
Abstract
Gastrointestinal dysfunction is one of several physiologic complications in patients with traumatic brain injury (TBI). TBI can result in increased intestinal permeability resulting from apoptosis of intestinal epithelial cells, which contain a large number of mitochondria for persisting barrier function. Autophagy of damaged mitochondria (mitophagy) controls the quality of the mitochondria and regulates cellular homeostasis. However, the exact mechanism of mitophagy that underlies the pathological changes induced by TBI is unknown. Here, we report that mitophagy decreases the intestinal epithelial cell damage and apoptosis that are activated in a rat model of controlled cortical impact (CCI). CCI-induced mitophagy is associated with an increase in 3-nitrotyrosine and 4-hydroxynonenal, indicating that oxidative stress may increase in response to mitochondrial disturbance. CCI also results in the expression of the tight junction proteins zonula occludens-1 (ZO-1) and occludin, which may regulate the in vivo intestinal hyperpermeability induced by CCI. Additionally, CCI-induced mitophagy was shown to be mediated by the oxidative stress-related extracellular signal-regulated kinase (ERK)/nuclear factor-erythroid2-like2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathway, which may serve to reduce the apoptosis induced by oxidative stress. These results suggest that CCI-induced mitophagy serves to diminish apoptosis-mediated intestinal epithelial cell damage and to improve intestinal permeability, via ERK/Nrf2/HO-1 signaling. These findings may be useful in the design of rational approaches for the prevention and treatment of symptoms associated with TBI.
Introduction
T
When vital organs such as the heart and brain are perfused, ischemia or hypoperfusion of the intestinal mucosa often occurs, and intestinal mucosal epithelial cells are very sensitive to ischemia and hypoxia because their energy store is small after TBI. As a result, a large number of free radicals are generated after TBI, which may destroy intestinal mucosal barrier function. 10,14 –16 Mitochondria are vital storehouses for oxidative phosphorylation and energy. In particular, dysfunctional or damaged mitochondria trigger a destructive cycle of mitochondrial damage, and more reactive oxygen species (ROS) are generated through the electron transport chain, which is detrimental to cell survival. 17 Evidence suggests that rat enterocyte mitochondrial respiratory function and enzyme activities are inhibited after TBI, which may indicate that mitochondria are the primary target of oxidative and mitochondrial dysfunction in TBI-induced gastrointestinal dysfunction. 14 However, intestinal epithelial cells play an important role in absorbing nutrition and regulating barrier functions, which heavily depend on mitochondrial function. 18,19 Mitochondria serve as platforms to activate inflammation and immune response signaling, 20 –24 and also to regulate apoptosis by releasing ROS in response to varying energy requirements and environmental conditions. 22,25 Therefore, mitochondria are important in maintaining the cell balance. 26,27 Mitophagy, a selective form of autophagy that is specific to mitochondrial degradation, is important for mitochondrial morphology and homeostasis. 28,29 The clearance of damaged mitochondria by mitophagy may counteract apoptosis. 30,31 However, the mechanism of intestinal dysfunction and mitophagy following TBI has not yet been determined.
In this study, we evaluated the mitophagy induced in rats by controlled cortical impact (CCI), which provides an in vivo model of TBI. Our results show that mitophagy induced by CCI attenuates intestinal epithelial cell apoptosis and alleviates intestinal high permeability, which is associated with decreased expression of tight junction protein zonula occludens-1 (ZO-1) and occludin. This study provides direct biochemical evidence for mitophagy in intestinal epithelial tissue after CCI, and may reveal the molecular mechanisms underlying the effect of mitophagy on TBI-induced apoptosis and intestinal dysfunction.
Methods
CCI model
All animal experiments were approved the Institutional Animal Care and Use Committee of Nanjing Medical University and were performed according to the guidelines of the Principles of Laboratory Animal Care (National Institutes of Health Publication No. 85–23, revised 1996). Seventy male Sprague–Dawley (SD) rats (weighing 240–260 g) were provided by the animal center of Nanjing University and received food and water ad libitum in the university animal facility on a 12/12 h light/dark cycle. Five animals were used in each experimental group at each time point. The CCI model was performed as previously described 32 using the Pneumatic (Cortical) Impact Device (Model: AMS 201). Rats were anesthetized with 4% isoflurane in 70% N2O and 30% O2 and were maintained under anesthesia using 2% isoflurane. The heads were fixed in a stereotactic device. A craniotomy of 10 mm in diameter was made over the left parietal cortex with a dental drill, using the coronal and interparietal sutures as margins. The brain temperature was monitored with a microprobe (Physitemp Instruments, Clifton, NJ) inserted through a burr hole into the left frontal cortex and was maintained at 37 ± 0.5°C using a heat lamp. Isoflurane was decreased to 1% and was allowed to equilibrate (30 min). For all studies, rats were subjected to CCI with a 6 mm metal pneumatically driven impactor tip at a velocity of 6.0 ± 0.2 m/sec, with 2.5 mm depth of penetration and 50 ms duration of deformation. Before the operation, rats were injected intraperitoneally with 3-methyladenine (3-Ma) (30 mg/kg), rapamycin (Ra) (2 mg/kg), or PD98059 (10 mg/kg, ERK inhibitor, purchased from Sigma-Aldrich Incorporation, St. Louis, MO). After the operations, the bone flaps were replaced and the rats were returned to their cages.
The rats were euthanized with inhaled isoflurane, and the terminal halves of the ileums were collected and fixed in 10% buffered formalin overnight for tissue section preparation. The other halves of the ileums were stored at −80°C for intestinal lysate preparation for immunoblotting. Hematoxylin and eosin (H&E) stained sections were assessed histologically for ileum damage.
Immunohistochemistry (IHC)
Paraffin intestinal sections were dewaxed, immersed in xylene three times and then hydrated with ethanol (two times with 100%, two times with 95%, and one time with 75% ethanol) for 5 min each. Immunohistochemical staining was performed as described previously. 33 Intestinal sections were incubated overnight with antibodies against microtubule-associated protein 1 light chain 3 (LC3) (1:1,000; Cell Signaling Technology, catalog no. 2775s), p62 (1:1,000; Abcam, catalog no. ab56416), cleaved-caspase-3 (1:1,000; Cell Signaling Technology, catalog no. 9664s), Atg5 (ab108327), Beclin-1 (ab55878), 3-nitrotyrosine (3-NT) (ab61392), or 4-hydroxynonenal (4-HNE) (ab46545). Positive staining was visualized and quantified using Plus6.0 Image-Pro software. Each section was imaged randomly by scanning five to eight fields in each quadrant. Using Plus6.0 Image-Pro software to select the same brown color as the uniform standard for all photos, the integral optical density (IOD) of each photo was analyzed.
Apoptotic cell death was examined using the terminal deoxynucleotidyl transferase (2'-deoxyuridine 5'-triphosphate (dUTP) nick-end labeling (TUNEL) assay as described by the manufacturer (Roche, Germany). The apoptotic cells from paraffin intestinal tissue sections showed brown staining as detected by TUNEL staining.
Immunofluorescence (IF)
Immunostaining of rat intestinal samples was performed as previously described. 34 Intestinal sections were incubated with LC3 antibody (1:1,000; Cell Signaling Technology, catalog no. 2775s) and p62 antibody (1:1,000; Abcam, catalog no. ab56416). Double immunofluorescent staining was performed on parallel sections with rabbit anti-COXIV polyclonal antibody (Cell Signaling Technology) and mouse anti-LC3 II polyclonal antibody (Abcam). Counterstaining was performed with the nuclear probe DAPI (Beyotime Biotechnology). The cover-slips were observed and quantified under an Olympus fluorescent microscope. Each section was imaged randomly, scanning five to eight fields in each quadrant.
Histological evaluation
The 10% buffered formalin-fixed distal ileum was embedded in paraffin, sectioned at 4 μm thickness with a microtome, and stained with H&E. Using a light microscope, images were obtained at varying magnifications. Each ileum specimen was examined and scored using a modified histopathologic score. 35 A scale of 0–3 was used to assess intestinal damage: 0 = normal, no damage; 1 = mild, focal epithelial edema and necrosis; 2 = moderate, diffuse swelling or necrosis of the villi; and 3 = severe, diffuse necrosis of the villi with evidence of neutrophil infiltration in the submucosa or hemorrhage.
Preparation of enterocyte mitochondria
Enterocyte mitochondria were isolated as previously described. 36 Briefly, one segment of the ileums of the experimental rats was removed for subsequent procedures. The intestinal contents were washed with ice-cold phosphate-buffered saline (PBS) (0.01 mol/L), and the intestinal wall was turned over to expose the intestinal mucosa. Intestinal epithelial cells were shaved off and placed in 50 mL of ice-cold buffer (containing 0.01 mol/L PBS and 10 mmol/L ethylenediaminetetraacetic acid [EDTA]), the supernatant was discarded, and the pellet was washed once with isolation buffer containing 25 mmol/L sucrose, 10 mmol/L Tris, 2 mmol/L EDTA, pH 7.4 by centrifuging at 3500 r/min for 2.5 min. The buffer was centrifuged at 3500 r/min for 2.5 min, the pellet was washed twice with isolation buffer, and then the homogenates were diluted to 50 mL and centrifuged at 1700 r/min for 10 min. The supernatant was collected and centrifuged at 10,500 r/min for 8 min. The mitochondrial pellet was suspended in isolation buffer. The procedures described were performed at 4°C.
Measurement of mitochondrial respiratory chain complex I-IV activities
The activities of mitochondrial respiratory chain complex I-IV were detected according to the instructions provided with their respective kits (Beijing Solarbio Science & Technology Co., Ltd). Briefly, complex I activity was assayed by monitoring the decrease in nicotinamide adenine dinucleotide (NADH) at 340 nm for 2 min. Complex II activity was assayed by monitoring the decrease in 2,6-Dichloroindophenol (DCIP) and scanned at 605 nm for 2 min. Complex III activity was assayed by monitoring the increase in reductive cytochrome C with the increase in absorption at 550 nm recorded for 2 min. Complex IV activity was assayed by monitoring the decrease in reduced cytochrome C at 550 nm.
Transmission electron microscopy (TEM)
The ileum tissues for electron microscopy were fixed in phosphate-buffered glutaradehyde (2.5%) and osmium tetroxide (1%). Dehydration of the mucosa was accomplished in graded acetone solutions. The sample was embedded in an epoxy resin. Semi-thin (1 mm) sections through the mucosa were then prepared and stained with toluidine blue. Then 600 angstrom-thin sections were prepared from a selected area of tissue defined by the semithin section, and these sections were stained with lead citrate and uranyl acetate before being observed under TEM (Hitachi HT7700).
Western blot analysis
Intestinal tissues from each rat were excised, dissolved in radioimmunoprecipitation assay (RIPA) buffer, and centrifuged at 1000g for 15 min at 4°C. Proteins were separated by electrophoresis and then transferred to polyvinylidene difluoride (PVDF) membranes by electroblotting according to standard immunoblot procedures. The protein bands were visualized using a G:BOX chemiXR5 (SYNGENE G:BOXChemiXR5, UK) and analyzed by Gel-Pro32. The antibodies used were as follows: LC3 (1:1,000; Cell Signaling Technology, catalog no. 2775s), p62 (1:1,000; Abcam, catalog no. ab56416), Beclin-1 (ab55878), TOM40 (1:1,000; Abcam, catalog no. ab51884), COXIV (1:1,000; Cell Signaling Technology, catalog no. 11967), MnSOD (1:1,000; Cell Signaling Technology, catalog no. 13141), GAPDH (1:1,000; Cell Signaling Technology, catalog no. 5174), cleaved-caspase-3 (1:1,000; Cell Signaling Technology, catalog no. 9664s), BAX (1:1000, ab200478, Abcam32503), Occludin (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), ZO-1 (1:500; Zymed, Carlsbad, CA), pERK (1:1,000; Cell Signaling Technology), HO-1 (1:500; Stressgen Biotech, San Diego, CA), and Nrf2 (1:500; Santa Cruz Biotechnology Inc. Santa Cruz, CA).
Real-time polymerase chain reaction (PCR)
Total cellular DNA was extracted with a DNeasy Blood and Tissue kit (Qiagen, 69506). For intestinal tissues, after 24 h, rats were subjected to TBI and euthanized by choral hydrate administration. Ileum tissues were separated and homogenized in ice-cold PBS (pH 7.4), and then the DNA was isolated. Aliquots of 20 ng total DNA were used for PCR reactions. PCR amplification was performed as follows: denaturing at 95°C for 45 sec, annealing at 60°C for 45 sec, and extension at 72°C for 30 sec. The primer set for rat mt-cytochrome C oxidase subunit II (COII) was 5′-TGAGCCATCCCTTCACTAGG-3′ (forward) and 5′-TGAGCCGCAAATTTCAGAG-3′ (reverse), and the primer set for rat Rpl13 was 5′-CAC AAG AAA ATG GCA CGC AC-3′ (forward) and 5′-GAG CAG AAG GCT TCC TGG G-3′ (reverse). All data analyses were performed using an ABI Prism 7900HT system (Applied Biosciences). CT values were obtained automatically. The level of gene expression was calculated and normalized using a standard curve. Relative expression was presented as mt-COII/Rpl13.
Statistical analysis
Data were expressed as mean ± standard deviation (S.D). Statistical analysis was performed using the two tailed Student's t test for paired data or the ANOVA test with SAS statistical software (SAS Institute, Cary, NC). Results were considered statistically significant at p < 0.05. Each experiment was performed at least three to five times.
Results
Intestinal mitophagy is activated by damaged mitochondria after TBI
To verify that TBI induces intestine mitochondrial dysfunction, we assessed the activities of mitochondrial complex proteins. As shown in Figure 1A, the activities of mitochondrial complexes I and II were significantly decreased at days 1 and 3 after CCI. Complex III and IV activities did not differ in the control and CCI groups. No obvious morphological changes in the mitochondria were observed in the intestinal epithelial mucosa in the control, sham, and 6 h groups. In contrast, reduction of the mitochondrial matrix, disruption of cristae, and mitochondrial swelling were observed at 1 and 3 days after CCI, with clear signs of autophagy (Fig. 1B). These results are consistent with other reports suggesting that damaged mitochondria can activate mitophagy. 31 To further examine whether TBI induces mitophagy, we performed immunohistochemical examination of intestinal samples from rats after TBI was induced by CCI. The expression of the autophagy markers Beclin-1, Atg5, and LC3B increased from 6 h to 7 days, with maximum levels at 1 day after CCI (Fig. 2A and B). These results suggest that TBI causes increased levels of autophagy.
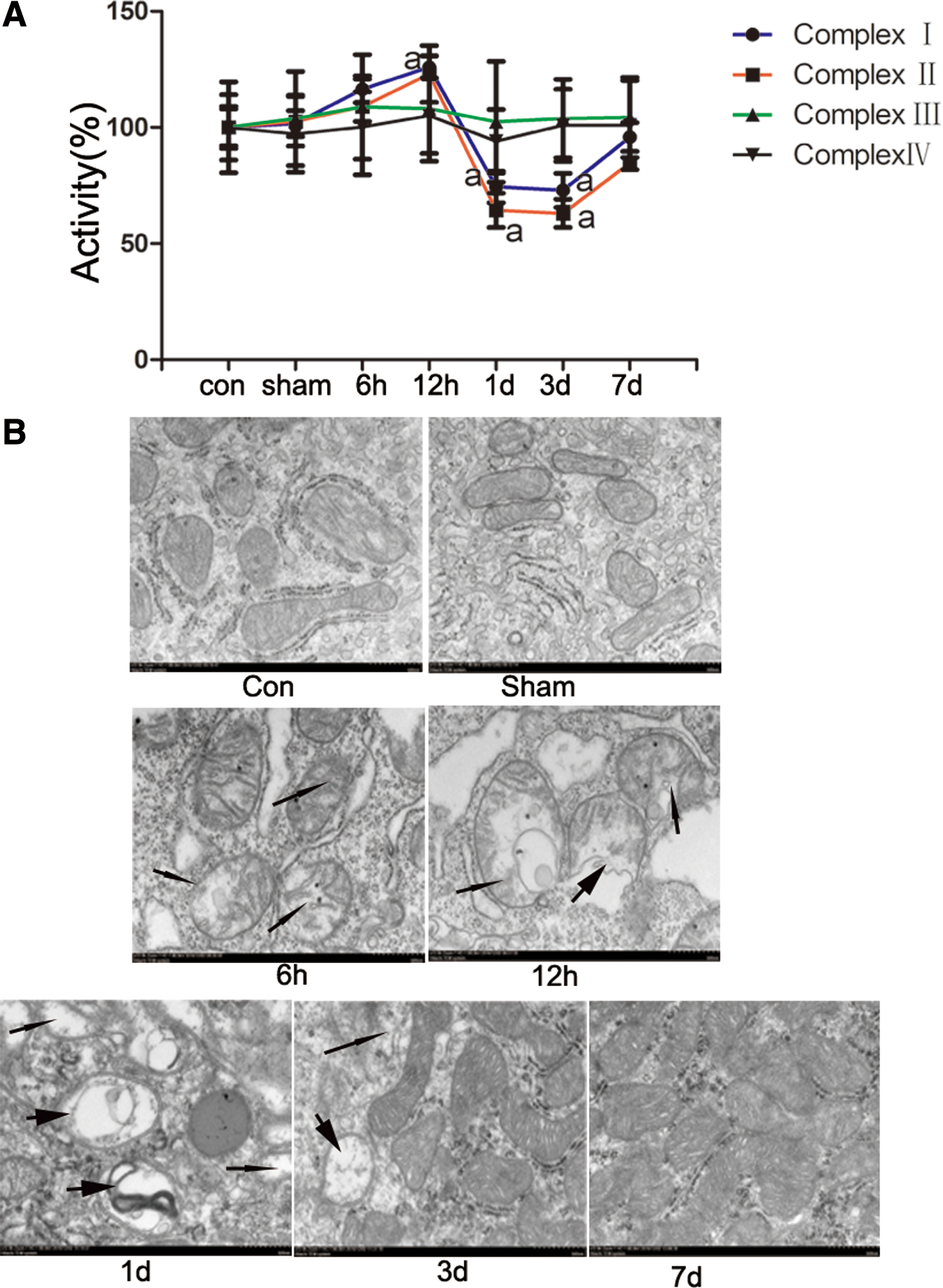
Damaged mitochondria were induced after controlled cortical impact (CCI) in rat intestines.

Autophagy is activated after controlled cortical impact (CCI) in rat intestines.
During autophagy, LC3-I is transformed to a phosphatidylethanolamine-conjugated LC3-II form, 37 Beclin-1 expression is activated, and the ubiquitin-binding scaffold protein p62 becomes degraded. 38 To confirm that intestinal autophagy is increased after CCI, the conversion of LC3 and the protein expression of Beclin 1 and p62 were analyzed by Western blotting of small intestine lysates harvested from surviving rats after CCI (Fig. 2C). Densitometric quantification of the protein bands demonstrated a significant increase (p < 0.05) in the ratio of LC3-II/GAPDH and Beclin1/GAPDH and a significant decrease (p < 0.05) in the ratio of p62/GAPDH in injured animals compared with the sham group (Fig. 2D). The peak in protein increase or decrease for each of these proteins occurred at 1 day after CCI.
We repeated Western blotting assays to examine the expression of mitochondria-specific autophagy proteins in the intestinal samples after CCI. The expression of immune-mediated myositis (IMM) (COXIV), outer mitochondrial membrane (OMM) (TOM40), and matrix (MnSOD) was markedly decreased 1 day after injury, and started to rise again 3 days after CCI (Fig. 3A and B). To assess whether LC3 puncta, which are indicative of autophagosome formation, 39 co-localize with mitochondria, we performed confocal microscopy. Increased levels of co-localization were detected between COXIV and LC3 at 24 h after CCI (Fig. 3C and D), verifying that mitophagosomes were formed. For additional confirmation that autophagy occurred at the level of the mitochondria, the ratio of mitochondrial to genomic DNA (mtDNA:gDNA) was calculated by real-time PCR as a measurement of the relative mitochondria level. The results demonstrate that the mtDNA content was reduced at 24 h in the CCI group compared with the sham group (Fig. 3E). These results suggest that intestinal mitophagy was activated by TBI.

Intestinal mitophagy is activated after controlled cortical impact (CCI).
Mitophagy alleviates TBI-induced intestinal epithelial cell damage by mitigating cell apoptosis
To investigate the effect of mitophagy on intestinal epithelial cells after TBI, rats were treated by intraperitoneal injection with the autophagy inhibitor 3-Ma or the autophagy activator Ra prior to CCI treatment. Intestinal sections at 1 day after TBI were examined by ICH. As expected, the levels of autophagy, as indicated by increased LC3B and decreased p62 levels, were activated by CCI exposure, partially reversed by 3-Ma, and further enhanced by Ra (Fig. 4A and B). Examination of the stained sections also revealed that in the absence of CCI, animals had normal-looking villi with consistent villous height; however, the villi of CCI-treated animals had architectural deformity with blunted villi, which were further damaged by treatment with 3-Ma and ameliorated by treatment with Ra (Fig. 4A and C). These effects of 3-Ma and Ra on mitophagy and intestinal damage were further verified by immunofluorescence staining (Fig. 4D) and Western blotting (Fig. 4E and F). These findings suggest that with CCI, autophagy and intestinal damage are induced in rats, and that the decreased levels of mitophagy with 3-Ma treatment lead to further intestinal damage, whereas the increased levels of autophagy with Ra treatment protect rats from the intestinal damage.
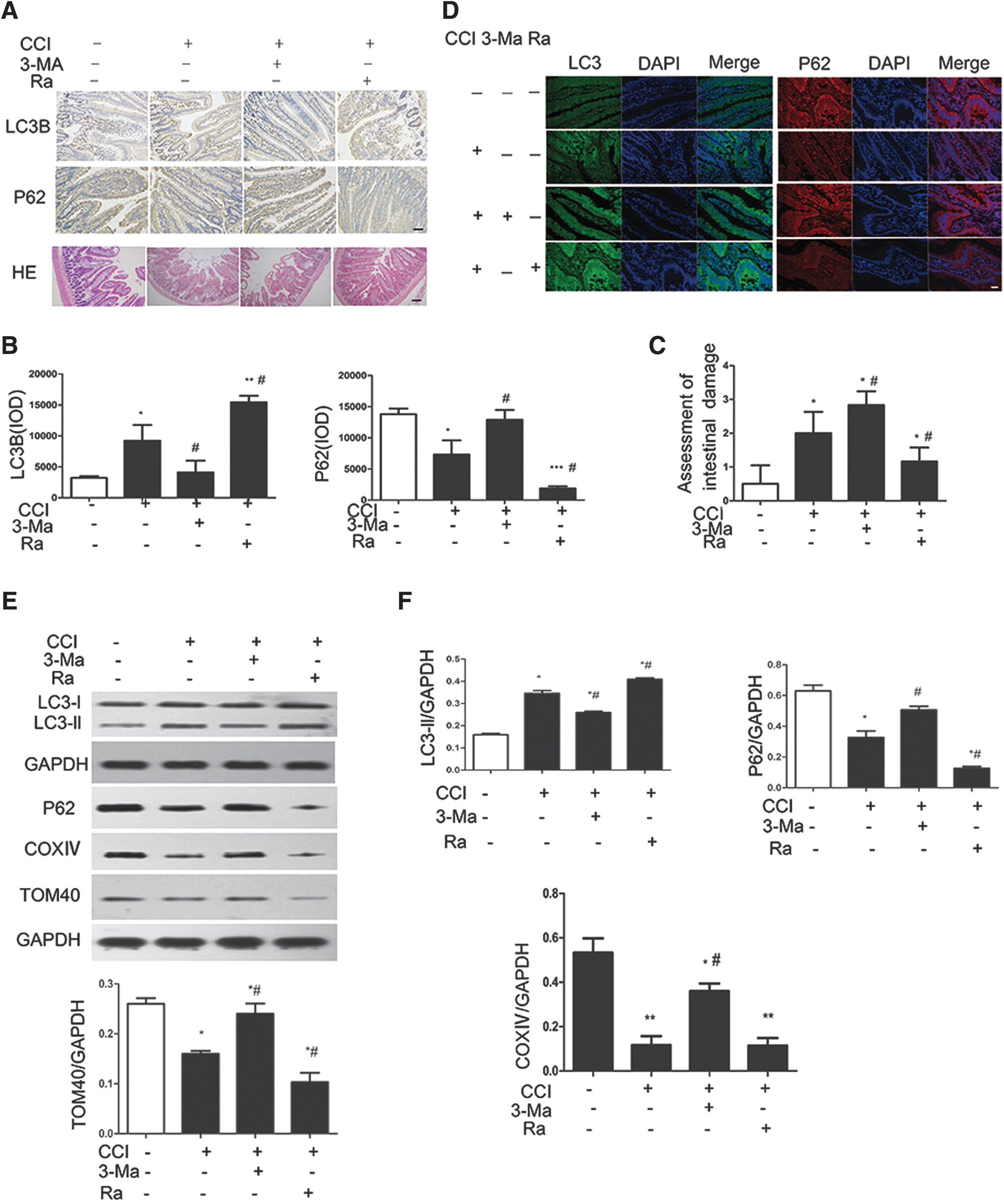
Mitophagy alleviates controlled cortical impact (CCI)-induced intestinal epithelial cell damage.
The intestinal mucosa is thought to deteriorate after TBI, leading to damaged barrier function in response to apoptosis of cells within the intestinal epithelium. To verify the protective effect of autophagy and to determine whether the intestinal damage with CCI is accompanied by increased levels of apoptosis, we performed TUNEL staining and immunohistochemical staining for cleaved caspase-3. TUNEL-positive enterocytes and caspase-3-positive enterocytes were rare in the ileum of control rats, but were more frequent in rats subjected to CCI; further, TUNEL-positive and caspase-3-positive staining at 1 day after CCI was enhanced by 3-Ma treatment of the rats, whereas Ra had the opposite effects (Fig. 5A and B). To further verify these findings, we performed Western analysis of small intestine lysates harvested from surviving rats 1 day after CCI. The levels of cleaved caspase-3 and the pro-apoptotic protein BAX increased after CCI, confirming that apoptosis of cells from the intestinal epithelium was induced. Further, the increased levels of these proteins were enhanced by 3-Ma and attenuated by Ra (Fig. 5C and D). Collectively, our results indicate that the enhanced mitophagy after TBI treatment alleviates epithelial cell damage associated with apoptosis.
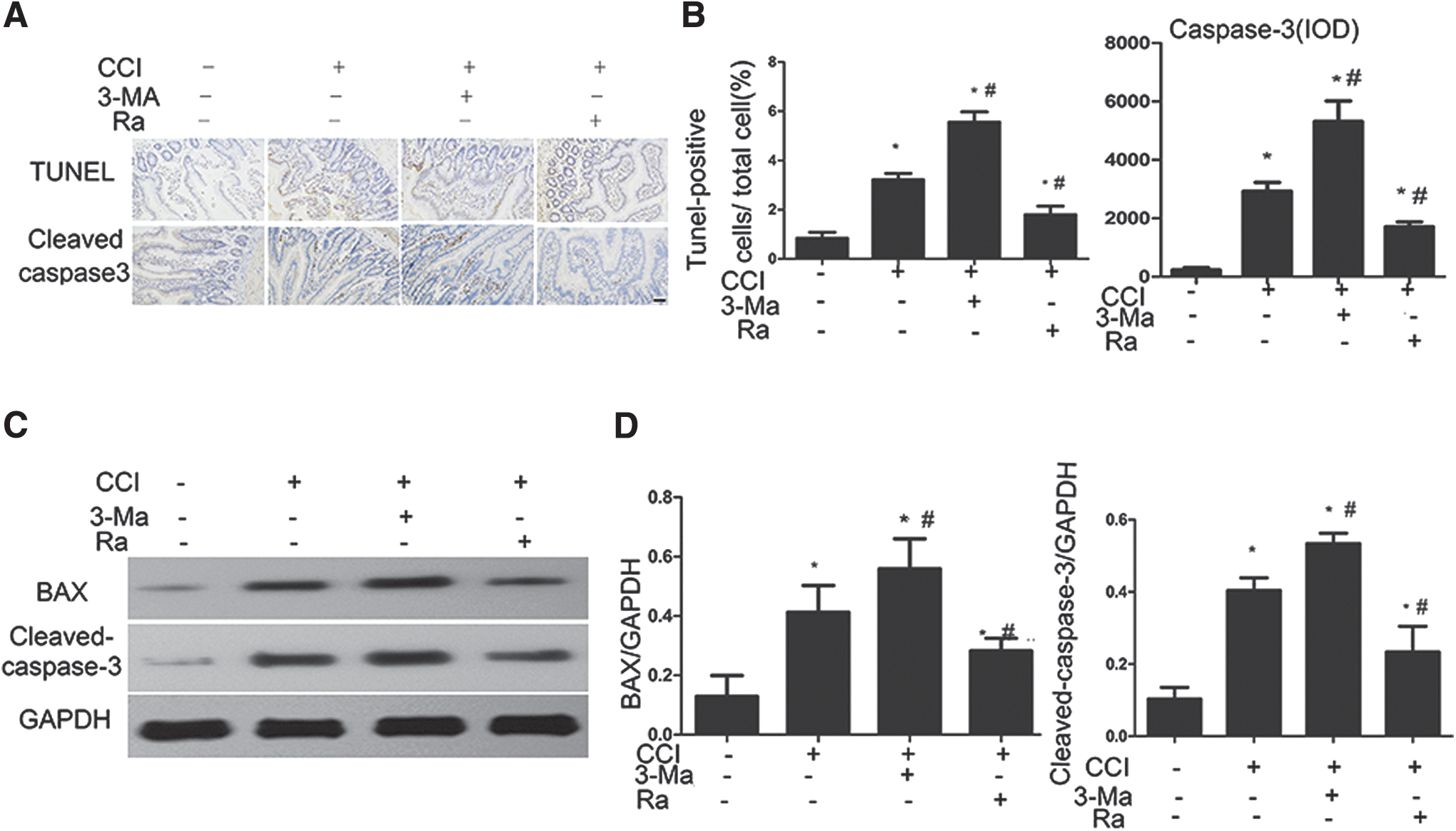
Mitophagy alleviates controlled cortical impact (CCI)-induced cell apoptosis.
Mitophagy activated by the extracellular signal-regulated kinase (ERK)/nuclear factor-erythroid2-like2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathway ameliorates TBI-induced intestinal barrier hyperpermeability and oxidative stress
Mitophagy plays an important role in the maintenance of cell homeostasis during nutrient deprivation, oxidative stress, and endoplasmic reticulum stress. 40 –42 To examine the mechanism by which mitophagy is induced after TBI treatment, we evaluated the effects of CCI on oxidative stress. The oxidative stress markers 3-NT and 4-HNE were significantly increased in CCI-treated rats compared with sham rats; further, Ra attenuated this increase, whereas 3-Ma enhanced the effects of CCI on both markers (Fig. 6A and B). These results indicate that autophagy may decrease the oxidative stress caused by mitochondrial disturbance after TBI.
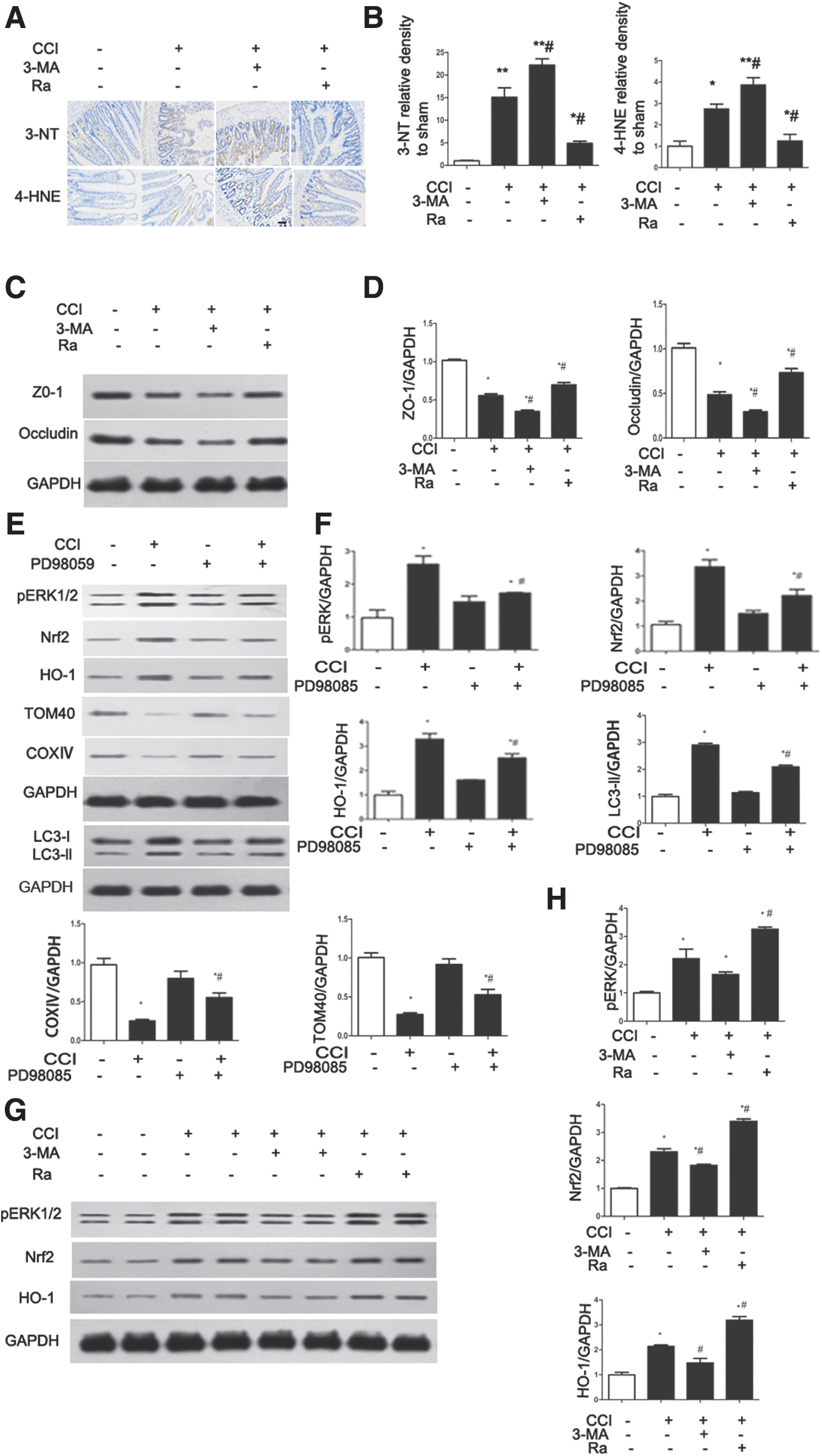
Mitophagy activated by extracellular signal-regulated kinase (ERK)/nuclear factor-erythroid2-like2 (Nrf2)/heme oxygenase-1 (HO-1) signaling ameliorates controlled cortical impact (CCI)-induced intestinal barrier hyperpermeability and oxidative stress.
The expression of the tight junction proteins ZO-1 and occludin is associated with intestinal epithelial permeability in response to oxidative stress. 43,44 To evaluate the levels of these proteins after CCI, we performed Western blotting. The expression of both proteins was reduced by CCI; further, the reduction in expression was reversed by Ra treatment and enhanced by 3-Ma (Fig. 6C and D). These results indicate that mitophagy ameliorates TBI-induced intestinal barrier hyperpermeability.
It has previously been reported that the expression of HO-1 is increased with oxidative stress-induced intestinal epithelial barrier dysfunction. 43 Further, HO-1 protects against hepatocyte cell death in experimental sepsis in vivo via the induction of autophagy. 45 To determine whether the ERK/Nrf2/HO-1 pathway might be activated by TBI and whether the ERK/Nrf2/HO-1 pathway can activate mitophagy after TBI, we performed Western blotting after CCI treatment. As shown in Figure 6E and F, ERK phosphorylation, Nrf2 expression, and HO-1 expression were upregulated, and mitophagy was suppressed by the ERK inhibitor PD98059 after CCI. Interestingly, ERK phosphorylation, Nrf2 expression, and HO-1 expression were further upregulated by Ra and suppressed by 3-Ma (Fig. 6G and H). Thus, the ERK/Nrf2/HO-1 pathway could activate mitophagy as a mechanism of protection against oxidative stress-induced cell death, resulting in the reduction of intestinal mucosa damage in rats after CCI.
Discussion
TBI causes significant impairment of the gastrointestinal system and is associated with ischemia of the intestinal mucosa, enhancement of vascular permeability, and damage to the tight junction of the intestinal epithelia. 4,10,11 These serious clinical events eventually may lead to high morbidity and mortality, 4,46 –48 and many patients with severe TBI often die as a result of multiple organ disfunction syndrome (MODS), rather than from the TBI itself. 9 For these reasons, gastrointestinal dysfunction caused by TBI is an increasingly recognized phenomenon and is an important current focus of research in the biomedical field. Evidence suggests that apoptosis of intestinal mucosal epithelial cells and attack by oxygen free radicals may contribute to stress damage of the intestinal mucosal barrier in the early stage of TBI. 49,50 Mitochondria are an important part of oxidative phosphorylation and energy supply, whereas oxygen radicals generated in mitochondria during oxidative phosphorylation and electron transport are in equilibrium under normal condition. Vast amounts of free radicals are produced through strong lipid peroxidation, which destroys the relative balance after TBI. In turn, increased oxidant production may further aggravate mitochondrial lesions, which lead to the generation of more oxygen radicals, which may explain why ERK phosphorylation, Nrf2 expression and HO-1 expression were further upregulated by Ra and were suppressed by 3-Ma, 16,22,51 which leads to the activation of the apoptotic signaling pathway. 52 Our study revealed the reduction of the matrix of the mitochondria, disruption of their cristae, and changes in respiratory chain complex I-IV activities following TBI, which are features of mitochondrial dysfunction. The activities of mitochondrial complexes I and II increased in the early stage of TBI, which may constitute a compensatory mechanism for energy requirements. It has been confirmed that mitochondrial complex III and IV activities decrease with age, 53,54 which probably explains why there were no differences in complex III and IV activities between the control and TBI groups. Additionally, some reports suggest that intestinal dysfunction is associated with damaged mitochondria and autophagy, 14,33,34,55 and damaged mitochondria can activate mitophagy. 29,31 However, little is known about the relationship between intestinal dysfunction and mitophagy induced by TBI.
In this study, we provide the first demonstration that TBI can induce mitophagy in ileum tissue, which is especially evident 24 h after CCI induction in rats. We also show for the first time that mitophagy attenuates TBI-induced intestinal damage, intestinal epithelial barrier dysfunction, and levels of apoptosis in the intestine. Finally, we provide evidence that the ERK/Nrf2/HO-1 pathway may play an important role in mitophagy-mediated protection during TBI-induced intestinal mucosal damage and intestinal epithelial barrier dysfunction. To the best of our knowledge, this is the first study exploring the role of mitophagy in regulating these key pathogenic events of oxidative stress and apoptosis in TBI-induced intestinal mucosal damage and intestinal epithelial barrier dysfunction.
Recent evidence suggests that mitophagy may serve as an adaptive response to stress, promoting cell survival and homeostasis by regulating energy generation and by clearing dysfunctional mitochondria that release pro-apoptotic factors, such as ROS and cytochrome c, after injury. 31,56 Damaged mitochondria in cells are degraded by a specific autophagy-lysosome pathway. 27 In our in vivo experiment, CCI is likely to induce mitophagy in response to the increase in dysfunctional mitochondria. 14,31 This induction of mitophagy may reduce TBI-induced intestinal mucosal damage and intestinal epithelial barrier dysfunction, protecting cell survival and barrier function, which is consistent with the effects on intestinal epithelial structure and apoptosis demonstrated in our study.
To examine the mechanism of mitophagy induction, we evaluated the effects on signaling pathways using Ra or 3-Ma to modulate the levels of CCI-induced mitophagy. As expected, the oxidative stress markers 3-NT and 4-HNE were increased in intestinal tissue after CCI. Further, the expression of these proteins was suppressed by autophagy activation with Ra, and enhanced by mitophagy inhibition with 3-Ma. Because mitochondria are regarded as a control point in the apoptosis pathway, 57 it is reasonable to postulate that mitophagy may regulate apoptosis. The mitochondria constitute the main ROS factory, and act as signaling mediators by oxidizing redox-sensitive cysteine residues and inhibiting protein phosphorylation. 58 Because high ROS levels can result in cell death, 59 oxidative stress caused by TBI may explain the apoptosis and epithelial structural damage induced by CCI in our study. Conversely, activation of mitophagy may serve to counteract CCI-induced intestinal damage and apoptosis by removing old and damaged mitochondria and, consequently, suppressing oxidative damage.
Oxidative stress after TBI also is reported to lead to intestinal epithelial hyperpermeability, which is associated with decreased expression of the tight junction proteins ZO-1 and occludin. 44,60 Intestinal permeability permits translocation of intestinal bacteria and endotoxin, which ultimately leads to SIRS and sepsis with subsequent multiple organ failure. We demonstrated that CCI leads to decreased expression of both ZO-1 and occludin, and that Ra attenuates this decrease whereas 3-Ma enhances this decrease. Therefore, the decrease in intestinal permeability may be a response to TBI-induced oxidative stress, and mitophagy may ameliorate the hyperpermeability by stabilizing ZO-1 and occludin expression.
The ERK/Nrf2/HO-1 signaling pathway is also known to be upregulated in situations of oxidative stress, 61,62 and, therefore, we further examined whether CCI regulates molecules within this pathway. Our results demonstrate that ERK phosphorylation is increased and the expression of Nrf2 and HO-1 are upregulated after CCI. Moreover, the levels of activation were proportional to the level of mitophagy, which may occur because more damaged mitochondria were generated by inhibiting mitophagy after CCI, leading to the production of more ROS that may conversely inhibit the ERK/Nrf2/HO-1 signaling pathway. 16,22,51 To determine if this pathway is upstream of mitophagy, animals were pretreated with an ERK inhibitor prior to CCI. The results suggest that CCI-induced mitophagy is mediated by the oxidative stress-related ERK/Nrf2/HO-1 signaling pathway. These findings also raise the possibility that mitophagy activated by the ERK/Nrf2/HO-1 pathway makes the intestinal mucosal more resistant to oxidative stress. Nrf2 is activated through upregulation of ERK phosphorylation 51 and plays an important role in cell protection by the induction of antioxidative enzymes such as HO-1, as well as of detoxifying enzymes such as glutathione-S-transferase mu1. 63 Therefore, Nrf2 activation might be the most important protective effect of mitophagy, and its altered levels support its role and potential consideration as a therapeutic target.
Conclusion
In conclusion, we have provided evidence for the involvement of mitophagy in the pathogenesis of gastrointestinal diseases induced by TBI in vivo. Our study demonstrates for the first time that mitophagy activated by the ERK/Nrf2/HO-1 pathway after TBI may alleviate intestinal mucosal damage and epithelial barrier dysfunction. The possible mechanism is depicted in Figure 7. Oxidative stress might be one of the most important factors associated with TBI-induced intestinal mucosal damage and barrier dysfunction. Therefore, activation of antioxidant signaling systems such as the ERK/Nrf2/HO-1 pathway could provide a rational therapeutic target for protecting the small intestine against TBI-induced injury.
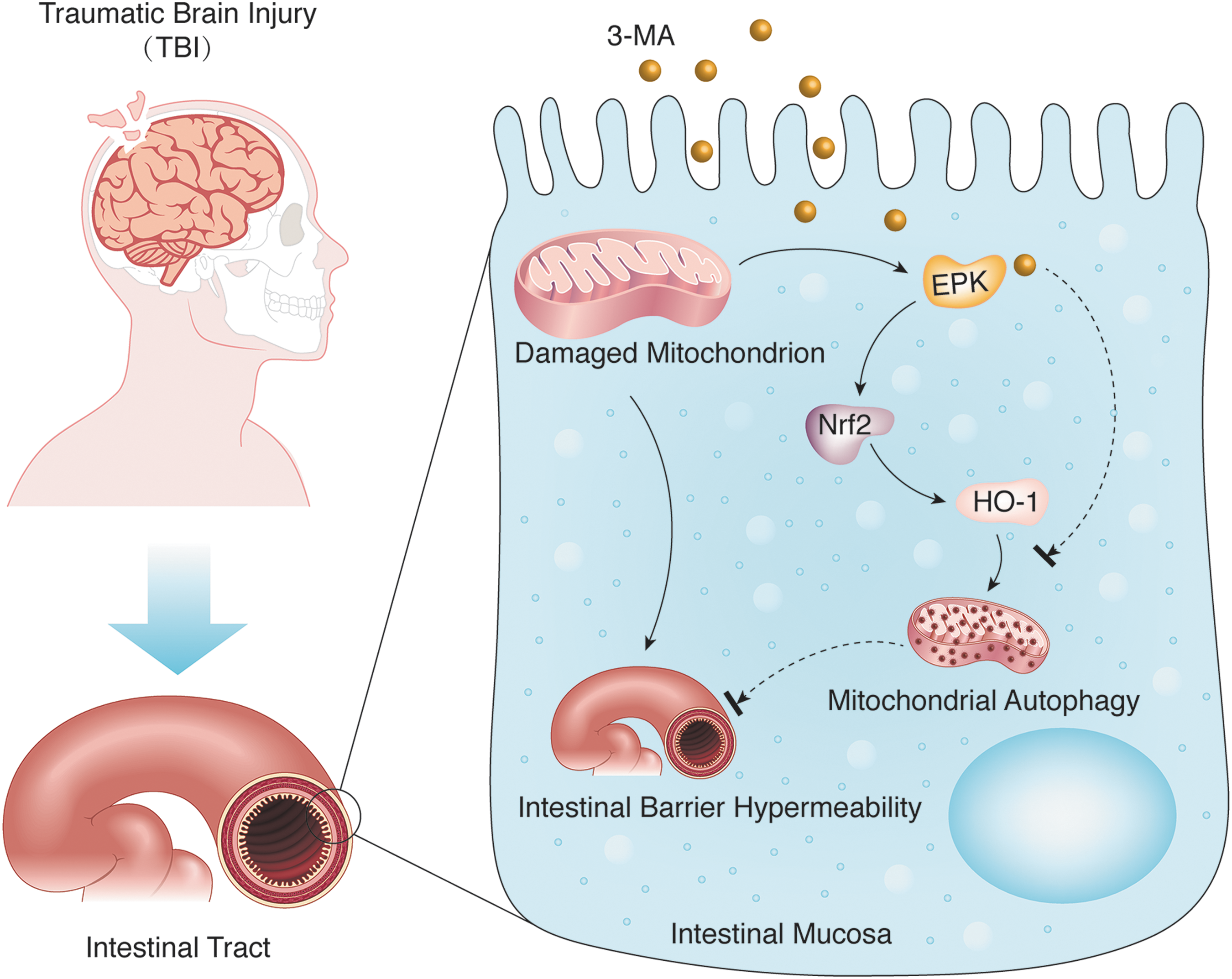
Model of intestinal mitophagy after traumatic brain injury (TBI). TBI induces intestinal mitochondrial injury. The damaged mitochondria activates the extracellular signal-regulated kinase (ERK)/nuclear factor-erythroid2-like2 (Nrf2)/heme oxygenase-1 (HO-1) pathway, which mediates selective autophagy to eliminate injured mitochondria. Our results suggest that mitophagy activated by the ERK/Nrf2/HO-1 pathway after TBI may alleviate intestinal mucosal damage and epithelial barrier dysfunction. Color image is available online at
Footnotes
Acknowledgments
This study was supported by grants from Jiangsu Province's Key Discipline of Medicine (XK201117). Jiangsu Province and the Priority Academic Program Development of Jiangsu Higher Education Institutions (2016, PAPD), Outstanding Youth of Jiangsu Province (BK20160047 and BK20160044), and National Natural Science Foundation of China (81471269, 81300998, and 31570881).
Author Disclosure Statement
No competing financial interests exist.