 FJP cells, p < 0.05 vs. respective sham) (Fig. 7 A–E). The majority of neuronal death was concentrated within the sensorimotor cortex in the ipsilateral hemisphere; however, some animals did show contralateral FJB-positive neurons in the contralateral cortex and cerebellum. Sham and rSHAM rat brain tissues were devoid of positive FJP staining.

Repeated mild traumatic brain injury (mTBI) increases Fluoro Jade B (FJB) and glial fibrillary acidic protein (GFAP) brain levels at 24 h post-injury.
For mTBI rats, GFAP immunostaining revealed evidence of mild reactive astrogliosis in regions of the cortex and hippocampus underlying the impact site (Fig. 7H, I). Importantly, increases in GFAP levels were most salient on the impact side. High resolution images of brain tissue from mTBI and rmTBI rats showed notable increases in cortical GFAP immunoreactivity (∼10–20% greater) compared with respective sham controls. Overall, the cumulative increase of GFAP expression across the cortex was greater in rmTBI animals (SHAM 20 ± 2% vs. mTBI 30 ± 4%; p < 0.05; rSHAM 15 ± 2% vs. rmTBI, 37 ± 3%, p < 0.05, Fig. 7J). Similar changes were present in the hippocampus; however, significant elevations of GFAP levels were limited to rmTBI rats only. In the hippocampus, there was no evidence of increased immunoreactivity in rats subjected to sham or single mTBI (sham 13 ± 2% vs. mTBI 18 ± 4%, p > 0.05). By contrast, increased hippocampal GFAP expression was observed in rmTBI rats compared with rSHAM (rSHAM, 10 ± 2% vs. rmTBI, 20 ± 3%, p < 0.01, Fig. 7K). Samples of CSF taken 24 h after single or repeated head impacts showed significantly elevated GFAP levels (Fig. 7L). CSF GFAP levels were 0.95 ± 0.02 ng/mL after SHAM, 1.04 ± 0.02 after rSHAM, 1.14 ± 0.05 after mTBI, and 1.23 ± 0.05 ng/mL after rmTBI. In contrast, GFAP was not detectable in serum after SHAM, rSHAM, or single mTBI. Serum GFAP was detectable at ∼0.08 ± 0.04 ng/mL after rmTBI, but this elevation was not statistically different from matched controls (data not shown). Quantification of soluble tau protein from CSF samples of rmTBI rats showed robust increases of 20-fold compared with rSHAM (rmTBI, 91.7 ± 14.5 ng/mL vs. rSHAM, 4.76 ± 0.20 ng/mL, p < 0.0001, Fig. 7M), whereas CSF tau levels rose slightly from 2.83 ± 0.025 ng/mL in SHAM animals to 4.19 ± 0.20 ng/mL among single mTBI cohorts. Tau was not detectable in serum of these cohorts (data not shown).
Chronic functional deficits and histopathological damage after single and repeated mTBI
We tracked a number of animals from time of injury to 3 months post-injury, and used a battery of examinations to determine chronic neurological, sensorimotor, and functional impairment (Fig. 8). Notably, both mTBI and rmTBI groups showed elevated NSS-R scores compared with SHAM controls (1.5 and 2.5-fold, respectively) indicating a continuation or re-emergence of previously resolved neurological changes. To examine chronic motor deficits, we assessed animals using a rotarod testing paradigm designed to mirror an exercise stress test that is often used to evaluate post-concussion cardiovascular fitness. Rats were challenged on the accelerated rotarod with minimal recovery time between tests. Although both rmTBI and rSHAM groups initially showed similar performance levels, rmTBI-injured groups failed to maintain rotarod latencies comparable to sham animals (an ∼35% decrement), specifically on the final trial set (trials 11–15), where increased fatigue would be expected to emerge. In contrast, single mTBI animals showed no significant differences from corresponding controls (Fig 8E, F). Trials were collapsed into three sets and compared between injury and control groups. Whereas single mTBI performance was indistinguishable from SHAM, rmTBI-injured animals significantly underperformed rSHAM animals (rmTBI vs. rSHAM, p < 0.05; mTBI vs. SHAM, p > 0.05, Fig. 8G).
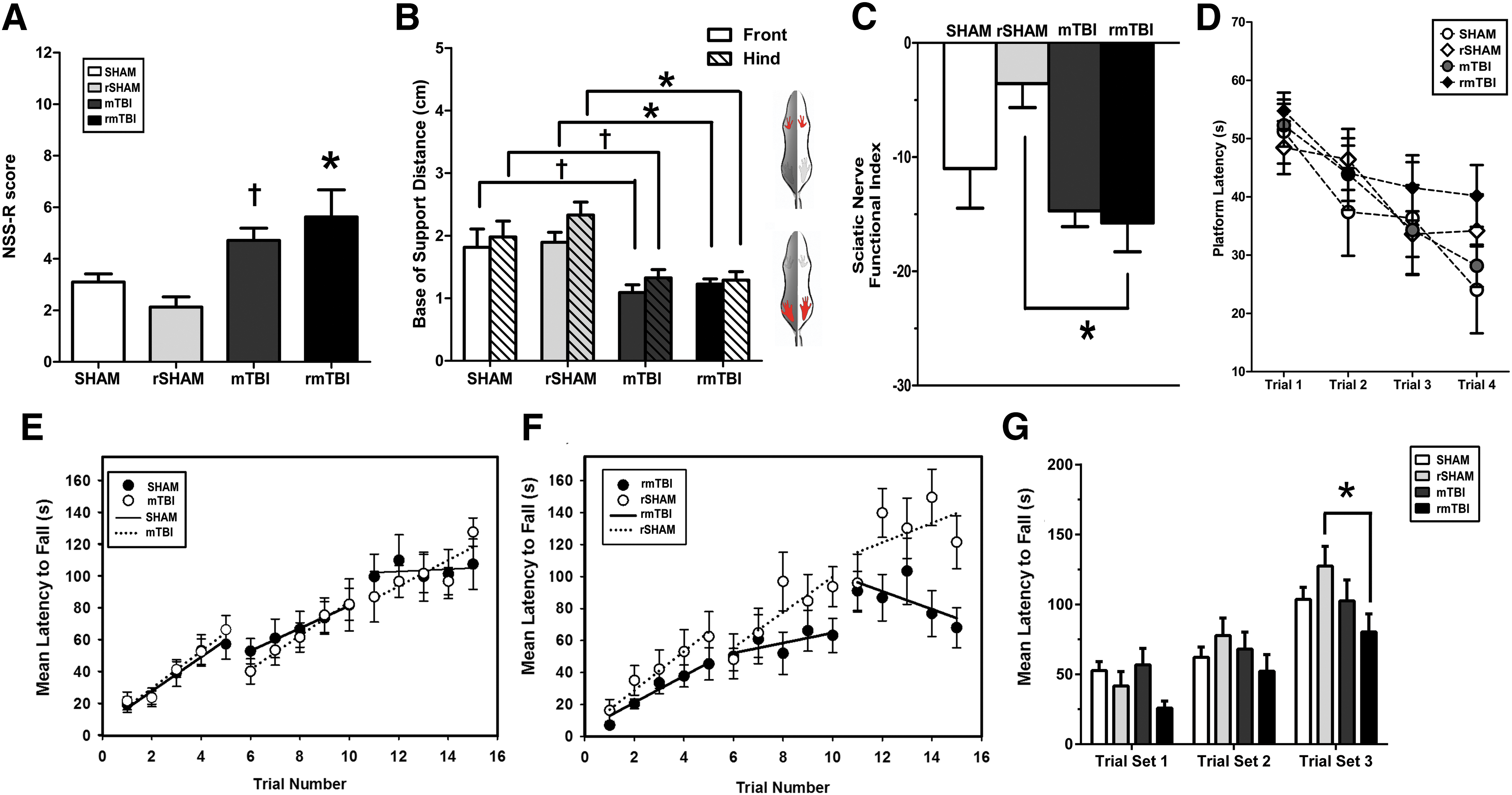
Single and repetitive mild traumatic brain injury (rmTBI) result in chronic neurological damage and functional impairment at 3 months post-injury.
Chronic gait analysis 3 months following head impact showed few differences in >150 different gait parameters between single and repeated brain injury cohorts and respective groups. However, we did find that both mTBI and rmTBI rats showed persistent reductions in BOS in both the front and hind paws (Fig. 8B). These findings in conjunction with abnormal reflex, vestibular, and sensorimotor function detected with the NSS-R, and poorer performance during challenging vestibular-motor tasks prompted us to conduct additional analyses of more subtle gait parameters using our acquired CatWalk data. By analyzing changes in toe placement relative to the body axis, we found that multiple impacts resulted in chronic changes in the Sciatic Functional Index (SFI), a measure for the functionality of the sciatic nerve, which innervates the hind paws and can only be detected with advanced gait analysis. Repeated injury resulted in a lower SFI for rmTBI rats compared with control animals (Fig. 8C).
We evaluated histopathological changes and astroglial markers in brain tissue to determine if the chronic deficits seen in both single mTBI and rmTBI-injured rats had manifested in response to sustained neuronal damage and gliosis. Changes were evident only in animals that had sustained multiple head impacts. Animals with repeated head trauma, 3 months prior, showed significantly increased levels of astrocytic GFAP immunoreactivity in the ipsilateral cortex compared with sham and single mTBI rats (p < 0.05 vs. rSHAM, p < 0.05 vs. mTBI, Fig. 9A, C, D). These animals showed evidence of white matter atrophy and reduced corpus callosum thickness (Fig. 9B). Notably, decreased corpus callosum thickness correlated with higher levels of GFAP in the ipsilateral cortex and hippocampus (Pearson's correlations, r = -0.575, p = 0.003, and r = -0.556, p = 0.003). However, we did not detect enlargement of the lateral ventricles in any group. To complement histological studies, levels of neuroinflammatory cytokines were assessed in the CSF; however, there was no difference between injured and sham cohorts (data not shown). A small number of animals were carried through 6 months post-injury to determine residual changes in behavior or if chronic markers of neurodegeneration were apparent. Although these animals displayed no differences in cognitive ability during MWM testing (Fig. 8F), we did find a trend toward higher levels of the neuropathological protein, tau, and phospho-tau levels in the cortex (Fig. 9E–G).
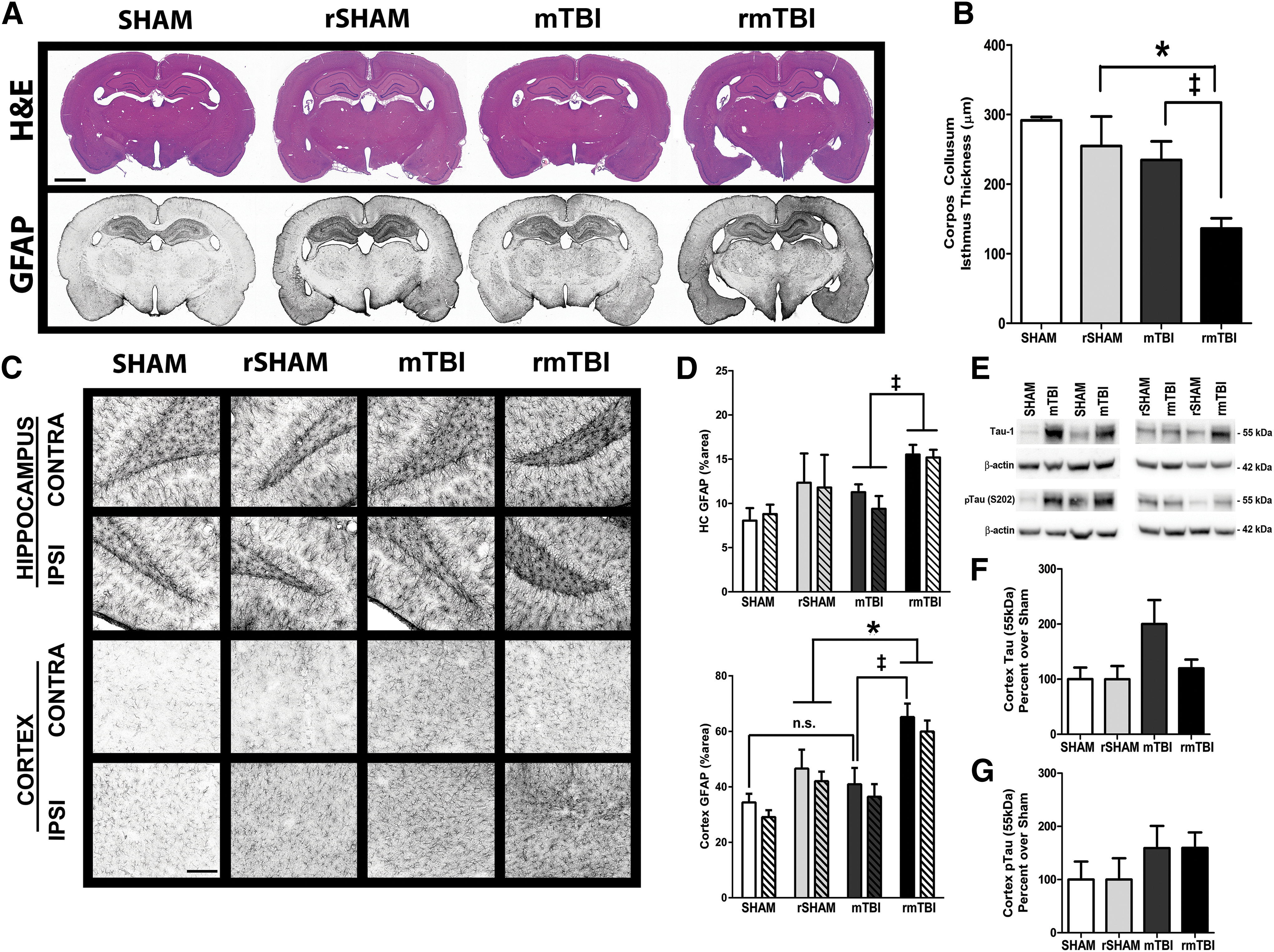
Repeated brain injury resulted in chronic neuropathological damage. At 3 months post-injury, the repetitive mild traumatic brain injury (rmTBI) group showed increased GFAP in the ipsilateral cortex
Sensorimotor and molecular correlates
We determined the relationship between loss of consciousness, a routinely measured indicator of concussion, and deficits in functional and molecular outcome metrics following single and repeated mTBI. In our study, RR times (range of 5–15 min) met the criteria for mTBI. We found that increased RR also correlated with a number of acute changes as well as chronic abnormalities. RR was associated with worsened NSS-R scores, higher serum cytokine levels (1 h and 24 h), higher GFAP brain levels, and poorer sensorimotor ability. Overall, RR consistently correlated with poor rotarod performance, suggesting that longer times to regain consciousness may be associated with consistently worsened vestibular ability. Additionally, higher RR was associated with chronic gait alterations, including increased foot width, lower BOS, and lower Sciatic Nerve Index (SNIndex) (Table 1).
The relationship between righting reflex and molecular markers and functional abnormalities were determined using Pearson's correlation. Individual r and p values at 1 h, 24 h, and 3 months are presented. Metrics are organized into serum, sensorimotor, vestibular, and sensorimotor categories.
p < 0.05; ** p < 0.01.
r, Pearson's correlation coefficient; CORT, corticosterone; CINC-1, cytokine-induced neutrophil chemoattractant 1; CXCL-3, chemokine (C-X-C motif) ligand 3; CNTF, ciliary neurotrophic factor; GMSF, ; IFN, interferon; IL, interleukin; VEGF, vascular endothelial growth factor; GFAP, glial fibrillary acidic protein; FJ, Fluoro-Jade; NSS-R, Neurobehavioral Severity Scale Revised; SNIndex ; BOS, base of support; CC, corpus callosum.
We additionally correlated GFAP reactivity with several outcome metrics at acute (24 h) and chronic (3 month) phases of injury (Table 2). At 24 h post-injury, higher GFAP levels in the cortex and hippocampus were correlated with longer RR times, higher microRNA levels (miR145 and Let-7d), increased neuronal cell death (FJB-positive neurons), elevated cytokines (TIMP-1, CINC-1, IL-4, IL-10, chemokine [C-C motif] ligand 9 [CCL9], RANTES, and vascular endothelial growth factor [VEGF]), and poorer rotarod performance. At 3 months post-injury, brain GFAP levels were associated with higher RR times, lower CORT levels, increases in chemokines CCL3 and CCL20, and decreased corpus callosum thickness.
The relationship between GFAP expression and molecular and functional abnormalities was determined using Pearson's correlation. Individual r and p values at 24 h are presented.
p < 0.05, ** p < 0.01.
GFAP, glial fibrillary acidic protein; FJ, Fluoro-Jade; CINC-1, cytokine-induced neutrophil chemoattractant 1; IL, interleukin; CCL9, chemokine (C-C motif) ligand 9; VEGF, vascular endothelial growth factor.
Discussion
In the absence of mTBI-positive imaging modalities, translational studies have aimed to identify injury-specific physiological markers and aberrant molecular mechanisms that correspond to acute and chronic brain injury-induced functional abnormalities. We sought to characterize the multidimensional symptoms following a single concussive injury (mTBI) in rodents as well as cumulative damage resulting from repeated head trauma. The PCI device was developed to consistently and reproducibly induce a noninvasive closed-head injury that models the constellation of physical, cognitive, and affective symptoms associated with concussion. We showed that after mTBI, a state of transient vulnerability exists during which subsequent concussions within hours of the first injury may lead to prolonged sensorimotor deficits, exacerbate neuro-motor impairment, increase neuronal damage, and elevate levels of inflammatory markers and gliosis. Following a single mTBI using the PCI model, animals displayed acute changes in a number of different outcome metrics including neurological impairment, vestibular dysfunction, gait alterations, neuroinflammation, and astrogliosis similar to what has been shown in published studies. 42,59 Similar yet more robust changes were detected following multiple head impacts, which resulted in disproportionally severe acute outcomes, including death in a small number of animals (< 2% of animals)
The PCI model was designed as a reproducible injury paradigm that could be used to model the pattern of post-injury symptoms often diagnosed following human concussive head trauma. 52 Currently, limited data are available on the exact biomechanical forces that are classified as “mild” or concussion inducing; however, comprehensive data from studies of football players with telemetry-implanted helmets has determined that concussion is correlated with g forces >100g. 60 In our study, calculated impact forces were, on average, 7.5g during the initial impact, which dissipated within the helmet to 4.08g on the inner surface. These values fall within the “mild” range (g force = F/9.8 m/sec2; F = 74 N and 40 N). Importantly, although the injury target was over the right sensorimotor cortex, we detected an ∼8-fold increase in the force contact area on the inner side of the helmet as a result of the energy dissipation pattern through the material. A comprehensive analysis of the material testing, projectile impact energy/head kinematics, and impact location using this model has been published. 52 Those results indicated that, to a limited degree, a small portion of injury extended over the midline area onto the contralateral side, and likely explains increases in GFAP-positive staining over the opposite hemisphere.
Notably, one of the caveats in producing a model of mTBI is that the injurious signals caused by a single impact to the head may be too subtle to detect on select outcome measures. On the other hand, more robust changes in physiological markers can indicate a more moderate-severe injury and would not fall within the criteria for mild TBI. For this reason, we previously conducted initial pilot studies to determine the maximum number of impacts that could be sustained by the rat without causing any skull fractures or subdural bleeding. 52 Based on the data generated by those pilot experiments, the repeated injury model used for the present study consisted of exposing the animals to four consecutive head impacts spaced 1 h apart. Additional experiments are currently underway that evaluate select changes in sensorimotor and biochemical markers following one, two, three, and four injuries with varying inter-injury intervals ranging from days to weeks apart.
One distinctive sign of concussion is acute vestibular/balance dysfunction, which manifests as subtle balance difficulties or overt uncoordinated stumbling motions. Dynamic balance requires coordination of center mass while in motion (gait). Impairments in gait and postural instability may occur as a result of impairment of the brain centers responsible for central integration of vestibular, visual, and somatosensory information. 61,62 Longitudinal postural stability testing has become a routine metric in managing sports-related concussion. 63,64 Consistent with pre-clinical data and studies from concussed athletes, our rmTBI rats showed acute imbalance at 15 min post-injury that normalized by 24 h with repeated testing. 37,65 Conversely, deficits were apparent at 24 h post-injury if animals were tested one time. The conflicting recovery time between our testing paradigms highlights the strong influence of the learning component of repetitive testing, which may mask functional deficits still present after injury. Repetitive testing induces “practice effects” in which conscious effort is decreased and the motion becomes an automatic response. Therefore, caution must be employed with repetitive testing to ascertain recovery from injury, particularly following multiple concussive events, because the brain may still be vulnerable to additional insult.
Coordination and balance involve a complex interaction between the sensory and musculoskeletal systems such that minor impairments in information integration can lead to significant disability. 66 Comprehensive gait analysis detailed subtle yet significant injury-induced changes following brain injury that are consistent with previous studies. 53,56 Following single and repeated mild brain injury, gait abnormalities appeared within hours in all four limbs, and persisted through 1 month post-injury. The magnitude and duration of deficits appeared to be number dependent; repeated injury resulted in a cumulative impact with longer resolution profiles. Importantly, these changes align with reports from TBI patients who often report “imbalance” and “unsteadiness” while walking. 67 Such patients exhibit slower walking speeds, reduced cadence, increased stance and swing durations, and increased double limb support. 67 –69 In our study, mTBI resulted in longer swing durations, lower swing speeds, fewer steps at a time, and reduced stand index, a measure of the speed at which the paw loses contact with the walkway surface. Taken together, these parameters describe a slower walking pattern with resulting unsteadiness between the foot and contact surface. Human TBI patients can adopt gait patterns with increased double support time (the overlapping period of time during simultaneous contact of both feet with the ground) compared with time spent on a single limb. Time spent in double support directly reflects postural instability, because subjects with poor balance often spend more time with both feet on the ground. 63,70 Adapting the same rationale to quadruped rodents, we found that injured rats showed an increased reliance on tri-limb support (simultaneous contact of three limbs) while reducing double limb support time, suggesting a similar support instability seen in TBI patients.
Although we inflicted a unilateral (right hemisphere) focal brain injury, it was surprising that the majority of motor deficits were not detected in limbs of the contralateral (left) side. Injured rats showed bilateral gait impairment with more pronounced deficits in the right hindlimbs (ipsilateral side), including external paw rotation and lack of toe clearance when assessed by the BBB Locomotor Rating Scale. Repeated injury worsened BBB scores. To our knowledge, this is the first time that this test, which is typically used in spinal cord injury (SCI) models, has been used to assess locomotor movement following TBI. These results present an intriguing possibility about the nature and progression of the injury in our model. One possibility is that ascending sensory pathways (spinocerebellar pathways) were more damaged following mTBI than neurons of the major descending motor pathways. Descending motor pathways, including the corticospinal tract and corticorubrospinal tract, originate in the cortex and initially travel ipsilaterally, and subsequently decussate and descend on the contralateral side to target motor neurons in the spinal cord. 71 Trauma to neurons in the cortex that project to these pathways would be expected to disrupt transmission of motor impulses from the cerebrum to the spinal motor neurons, leading to improper activation of muscles, and affecting limbs on the opposite side the body. In contrast, we detected additional deficits in fore- and hindlimbs on the same side as the injury. Spinocerebellar pathways synapse in the cerebellum and transmit impulses from the trunk and lower limb proprioreceptors on one side of the body to the same side of the cerebellum for subconscious proprioreception. 72,73 Damage to these circuits would likely result in subtle, unilateral changes in locomotion of the lower limbs and reduced awareness of limb and foot placement, and may likely explain changes in BBB scores, including paw rotation and reduced toe clearance during normal gait. As previously noted, although the PCI injury is a direct focused impact, the head of the animal is unrestrained and allows free rotation and head movement following injury. In addition, we showed that the protective helmet absorbs a portion of the kinetic energy of impact but spreads the size of the contact area, albeit at a lower impact force. Therefore, it is plausible that additional areas of the brain were injured because of an expanded injury site and coup-contrecoup forces. Whereas the coup injury would result from the primary insult and consequential collision of the brain with the skull, a contrecoup would be expected on the opposite side after the brain impacts the skull. 74 We detected neuronal damage (FJB-positive neurons) in both the sensory motor cortex, likely from the initial direct impact, and in the cerebellum, an area on the opposite side of the brain. Notably, the cerebellum is the area responsible for coordination of voluntary motor movement, balance and equilibrium, and muscle tone. Damage to these cerebellar circuits would result in slow uncoordinated movements, staggered walking patterns, and postural instability as seen during gait analysis and rotarod testing. 75,76 However, these statements are speculative, and a series of carefully designed anterograde and retrograde tracing studies of distinct spinal networks and histopathological assessment would be necessary to confirm such conclusions.
The direct cause of balance/gait alterations in our PCI model is likely multifaceted. Damage to cerebellar circuits, cerebello-cortical networks, and sensory system connections can cause gait and balance ataxia. 70 Indeed, FJB-positive staining indicated cortical neuronal death as early as 24 h post-injury, which correlated with worsened vestibular function. Increased GFAP expression in the cortex and hippocampus emerged within 24 h of injury as accompanied by local increases in pro-inflammatory cytokine levels in the circulating CSF. Cortical GFAP levels were highest in the impact area following repetitive injury, and corresponded to vestibular imbalance. These changes in GFAP expression and neuronal damage are in accord with previously published reports using this model and a separate experimental closed-head injury model in which higher GFAP expression and neuronal death were reported in the hippocampus and cortex acutely (24–48 h) following injury. 52,53,77 Importantly, in our study, we detected differences in GFAP expression between mTBI and SHAM (10–25%), which were smaller than GFAP changes typically seen in other more severe TBI models. 78,79 Soluble GFAP increased in the CSF following single and rmTBI, whereas only repeated injury significantly upregulated tau expression. It is plausible that both cortical cellular death leading to network disruption and local astrocytic and inflammatory changes synergistically contribute to both acute and progressive sensorimotor dysfunction. The greater degree of ataxia following repeated concussion, again, highlights the brain vulnerability in which successive concussions result in cumulative damage with a protracted recovery period.
Neuroinflammation has been associated with pathological sequelae from neurodegenerative conditions and brain insults. 80 In-depth reviews of the changes in acute inflammatory markers in pre-clinical and clinical TBI studies have been published. 81,82 In particular, altered concentrations of various CSF components, such as IL-6, IL-8, IL-10, and TNF-α are detectable in response to severe head trauma, and often correlate with patient outcome. 83 –89 Because multiple cytokines operate in a large network where the action of one cytokine is regulated by the presence or absence of other cytokines, we used a rapid and sensitive method to screen a panel 29 different inflammatory markers and simultaneously profile the relative levels of multiple cytokines and chemokines between samples. We found that our model induced rapid upregulation of local and systemic pro-inflammatory cytokines, notably in CINC-1, TIMP-1, and L-selectin. In CSF, fold changes in inflammatory markers were more dramatic compared with peripheral blood levels, likely resulting from membrane degradation and unregulated transit of immune molecules in through the blood–brain barrier. Hinson and colleagues have noted that the patterns of inflammatory markers after TBI detected in the peripheral blood tend to be echoed in CSF; however, CSF levels are often orders of magnitude more pronounced. 90
CINC-1 represents a major chemokine involved in CNS neutrophil recruitment following brain injury, ischemia, meningitis, and oxidative stress. 91 –93 Therefore, the rapid rise in CINC-1 levels should be expected. A gradient of local CINC-1 at the site of injury attracts circulating leukocytes and recruits neutrophils. 94 Such a scenario would explain the high levels of CINC-1 in the ventricular CSF. Serum and CSF CINC-1 levels were positively correlated, suggesting a localized CNS response and blood–brain barrier leakage with systemic blood. Notably, we found a positive correlation between brain levels of the astrocytic marker GFAP and systemic CINC-1 levels. Taken together, these data indicate that CINC-1 serum levels may be an acute marker indicative of impending neuronal brain damage.
The neuroinflammatory marker, TIMP-1, was elevated in response to PCI injury. Similar expression patterns were noted in the CSF of TBI patients, where TIMP-1 levels remained 7-fold higher than controls and persisted over 120 h. 95 Rat ischemia models have shown acute TIMP-1 expression by reactive astrocytes in injured brain tissue. 96 We found a positive correlation between brain GFAP and TIMP-1 expression following injury in the acute phase, which is consistent with pre-clinical rodent hypoxia models. 94 The high concentration of inflammatory markers in our model in both serum and CSF suggests that local inflammation following concussive impact may cause blood–brain barrier disruption and enable bidirectional access of inflammatory mediators between the CNS and systemic tissue, further exacerbating local inflammation. 97 Importantly, these markers (TIMP-1 and CINC-1) may serve as prognostic biomarkers in TBI patients.
In addition to traditional TBI biomarkers often present in brain tissue, serum microRNA profiling has become increasingly popular. miR145 is elevated in response to transient cerebral ischemia, closed-head injury, and astrogliosis. 63,98 –100 We found a positive correlation between serum miR145 and GFAP astrogliosis in the ipsilateral cortex and hippocampus. The dramatic increase in miR145 may serve as a prognostic indicator for the degree of inflammatory response and overall mTBI injury severity, warranting further study. In addition, another microRNA sequence, Let-7b, although not significantly upregulated following mTBI, did positively correlate with increased post-injury GFAP levels. The correlation between systemic microRNA and brain GFAP levels in response to TBI is intriguing, and will be a subject in a future study.
This model indicates that a more expansive panel of biomarkers may have translational utility for mTBI/concussion. GFAP and tau have been at the forefront of clinical mTBI biomarker studies. Although GFAP, a marker of astroglial stress and inflammation, is typically associated with intracranial lesions or mortality after severe TBI, recent cases of mTBI indicate that CSF or serum GFAP is also increased as a consequence of mild injuries, and may be capable of stratifying CT+ versus CT– patient populations. 101 –103 Like GFAP, tau is detectable in biofluids as late as 1–5 days after severe head trauma, 104 and has been associated with intracranial pressure and poor patient outcome. 105 Accordingly, tau is moving toward the forefront of biomarkers for mTBI/ concussion. Increased levels of tau have been detected in the CSF or blood of boxers 106 or hockey players with multiple, rather than single, concussive incidents. 107 Notably, in our model, rmTBI resulted in elevated tau expression in the CSF levels that was 20-fold greater than sham levels. Moreover, cytokines and chemokines provide a myriad of potential biomarkers. TNF-α and IL-6 are two of the most intensely studied pro-inflammatory proteins in TBI. Therefore, it was initially surprising that neither was increased in the biofluids after PCI; however, it must be noted that we did not screen brain tissue for cellular changes in these inflammatory markers. It is plausible that these cytokines (IL-10, IL-6, and TNF-α) were elevated in areas of the cortex that showed higher GFAP expression or sites of FJP cells, as has been noted other mTBI studies using brain tissue homogenates. 108
Alternatively, mTBI/concussion may lead to a unique inflammatory response. CINC-1, TIMP-1, and L-selectin may be indices of activation of inflammatory mechanisms, loss of cellular adhesion, and a systemic response to mild brain trauma. 93 In fact, patients with the mild SCI had little or no increase in IL-6, but did have an increase in IL-8 (CINC-1 in our study). 109 The association of TIMP-1 with repeated mTBI or concussion is not yet known. However, TIMP-1 is increased in the serum of patients with brain or spinal cord inflammation. 110 –112 Clinical relevance of miRNAs is an emerging field in TBI research. Sequences tested in this study have been observed in moderate to severe paradigms and pilot studies of mild injuries. 113 miR145, a sequence linked to apoptosis and infarct size in animal models, 114 is increased in the serum of patients within 1 day after stroke. 115 Let-7b, which was increased in the serum after rmTBI, has not yet been observed in TBI cases, but has been reported to be upregulated in patients diagnosed with Alzheimer's disease. 116 The observation of these miRs, specifically miR145, is novel to mild models and may be crucial to further development of biomarker metrics relevant to repeated concussive injuries. Overall, increases in these proteins and miRNA levels in the CSF or serum may be indicative of transient inflammation and cellular permeability after mTBI.
Changes in EEG activities can also serve as an electrophysiological marker of brain dysfunction after TBI. 117,118 Even mild brain injury can shift EEG power distributions that are readily detectable using spectral analysis. 119,120 In this study, qEEG analysis revealed moderate but significant increases in the delta activity within 24 h following mTBI. Similar increases in delta power and EEG slowing have been observed in experimental mTBI models and studies of mild TBI patients. 121 –124 It has been hypothesized that and white and gray matter damage after closed-head injury may lead to reduced excitatory input to the cortex, which subsequently enhances delta EEG frequencies. 125 EEG patterns from our mTBI and rmTBI rats showed similar power shift changes and recovery time courses, and the lack of discriminative sensitivity between single and repeated injury in our study was surprising. Previously, we had demonstrated that qEEG analysis was able distinguish between injury severity levels following severe penetrating brain injury. 118 In this study, the delta activity achieved ∼60% of the total power for both single and rmTBI and likely indicates an injury threshold level at which point additional impacts do not lead to additional increased delta activity. Despite the limitation of an indiscriminative power of the qEEG analysis applied to this study, the detection of early EEG slowing can provide a brain injury marker of functional impairment. Interestingly, Vyazovskiy and colleagues reported that during prolonged wakefulness, rodents show an increase in EEG frequencies (1–8 Hz) traditionally seen during sleep periods, which can intrude into normal waking EEG frequencies. This increased slow wave activity during wake periods results in an area of “local sleep,” and animals showed more errors in behavioral performance. 126 In our study, increases in delta power following single and repeated mTBI were recorded during wakefulness, and corresponded to time points of impaired functional performance. Although is it unknown if a similar “local sleep” phenomenon occurs following mTBI, focal damage to neuronal circuits and neuronal tiredness caused by synaptic overload 127 may result in the EEG slowing and behavioral changes seen after injury. Additional research on the mechanisms underlying EEG slow waves following mTBI is warranted.
The majority of concussive symptoms typically resolve within a few weeks post-injury; however, a portion of TBI patients show prolonged symptoms of headache, fatigue, vertigo, affective disorders, and difficulty with concentration and exercise. 128,129 Because the duration of behavioral impairments and the progression of histological damage in the PCI model were unknown, we examined chronic neuropathologies in a subset of mTBI and rmTBI-injured animals. In mTBI patients with post-concussion syndrome (PCS), symptoms can be exacerbated during exercise because of autonomic system (ANS) dysfunction. 129,130 A graded exercise test can identify mTBI patients with symptomatic PCS who exhibit low exercise tolerance compared with uninjured controls. 130,131 This low exercise tolerance likely explains the poor rotarod performance of rmTBI rats at 3 months-post injury, when subtle vestibular deficits emerged when animals were challenged with increased exertion and shorter recovery times. Similar impairments following repetitive experimental head trauma have been noted in testing paradigms with short inter-trial intervals. 33,132 In addition, we found that rmTBI rats showed reduced corpus callosum thickness, which persisted concurrently with sustained astrogliosis. Brain tissue atrophy has been documented in both clinical and pre-clinical studies using imaging modalities and postmortem examination. 51,133 Importantly, our animals subjected to rmTBI showed thinning of the corpus callosum at its midportion, which has been seen in boxers and football players who displayed cognitive deficits and balance impairment. 133
Overall, our findings suggest that the PCI model provides a true closed-head injury model producing a constellation of symptoms and cumulative short- and long-term changes in neurological responses, sensorimotor function, and brain neuropathology. These findings are consistent with clinical data indicating that a history of concussion is associated with prolonged recovery following subsequent concussions. The protracted recovery period may be indicative of a period of increased neuronal vulnerability during which recurrent brain injuries can result in cumulative impairments. 17 We recognize the limitations of our study with regard to the number and temporal duration of repeated impacts. The specific time points we have investigated here may correspond to a limited interval between injuries; however, our data specifically define quantitative molecular changes that directly correlate to clinically relevant functional anomalies. In addition, changes in the number of FJB-positive cells and GFAP expression were captured only at distinct points (24 h and 3 months post-injury). Future studies are needed to map the comprehensive profile of GFAP expression, neuronal death, and cytokine responses following single and repeated injury using this model. Additional studies are currently underway that will further characterize the time frame of enhanced vulnerabilities and potential therapeutic interventions to reduce cumulative neuronal damage.
Conclusion
The present study describes the cumulative effects of single and repeated head trauma in a rodent closed-head PCI model. A single concussive event produced transient functional deficits with residual effects still present at 3 months after injury. Compared with a single mTBI, repeated head trauma produced a constellation of symptoms and cumulative short- and long-term changes in neurological responses, sensorimotor function, and brain neuropathology. Repeated mild brain injury resulted in long-term balance deficits and white matter thinning. We found that longer periods to regain consciousness and balance impairments were correlated with higher GFAP levels in cortical tissue and neuroinflammation. Our findings are consistent with clinical data indicating that a history of concussion is associated with prolonged recovery, and indicate a number of molecular correlates that suggest a period of increased neuronal vulnerability.
Footnotes
Acknowledgments
This research was supported by United States Army grants W81XWH-12-2-0134 and H_024_2014_WRAIR. The authors are grateful to Weihong Yang, Ying Cao, Matthew Wise, and the Walter Reed Army Institute of Research Pathology Department for their technical assistance. This material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting true views of Department of the Army or Department of Defense.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.