
Abstract
Modulation of N-acylethanolamine-hydrolyzing acid amidase (NAAA) represents a potential alternative strategy in the treatment of neuroinflammation. Recent studies showed that pharmacological modulation of NAAA could be achieved with the oxazoline of palmitoylethanolamide (PEA; PEA-OXA). The aim of this study was to evaluate the neuroprotective effects of PEA-OXA in the secondary neuroinflammatory events induced by spinal and brain trauma in mice. Animals were subjected to spinal cord and brain injury models and PEA-OXA (10 mg/kg) was administered both intraperitoneally and orally 1 h and 6 h after trauma. PEA-OXA treatment markedly reduced the histological alterations induced by spinal cord injury (SCI) and traumatic brain injury (TBI) and ameliorated the motor function and behavioral deficits, as well. In addition, the expression of neurotrophic factors, such as glial cell line-derived neurotrophic factor, brain-derived neurotrophic factor, and neurotrophin-3 were increased by PEA-OXA treatment. Moreover, PEA-OXA also significantly decreased glial fibrillary acidic protein hyperexpression, the nuclear translocation of nuclear factor (NF)-κB, phosphorylation of Ser536 on the NF-κB subunit p65, and degradation of IκB-α, as well as diminished the expression of pro-inflammatory mediators such as cyclooxygenase-2 (COX-2), inducible nitric oxide synthase, tumor necrosis factor (TNF)-α and interleukin (IL)-1β. The modulation of intracellular NAAA by PEA-OXA treatment could thus represent a novel therapy to control neuroinflammatory conditions associated with SCI and TBI.
Introduction
T
The use of anti-inflammatory drugs to treat CNS neuroinflammation at the clinical level has proven disappointing until now. Given this large unmet medical need, new therapeutic strategies are called for. Some drugs may demonstrate their therapeutic potential by modulating neuroinflammation rather than by their usually ascribed actions only. ALIAmides are endogenous N-acylethanolamines (NAEs) whose levels are principally regulated by enzymes involved in their formation and degradation. 6 N-palmitoylethanolamine (PEA) is considered to be the prototype ALIAmide, known for its anti-inflammatory, analgesic, and neuroprotective properties. PEA exerts its anti-inflammatory effects by downregulating mast-cell degranulation via an autacoid local inflammation antagonism (ALIA) effect. In our earlier studies we demonstrated the beneficial effects of PEA alone or associated with antioxidants, using several experimental models of neuroinflammation such as SCI, TBI, and stroke. 4,5,7
The inhibition of PEA degradation by targeting its catabolic enzyme, NAE-hydrolyzing acid amidase (NAAA) is an alternative strategy in the treatment of neuroinflammation. A variety of selective NAAA inhibitors have been described, 8 –10 which modulate responses induced by inflammatory stimuli 8,11 and elevate PEA levels in vitro. 8,11 Pharmacological blocking of fatty acid amide hydrolase (FAAH) or NAAA with non-physiological agents to increase endogenous NAE levels (e.g., PEA) may have disadvantages from a metabolic point of view. Indeed, blocking FAAH can result in paradoxical effects. 12,13 These observations may be due to the fact that, to exert a regulatory effect on non-neuronal cells (e.g., glia and mast cells) PEA pleiotropic effects 14 should be strongly controlled by a mechanism allowing for its inactivation. Moreover, protracted blocking of FAAH could also alter the endocannabinoid system by decreasing the levels of 2-arachidonoylglycerol while increasing those of anandamide.
Thus, we propose that pharmacologically modulating—and not blocking—the specific amidases for NAEs and, in particular, NAAA could be a viable strategy that preserves the PEA function in keeping cellular homeostasis through its fast on-demand synthesis and equivalently rapid degradation. The patent application (WO2013121449 A1) reported that pharmacological modulation of NAAA can be achieved with the oxazoline of PEA (PEA-OXA). 15 The identification of natural plant sources of PEA-OXA, synthesis, and therapeutic use in a model of carrageenan-induced inflammation was recently reported. 16 Based on these findings, the aim of this study was to evaluate the beneficial effects of PEA-OXA in the secondary neuroinflammatory events induced by SCI and TBI in mice.
Methods
Animals
Male Adult CD1 mice (25–30 g, Envigo, Italy) were posted in a controlled location with standard rodent chow and water. Animal care conformed with Italian regulations on protection of animals for experimental and scientific purpose (D.M. 116192) as well as with the EEC regulations (O.J. of E.C. L 358/1 12/18/1986).
SCI
Mice were anesthetized under intraperitoneal (i.p.) ketamine and xylazine (2.6 and 0.16 mg/kg body weight, respectively). We performed the clip compression model described by Rivlin and Tator 17 with the same surgical procedure previously described in our studies. 4
TBI
Anesthesia was provoked by ketamine and xylazine (2.6 and 0.16 mg/kg body weight, respectively) administered i.p. A cortical contusion in the right hemisphere was made on the exposed cortex by the use of a controlled impactor device (Impact One™ Stereotaxic Impactor for CCI,
Experimental groups
For SCI, mice were randomized into several groups of 10 mice each:
SCI + vehicle group: mice were subjected to SCI plus administration of vehicle (carboxymethylcellulose (CMC) 2.5% p/p in water);
SCI + PEA-OXA: as for the SCI + vehicle group but PEA-OXA was administered i.p. at 10 mg/kg dissolved in CMC (2.5% p/p in water) 1 h and 6 h after SCI;
SCI + PEA-OXA: as for the SCI + vehicle group but PEA-OXA was administered orally (o.s.) at 10 mg/kg dissolved in CMC (2.5% p/p in water) 1 h and 6 h after SCI;
Sham+ vehicle group: mice were subjected to the surgical procedures as above except that the aneurysm clip was not applied (only laminectomy) and they were treated i.p. or o.s. with vehicle;
Sham + PEA-OXA: identical to sham+ vehicle group except that PEA-OXA was administered i.p. or o.s. 1 h and 6 h after SCI.
As described below, mice (n = 10 from each group for each parameter; respectively 10 mice for immunohistochemistry and histological analyses and 10 mice for Western blot analyses for each experimental group) were sacrificed 24 h after SCI. In a separate set of experiments another 10 animals for each group were observed until 10 days after SCI in order to evaluate motor score.
For the TBI model, all animals were randomized in the indicated groups (10 mice for each group):
TBI + vehicle group: mice were subjected to CCI plus administration of vehicle (CMC, 2.5% p/p in water);
TBI + PEA-OXA: as for the TBI + vehicle group but PEA-OXA was administered i.p. at 10 mg/kg in CMC (2.5% p/p in water) 1 h and 6 h after TBI;
TBI + PEA-OXA: as for the TBI + vehicle group but PEA-OXA was administered o.s 10 mg/kg dissolved in CMC (2.5% p/p in water) 1 h and 6 h after TBI;
Sham+ vehicle group: mice were subjected to the surgical procedures as above except that the impact was not applied and animals were treated i.p. or o.s. with vehicle;
Sham + PEA-OXA: identical to sham + vehicle group except that PEA-OXA was administered i.p. or o.s. 1 h and 6 h after TBI.
As described below, mice (n = 10 from each group for each parameter; respectively 10 mice for immunohistochemistry and histological analyses and 10 mice for Western blot analyses for each experimental group) were sacrificed 24 h after TBI. In another set of experiments, 10 additional animals for each group were used for behavioral testing: elevated plus maze (EPM; 1, 2, 3, 6, and 10 days post-TBI), rotarod test (24 h after TBI), and the elevated body swing test (EBST, 24 h after TBI).
The dose and route of PEA-OXA administration was chosen based on a dose-response carried out in a previous study. 19 The timing of PEA-OXA administration was chosen based on our previous studies on spinal cord and brain trauma. 20,21
Synthesis of PEA-OXA
The synthesis of PEA-OXA was described in our recent study. 16 Under a nitrogen atmosphere, 3.0 g of N-(2-hydroxyethyl)palmitamide was suspended at 0°C in 20 mL of thionyl chloride and stirred first for 30 min at 0°C and then at room temperature for 15 h. The solution obtained was taken to dryness under vacuum, and the residue purified by crystallization from 15 mL of tert-butyl methyl ether, isolated and vacuum dried. The crystallized product was suspended in 20 mL of anhydrous toluene and 1.3 g of potassium tert-butoxide was then added. This mixture was heated at 40°C for 2 h and then cooled at 4°C. The resulting solution was extracted 3 times with 6 mL water and the washes were discarded. The resulting organic layer was dried under vacuum and the final residue was distilled under high vacuum at about 0.5 mm Hg. The fraction that distilled at about 175°C was separated, solidified at room temperature, and stored under nitrogen. The final yield of PEA-OXA was about 92%, with the following characteristics: molecular formula C18H35NO; C = 76.81%, H = 12.53%, N = 4.94%, O = 5.68%; Mr 281.5; ESI-MS: 282 (MH+); uncorrected melting point 46–48°C; solubility: poorly soluble in water, >10mg/mL in ethanol.
Behavioral testing
EPM
Anxiety is a prevalent neurobehavioral condition after TBI; it was evaluated using the EPM system as previously described. 5,20 In this test, anxiety is revealed by escaping of the open arms of the maze. 22 Behavior was captured with a camera fixed over the EPM. The percent of time spent in the open arms was scored by a researcher blinded to the treatment.
Rotarod test
Motor function and balance were examined in a blinded manner using an accelerating rotarod as described in our previous studies. 5
EBST
The EBST was performed to measure a motor asymmetry parameter by managing the animal by its tail and observing the direction of the biased body swings. EBST was expressed in percent to determine the biased swing activity. 5,23 The test was performed in a blinded manner.
Grading of motor disturbance
Motor function of SCI mice was evaluated daily for 10 days after injury. Recovery from motor disturbance was graded using the Basso Mouse Scale (BMS). 24 The BMS was performed in a blinded manner.
Quantification of infarct area and volume
Quantification of infarct area and volume was detected by 2,3,5-triphenyltetrazolium chloride (TTC) staining as previously described.
20
Brain slices were photographed by a digital camera (HP Photosmart R707, Milan, Italy) and image analysis was performed with an image analysis software program (Image J for Windows; National Institute of Mental Health, Bethesda, MD). The corrected infarct volume was calculated as:
Values are given as mean ± standard error of mean (SEM). The total infarct volume was analyzed by summing the infarct area in each slice and multiplying by slice thickness (2 mm).
Light microscopy, lesion size, and histological score
Spinal cord and brain tissues from the peri-lesional area (in the area at the boundary between the necrotic core and the penumbra) were taken at 24 h following trauma. Tissue segments containing the lesion (1 cm on each side of the lesion) were paraffin embedded and cut into 5-μm-thick sections. Tissue sections (thickness 5 μm) were deparaffinized with xylene, stained with hematoxylin/eosin, and studied using light microscopy (Dialux 22, Leitz). Histological analyses were performed as previously described. 20,21 Sections were evaluated by an experienced histopathologist based on a 6-point scale: 0, normal; 1, 1–5 eosinophilic neurons within the gray matter area; 2, 5–10 eosinophilic neurons; 3, >10 eosinophilic neurons; 4, a small infarction (less than one-third of the gray matter area); 5, a moderate infarction (one-third to one-half of the gray matter area); and 6, a large infarction (more than half of the gray matter area). The scores from all slides were averaged to give a final score for each individual mouse. All histological studies were performed in a blinded fashion. For hematoxylin/eosin stained sections, lesions were measured (mm2) using a reticule and a micrometer bar posted into the ocular of a light microscope and the means were calculated.
Immunohistochemical localization of brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), neurotrophin-3 (NT-3), tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and glial fibrillary acidic protein (GFAP)
At 24 h after SCI and TBI, immunohistochemical localization of BDNF, GDNF, NT-3, TNF-α, IL-1β, and GFAP was performed as described earlier. 20,21 Sections were incubated overnight at 4°C with one of the following primary antibodies (all from Santa Cruz Biotechnology, Santa Cruz, CA): anti-BDNF (Cat. No. sc20981; 1:500 in phosphate buffered saline [PBS], v/v); anti-GDNF (Cat. No. sc328; 1:500 in PBS, v/v); anti-NT-3 (Cat. No. sc547; 1:500 in PBS, v/v); anti-GFAP (Cat. No. sc9065; 1:500), anti-TNF-α (Cat. No. sc1350; 1:500), and anti-IL-1β (Cat. No. sc7884; 1:500). Sections were washed with PBS and then incubated with secondary antibody. Specific labeling was detected with a biotin-conjugated goat anti-rabbit immunoglobulin G (IgG) and a vidin-biotin peroxidase complex (Vector; DBA, Milan, Italy). The counter stain was developed with diaminobenzidine (brown color) and nuclear fast red (red background). Positive staining (brown color) was found in the sections, indicating that positive immunoreaction. The photographs obtained (n = 5 photos from each sample collected from all mice in each experimental group) of the perilesional area were assessed by densitometry by using Leica QWin V3, U.K.). The immunohistochemical analyses were performed in a blinded manner.
Western blot analysis for IκB-α, nuclear factor-κB (NF-κB) p65, p-NF-κBp65Ser536, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2)
Cytosolic and nuclear extracts were prepared as previously described. 25 The following antibodies were used: anti-IκB-α (1:1000; Santa Cruz Biotechnology Cat. No. sc371), anti-COX-2 (1:500; Santa Cruz Biotechnology Cat. No. sc1746), anti-p-NF-κBp65Ser536 (1:500; Santa Cruz Biotechnology Cat. No. sc33020), anti-NF-κB p65 (1:1000; Santa Cruz Biotechnology Cat. No. sc8008), and anti-iNOS (1:200; BD transduction Cat. No. 610432) in 1 × PBS, 5% w/v non-fat dried milk, 0.1% Tween-20 (PMT) at 4°C overnight. Membranes were incubated with peroxidase-conjugated bovine anti-mouse IgG secondary antibody or peroxidase-conjugated goat anti-rabbit IgG (1:2000, Jackson ImmunoResearch, West Grove, PA) for 1 h at room temperature. To ascertain that blots were loaded with equal amounts of lysate, they were also probed with anti-β-actin or anti-lamin A/C antibodies. Relative expression of protein bands (IkB-α, 37 kDa; NF-κB p65, 65 kDa; iNOS, 130 kDa; COX-2, 72 kDa) was detected by enhanced chemiluminescence (ECL; Thermo, Waltham, MA), visualized with ChemiDoc XRS (Bio-Rad, Hercules, CA) and analyzed using Image Lab 3.0 software (Bio-Rad).
Materials
PEA-OXA was obtained from Epitech group S.p.A. All chemicals were purchased from Sigma-Aldrich Company Ltd. (Poole, Dorset, U.K.). All stock solutions were made in non-pyrogenic saline (0.9% NaCl; Baxter, Italy, U.K.).
Statistical analysis
All values are expressed as mean ± SEM of n observations. For in vivo studies, n represents the number of animals used. For histology or immunohistochemistry analyses, the figures presented are representative of at least three experiments (histological or immunohistochemistry coloration) performed on different days on tissue sections collected from all animals in each group. Results were analyzed by one-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test for multiple comparisons. For one-way ANOVA statistic testing, a single “F” value indicated as variation between sample means/variation within the samples was shown; p value of <0.05 was considered significant. BMS data were analyzed by two-way ANOVA followed by Bonferroni post-tests and considered significant when p was <0.05.
Results
PEA-OXA reduces severity of spinal cord trauma
Significant histological alterations were observed in mice subjected to SCI at 24 h. Spinal cord trauma, evaluated by hematoxylin/eosin staining displayed, in the peri-lesional area, edema, leukocyte infiltration, and white matter alterations when compared with sham animals (Fig 1A,B, Fig. 1A1,B1, and Fig. 1E, p < 0.001; F = 54.02). PEA-OXA treatment (10 mg/kg i.p. or o.s.) reduced histological damage in SCI mice (Fig.1C,C1 and Fig. 1D,D1, p < 0.01; F = 54.02). No significant differences were found between oral or intraperitoneal PEA-OXA administration in SCI mice (Fig. 1C,C1 and Fig. 1D,D1; histological score in Fig. 1E; F = 54.02). In addition, quantification of the lesion sizes showed lesions in SCI-subjected mice compared with controls (0.23 ± 0.013 mm2; Fig. 1F, p < 0.001; F = 129.2). PEA-OXA-treated mice showed less lesion sizes compared with vehicle groups (0.10 ± 0.003 mm2; 0.12 ± 0.008 mm2; Fig. 1F, p < 0.001; F = 129.2). Moreover, BMS score was used to assess whether histological injury to the spinal cord was correlated with loss of motor activity. Motor function was not impaired in sham groups, whereas mice subjected to SCI showed significant deficits in movement (Fig. 1G). PEA-OXA treatment (10 mg/kg i.p.) ameliorated the functional deficits induced by SCI (Fig. 1G, p < 0.05, p < 0.01). No significant difference was found between intraperitoneal or oral administration (Fig. 1G).
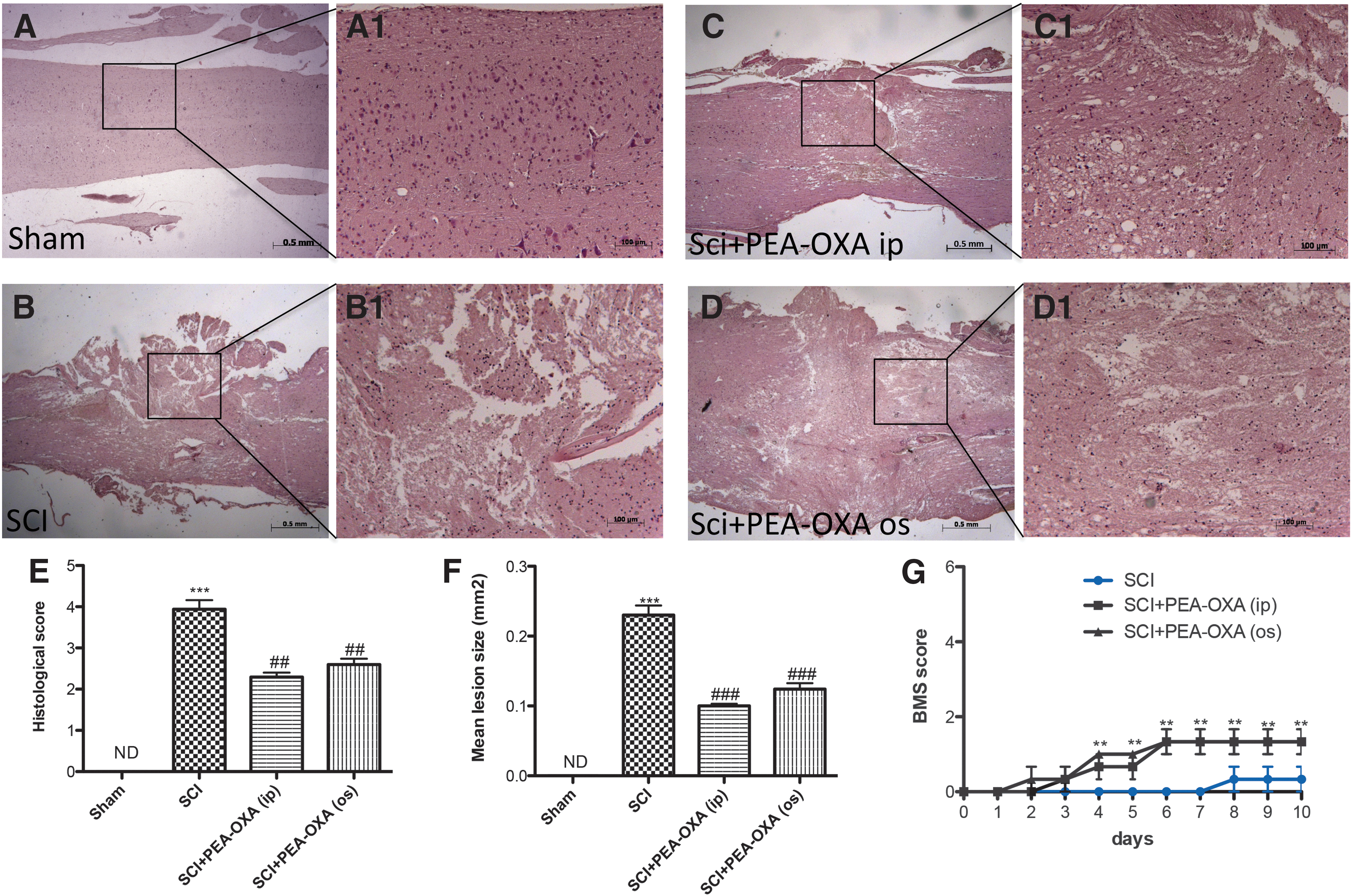
PEA-OXA reduces histological damage in SCI mice. No histological alterations were observed in spinal cord of sham-operated mice (
PEA-OXA reduces severity of brain trauma
Brain sections from TBI mice likewise showed, in the peri-lesional area significant tissue damage, ischemic changes, thickened blood vessels, and gliosis in the brain parenchyma (Fig. 2A,B, Fig. 2A1,B1, and Fig. 2E, p < 0.001; F = 127.1). PEA-OXA treatment (10 mg/kg i.p. or o.s.) reduced histological damage in TBI mice (Fig.2C,C1 and Fig. 2D,D1, p < 0.01 and p < 0.001; F = 127.1). However, the i.p. administration of PEA-OXA was more efficacious than when given orally (Fig. 2C,C1 and Fig. 2D,D1; histological score in Fig. 2E, p < 0.001; F = 127.1). For this reason we elected to limit the data obtained to those obtained by i.p. administration.
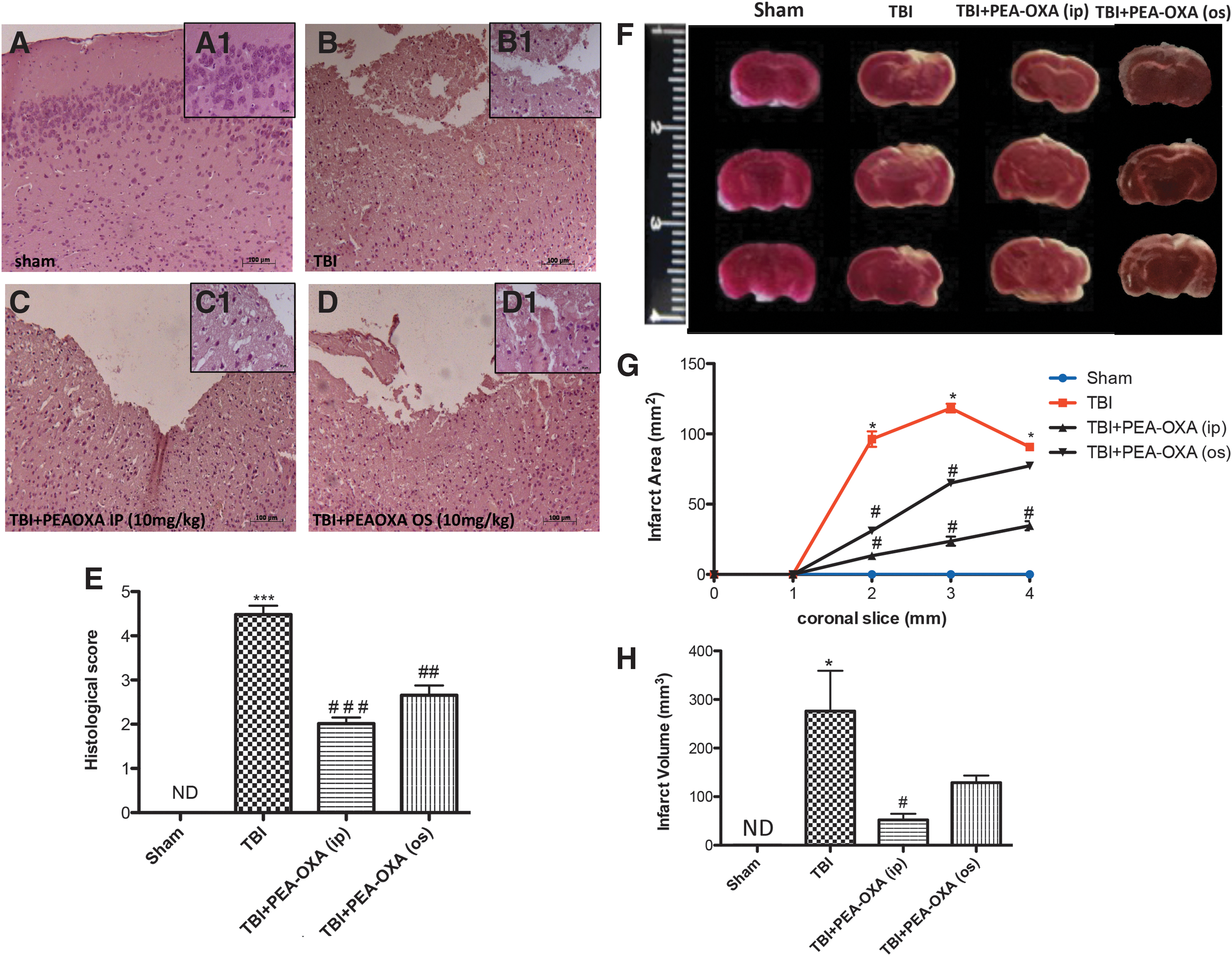
PEA-OXA reduces histological damage and brain infarct area and volume in TBI mice. Brain sections from TBI mice (
PEA-OXA reduced brain infarction following TBI
Measurement of brain infarct size is important when evaluating ischemic injury after trauma. To examine the effect of PEA-OXA on brain infarct after TBI, brain sections were subjected to TTC staining (Fig. 2F). At 24 h after TBI, infarct area (white color) was greater than in sham animals (Fig. 2F). Infarct area (Fig. 2G, p < 0.05) and volume (Fig. 2H, p < 0.05; F = 7.921) were reduced in a more significant way after i.p. treatment with PEA-OXA (Fig. 2G,H).
PEA-OXA ameliorates behavioral changes in TBI-subjected mice
To investigate whether histological brain damage was also correlated with behavioral changes, mice subjected to moderate brain injury were exposed to behavioral tests. In particular, to assess motor function, the animals were subjected to the EBST and rotarod test at 24 h after TBI. CCI-injured mice exhibited a range of impairments in locomotor tasks compared with sham (Fig. 3A, p < 0.01 and Fig. 3B, p < 0.05). Intraperitoneal treatment with PEA-OXA (10 mg/kg i.p.) clearly improved latency in comparison with the TBI + vehicle group in a more significant way compared with oral administration (Fig. 3A, p < 0.01 and Fig. 3B, p < 0.05). Because anxiety is considered a serious consequence of brain injury, CCI-injured mice were subjected to EPM at different time-points (Fig. 3C, p < 0.01). Daily intraperitoneal treatment with PEA-OXA markedly improved latency as compared with TBI-only mice in a more significant way than oral dose (Fig. 3C, p < 0.05).
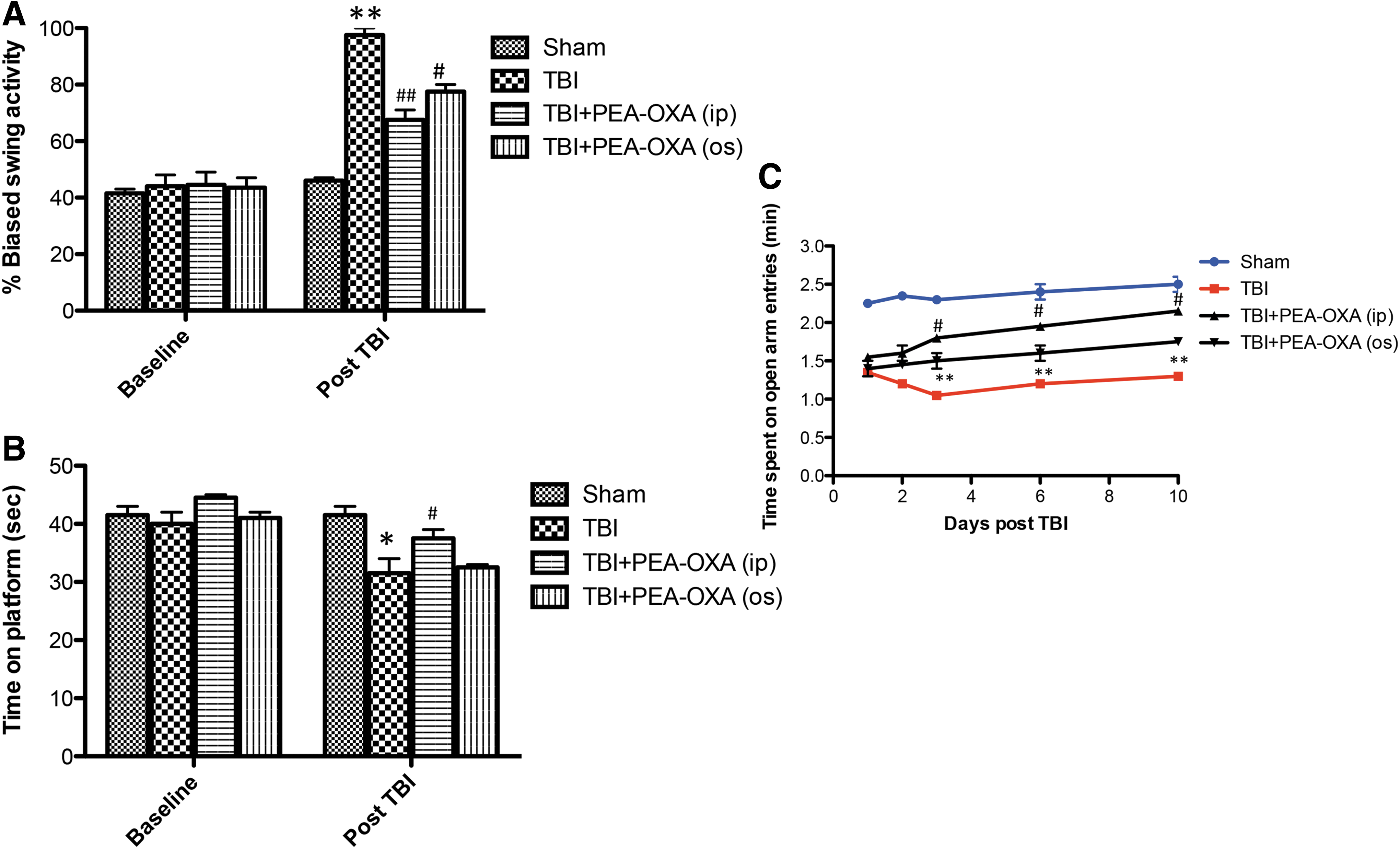
PEA-OXA ameliorates behavioral deficits in TBI mice. TBI caused a range of impairments in locomotor activity, as shown by biased swing activity
Effect of PEA-OXA on neurotrophic factor levels after spinal cord and brain trauma
Neurotrophic factors, such as GDNF, BDNF, and NT-3 have potent survival-promoting effects on various neuronal populations. We evaluated GDNF, BDNF, and NT-3 expression by immunohistochemical analysis in SCI and TBI mice. A positive staining for GDNF was found in spinal cord (Fig. 4 D,D1 and Fig. 4L, p < 0.01; F = 15.97) and in brains of sham mice (Fig. 5D,D1 and Fig. 5L, p < 0.001; F = 27.20). A positive staining for BDNF was found in spinal cord (Fig.4A,A1 and Fig. 4L, p < 0.01; F = 12.59) and in brains of sham mice (Fig. 5A,A1 and Fig. 5L, p < 0.001; F = 41.88). A positive staining for NT-3 was found in spinal cord (Fig.4G,G1 and Fig. 4L, p < 0.01; F = 17.71) and in brains of sham mice (Fig. 5G,G1 and Fig. 5L, p < 0.001; F = 48.11).
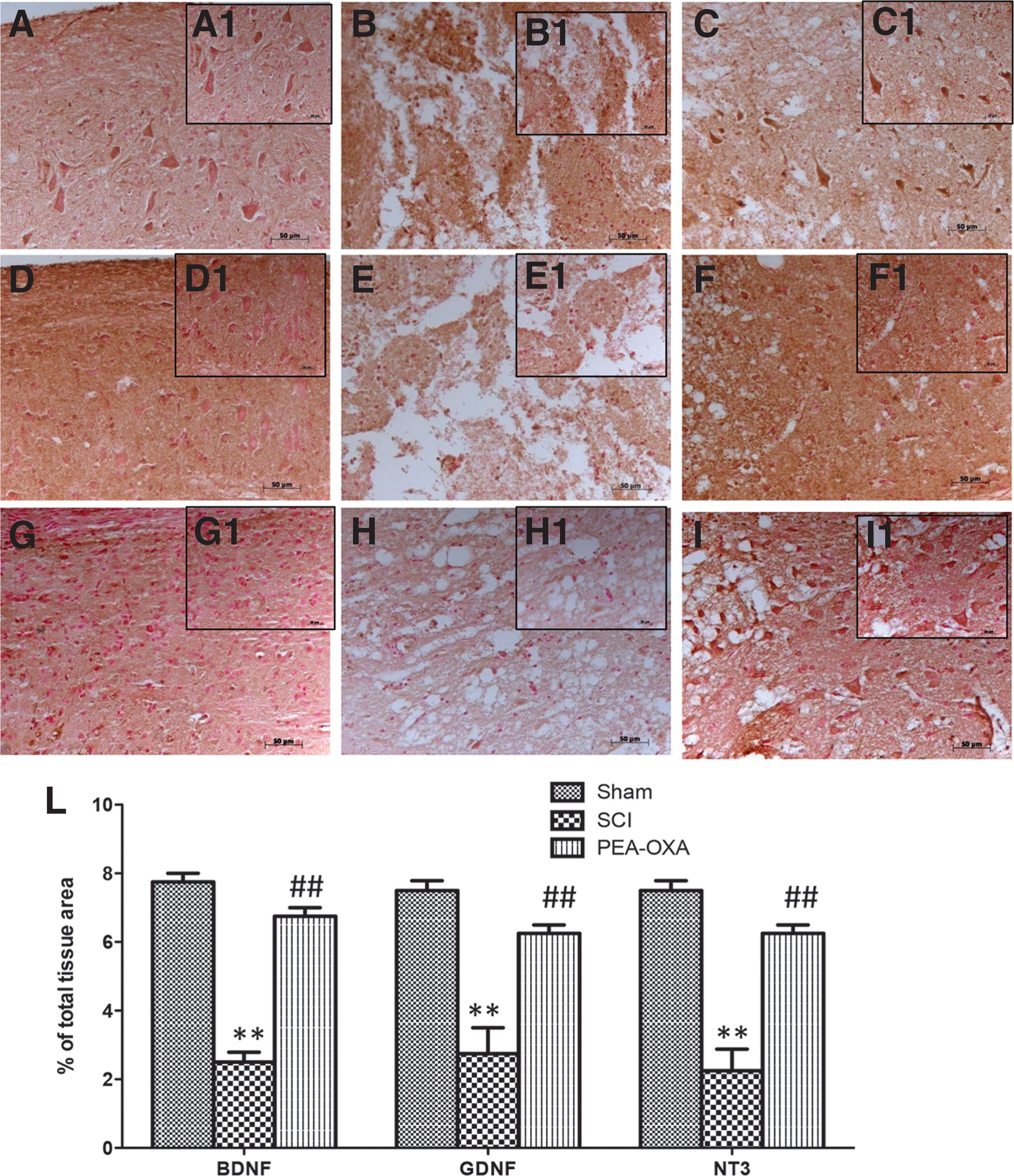
PEA-OXA increases BDNF, GDNF, and NT-3 expression after SCI. A reduction in neurotrophic factor expression, such as BDNF
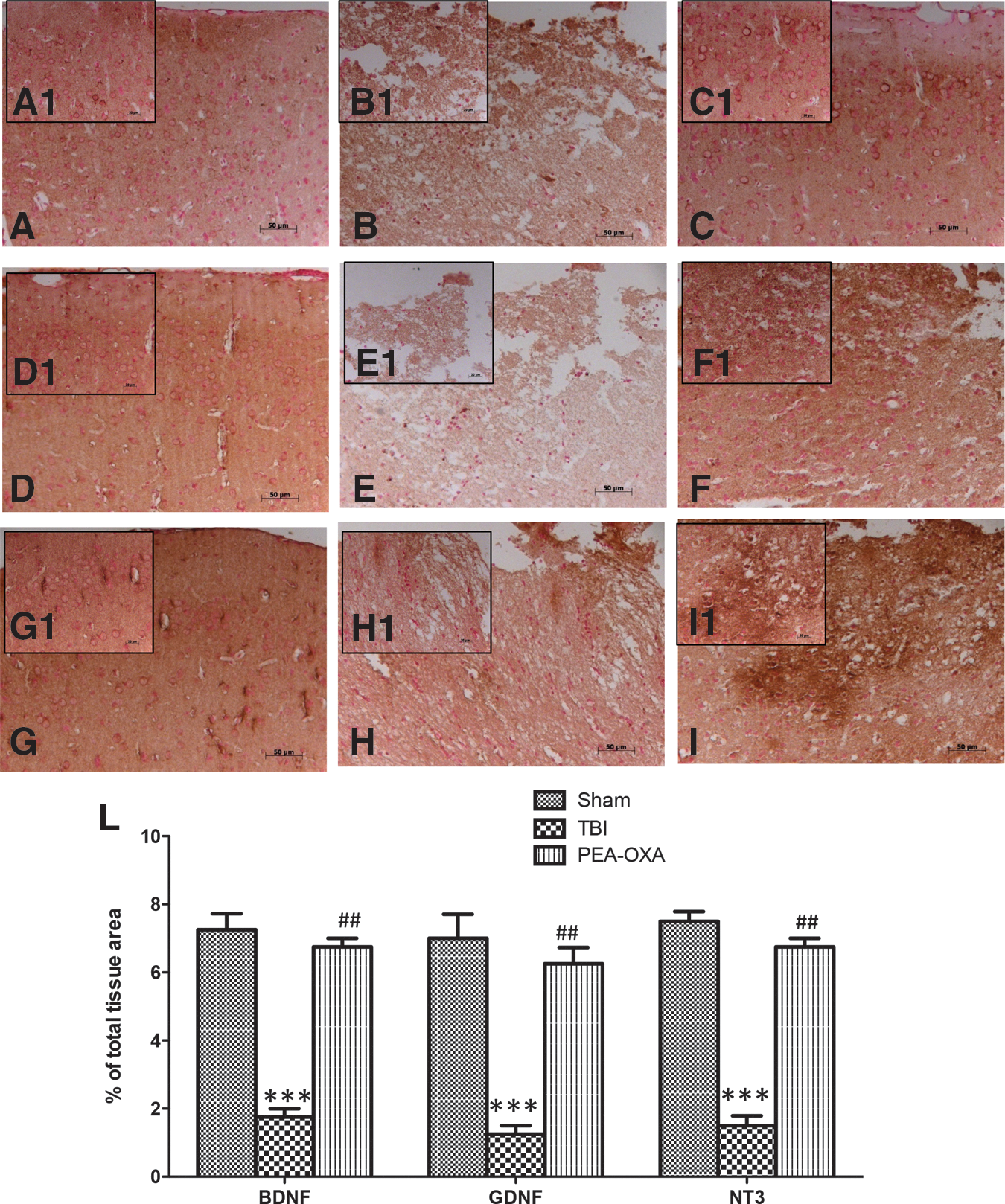
PEA-OXA increases BDNF, GDNF, and NT-3 expression after TBI. Reduced neurotrophic factor expression such as BDNF (
Spinal cord sections from SCI mice exhibited a reduction of about 4 times for immunostaining of GDNF, BDNF, and NT-3 compared with sham groups (Fig. 4B,B1, Fig. 4E,E1, Fig. 4H,H1, and Fig. 4L, p < 0.01; F = 15.97; F = 12.59; F = 17.71). Brain sections from TBI mice exhibited a reduction of about 4 times for immunostaining of GDNF, BDNF, and NT-3 compared with sham groups (Fig. 5B,B1, Fig. 5E,E1, Fig. H,H1, and Fig. 5L, p < 0.001; F = 27.20; F = 41.88; F = 48.11). PEA-OXA-treated mice displayed a significant increase of about 3 times for positive immunostaining of neurotrophic factors in the spinal cord sections (Fig. 4C,C1, Fig. 4F,F1, Fig. 4I,I1, and Fig. 4L, p < 0.01; F = 15.97; F = 12.59; F = 17.71) as well as an increase of about 4 times in brain sections (Fig. 5C,C1, Fig. 5F,F1, Fig. 5I,I1, and Fig. 5L, p < 0.01; F = 27.20; F = 41.88; F = 48.11).
Effect of PEA-OXA on GFAP expression after spinal cord and brain trauma
Astrocytes are the main glial cell population within the CNS. After activation, astrocytes can secrete various neurotoxic substances and express an increased level of GFAP, which is a marker for astrogliosis. 26 At 24 h after trauma GFAP, which has been implicated in the pathogenesis of spinal cord and brain trauma, was measured immunohistochemically. A significant increase of about four-fold in GFAP staining was found in tissue sections from mice subjected to SCI or TBI (Fig. 6B,B1, Fig. 6E,E1; see Fig. 6G, p < 0.001, F = 64.24 and Fig. 6H, p < 0.001, F = 167.7) compared with sham mice (Fig. 6A,A1, Fig. 6D,D1; see Fig. 6G,H). Positive immunostaining for GFAP was significantly decreased about 3 times in spinal cord and brain sections of PEA-OXA-treated mice (Fig. 6C,C1 and Fig. 6F,F1; see Fig. 6G, p < 0.01, F = 64.24, and Fig. 6H, p < 0.001, F = 167.7).

PEA-OXA increases GFAP expression after trauma. Increased GFAP expression was observed in mice subjected to spinal cord trauma (
Effect of PEA-OXA on pro-inflammatory cytokine levels after SCI and TBI
To test whether PEA-OXA could modulate inflammation by regulating secretion of pro-inflammatory cytokines, we analysed spinal cord and brain levels of TNF-α and IL-1β by immunohistochemical means. A substantial positive immunostaining of about 5 times for TNF-α and IL-1β was observed in spinal cord tissues from SCI mice (Fig. 7B,B1, Fig. 7E,E1, and Fig. 7G, p < 0.001; F = 37.77; F = 45.41) compared with controls (Fig. 7A,A1, Fig. 7D,D1, and Fig. 7G; F = 37.77; F = 45.41). In the same time, brain tissues from TBI mice 24 h after trauma showed an increased five-fold immunostaining for TNF-α and IL-1β (Fig. 8B,B1, Fig. 8E,E1, and Fig. 8G, p < 0.001; F = 43.17; F = 47.87) compared with sham mice (Fig. 8A,A1, Fig. 8D,D1, and Fig. 8G; F = 43.17; F = 47.87). Spinal cord of TNF-α and IL-1β (Fig. 7C,C1, Fig. 7F,F1, and Fig. 7G, p < 0.01; F = 37.77; F = 45.41) as well as brain levels of TNF-α and IL-1β (Fig. 8C,C1, Fig. 8F,F1, and Fig. 8G, p < 0.01; F = 43.17; F = 47.87) were significantly attenuated about 3 times by i.p. injection of PEA-OXA.
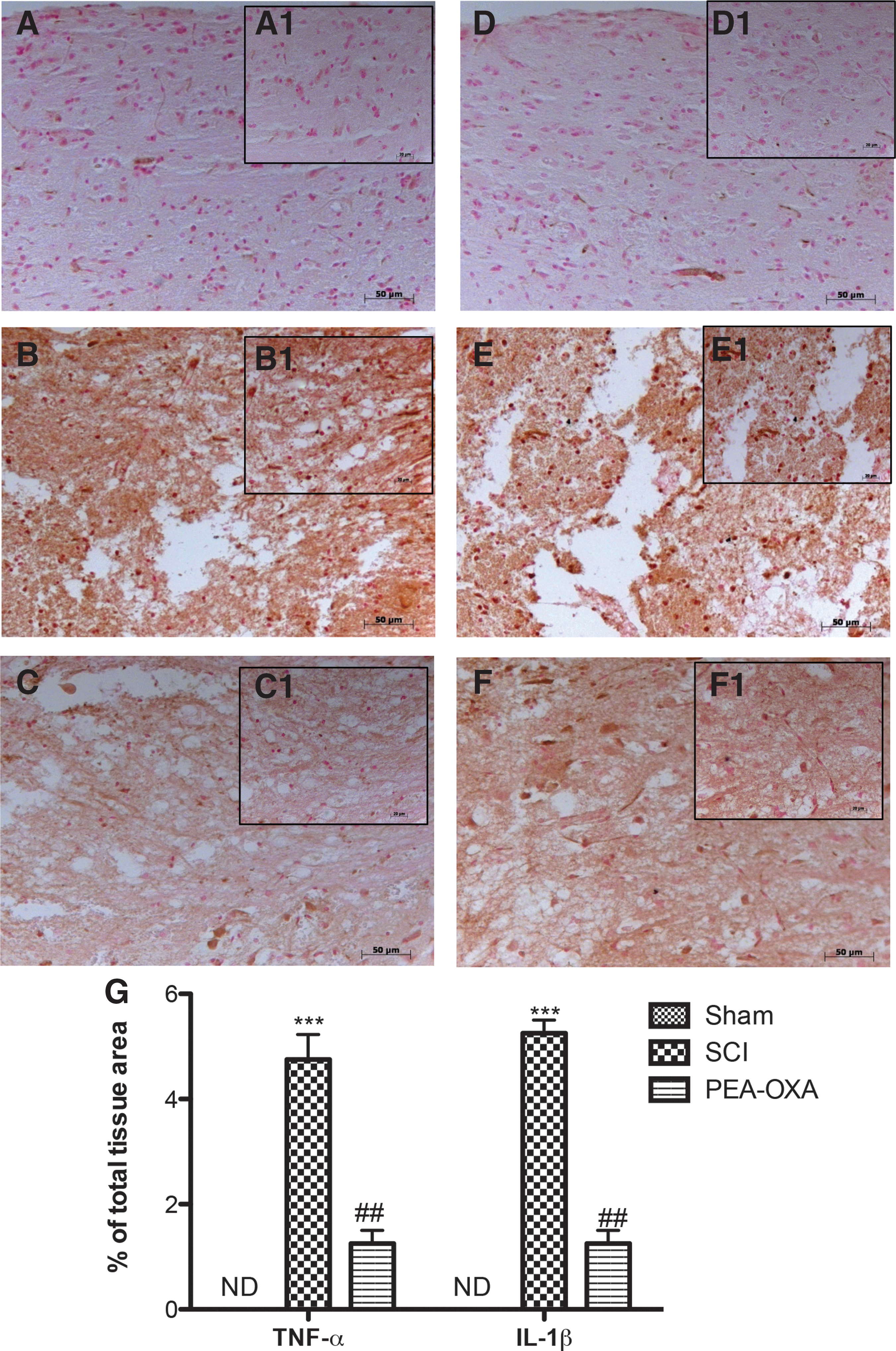
PEA-OXA reduces TNF-α and IL-1β expressions after SCI. An increased expression of TNF-α (
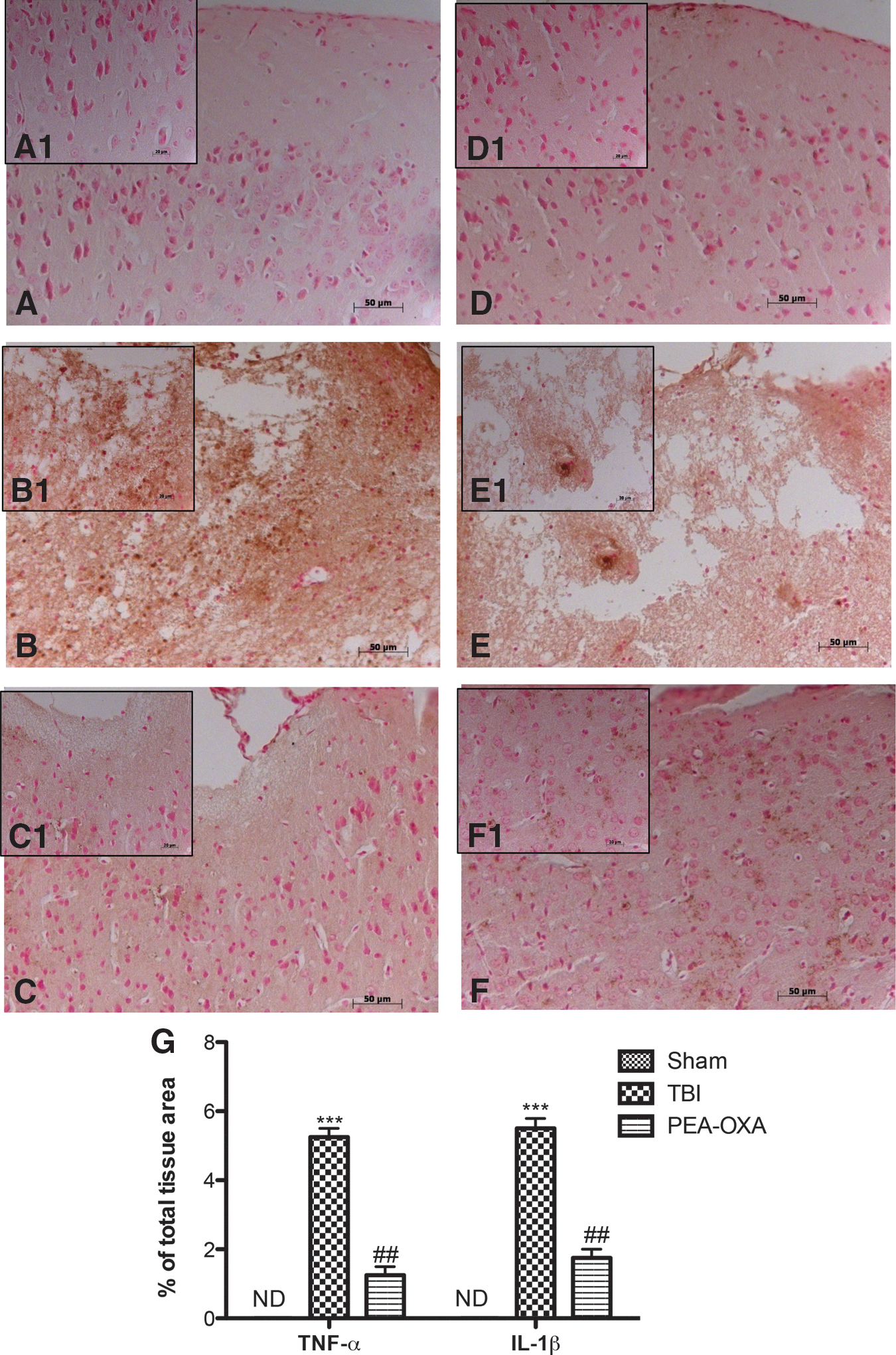
PEA-OXA reduces TNF-α and IL-1β expression after TBI. An increased expression of TNF-α (
Effect of PEA-OXA on IκB-α degradation and NF-κB p65 activation
By Western blot analysis, we evaluated the phosphorylation of Ser536 on the NF-κB subunit p65 and nuclear levels of NF-κB p65 to investigate the cellular mechanisms by which treatment may attenuate the development of SCI and TBI. SCI caused a significant increase in the phosphorylation of NF-κB p65 on Ser536 and increased nuclear levels of NF-κBp65 (respectively 80.85% and 60.73%) at 24 h after the injury compared with sham animals (Fig. 9B,C, p < 0.05; F = 11.56; F = 7.955). In addition, TBI also caused a significant increase in the phosphorylation of NF-κB p65 on Ser536 and increased nuclear levels of NF-κBp65 (respectively 75.55% and 68.97%) at 24 h after the injury compared with sham animals (Fig. 10B,C, p < 0.05; F = 10.41; F = 9.086). The treatment with PEA-OXA prevented the activation of NF-κB by reducing the phosphorylation of NF-κB p65 on Ser536 and nuclear levels of NF-κB p65 (respectively 52.42% and 50.72% in spinal cord tissues; 62.23% and 52.79% in brain tissues; Fig. 9 and Fig.10B,C, p < 0.05, F = 11.56, F = 7.955 and p < 0.05, F = 10.41, F = 9.086). In addition, a basal level of IκB-α was observed in the spinal cord and brain tissues from sham animals (Fig. 9 and Fig. 10A; F = 29.13; F = 17.58), whereas IκB-α levels were substantially decreased in SCI and TBI mice (respectively 66.95% and 47.97%; Fig. 9 and Fig. 10A, p < 0.001, p < 0.01; F = 29.13; F = 17.58) compared with sham animals. Intraperitoneal administration of PEA-OXA prevented trauma-induced IκB-α degradation by increasing IκB-α levels (respectively 32.85% and 34.46%; Fig. 9 and Fig. 10A, p < 0.05; F = 29.13; F = 17.58) compared with vehicle groups.
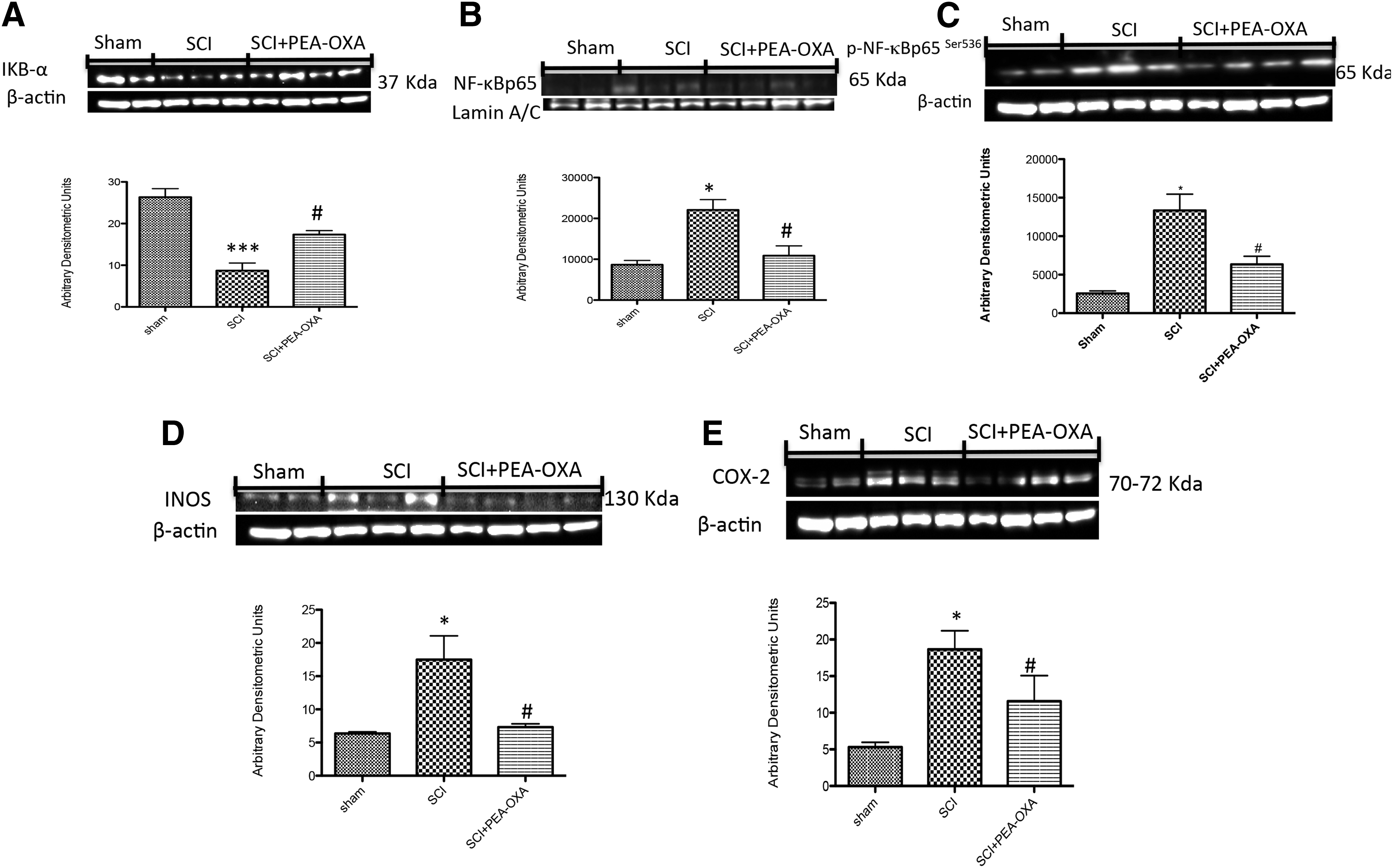
Effect of PEA-OXA on NF-κB pathway after SCI. The degradation of IκB-α was significantly blocked by PEA-OXA treatment in spinal cord tissues

Effect of PEA-OXA on NF-κB pathway after TBI. The degradation of IκB-α was significantly blocked by PEA-OXA treatment in brain tissues
Effects of PEA-OXA on iNOS and COX-2 expression
Many studies have demonstrated that NF-κB is involved in the regulation of COX-2 and iNOS expression. Several anti-inflammatory compounds have been shown to inhibit COX-2 and iNOS expression by blocking NF-κB activation. 27 In that regard, we also evaluated the expression of COX-2 and iNOS by Western blot analysis. At 24 h after SCI or TBI, the expression of iNOS and COX-2 in spinal cord and brain tissues was investigated by Western blot. iNOS and COX-2 levels were markedly increased (respectively 63.64% and 71.53%) in spinal cord tissues from mice subjected to trauma compared with sham animals (Fig. 9D,E, p < 0.05; F = 8.307; F = 10.69). Moreover, iNOS and COX-2 levels were also increased (53.62% and 88.11%) in brain tissues from mice subjected to trauma compared with sham animals (Fig. 10D,E, p < 0.01, p < 0.001; F = 21.35; F = 72.42). On the contrary, i.p. administration of PEA-OXA reduced (respectively 58.17% and 46.58%) the trauma-induced iNOS and COX-2 expression (Fig. 9D,E, p < 0.05; F = 8.307; F = 10.69) in spinal cord tissues compared with vehicle groups. PEA-OXA treatment also diminished (respectively 53% and 77.47%) iNOS and COX-2 expressions (Fig. 10D,E, p < 0.01, p < 0.001; F = 21.35; F = 72.42) in brain tissues compared with vehicle groups.
Discussion
Neuroinflammation plays a crucial role in the pathophysiology of pain, neurodegenerative diseases, stroke, SCI, brain trauma, and neuropsychiatric disorders. Tissue damage, stress, and their associated inflammatory response may trigger an endogenous program of resolution that encompasses the production of lipid mediators able to turn off inflammation and reestablish a homeostatic balance. 28,29 A number of molecules have been identified that take part in these protective mechanisms. Among these, the ALIAmide PEA, belonging to the family of NAEs, a class of naturally occurring lipid-signaling molecules, demonstrated beneficial effects alone and in combination in different models of inflammation, neuroinflammation, and pain. 4,30 Recent years have seen a growing number of works reporting the anti-neuroinflammatory and neuroprotective actions of PEA. 6,31 –33 In addition, modulating responses induced by inflammatory stimuli can be realized by increasing endogenous PEA levels by inhibiting its degradation, targeting either FAAH 34 or its principal catabolic enzyme, NAAA. 8,34 –37 Paradoxically, pharmacological or genetic manipulation of PEA catabolism may, in some instances, actually lead to undesirable effects. 12,13,38,39 For this reason, it has been suggested that pharmacologically modulating—and not blocking—the specific amidases such as NAAA could be important to preserving the role of PEA in sustaining cellular homeostasis through its rapid on-demand synthesis and so rapid degradation. 19 The patent WO2013121449 A1 described the pharmacological modulation of NAAA with the oxazoline of PEA (2-pentadecyl-2-oxazoline or PEA-OXA). 15 PEA-OXA, which occurs in nature, can be rapidly transformed through entirely physiological processes into PEA. The oxazoline (but not the analogous non-cyclic structure [PEA] is a weak inhibitor of NAAA with respect to other known blockers. It was recently demonstrated that in the rat paw carrageenan model, PEA-OXA had a greater beneficial effect compared with PEA. 16
Based on the above findings, we set out to examine the anti-neuroinflammatory and neuroprotective effects of PEA-OXA in two different experimental models of neuro-inflammation, namely, SCI and TBI in mice. We observed that SCI caused edema formation, infiltration of inflammatory cells, and alteration of white matter; TBI resulted in an ipsilateral injury with cortical contusion, hemorrhage, and blood–brain barrier disruption. In both injury paradigms, PEA-OXA treatment significantly ameliorated not only the histological alterations but also brought about an improvement in trauma-induced motor function and behavioral changes.
Glial cell activation is now recognized as playing a key role in the pathogenesis of CNS disorders. Microglia can provoke the recovery of injured CNS by scavenging dead cells and secreting neurotrophic factors. 40 Although activated astrocytes secrete neurotrophic factors that support neuronal cell survival, their rapid and robust activation augments/initiates an inflammatory response, leading to brain injury. 41 Neurotrophins such as BDNF and NT-3 have demonstrated the potential to promote neuronal development and regeneration. In particular, BDNF promotes neuronal cell survival and regeneration following mechanical injury. 42 In the present study we observed that in spinal cord and brain trauma, conditions associated with neuroinflammation, PEA-OXA treatment reduced astrocyte activation in terms of GFAP immunoreactivity and increased the release of neurotrophic factors such as BDNF, GDNF, and NT-3. These findings are in agreement with our previous studies where PEA was neuroprotective in models of SCI, TBI, and Parkinson disease. 5,43,44
Neuronal expression of NF-κB is induced by a variety of insults, including TNF-α and mechanical injury, and is associated with subsequent expression of inflammatory mediators such as iNOS and COX-2. 45,46 NF-κB is normally sequestered in the cytoplasm, bound to regulatory protein IκBs. In response to stimuli including oxidative stress, infection, hypoxia, extracellular signals, and inflammation, IκB is phosphorylated by IκB kinase. 47 This causes the release of the NF-κB dimer, which can translocate into the nucleus. In this pathway, the nuclear translocation of pp65Ser536 is critical for the regulation of target genes. The phosphorylation status of p65Ser536 has also been described as a critical factor for diverse biological functions, such as NF-κB transcriptional activity. 48 Accordingly, after TBI or SCI induction, we observed an increased IκB-α degradation with p-p65Ser536 phosphorylation on the p65 subunit and subsequent nuclear p65 translocation. PEA-OXA treatment significantly reduced IκB-α degradation, the phosphorylation of p65 on Ser536 and NF-κBp65 levels. Because NF-κB is important in inflammation, some enzymes that promote the production of reactive oxygen species (ROS) are also regulated as its targets such as iNOS and COX-2. 49 In the same time, NF-κB is also a critical transcription factor for the expression of many cytokines that are involved in the pathogenesis of inflammatory diseases. 50 Several of our studies also demonstrated that PEA treatment alone or in combination could ameliorate inflammatory conditions by inhibiting the NF-κB pathway, and iNOS and COX-2 expressions. 25,51 In that regard, in this study we observed that PEA-OXA was able to decrease the expression of iNOS and COX-2 as well as the release of pro-inflammatory cytokines TNF-α and IL-1β.
Conclusion
Neuroinflammation is seen as a prominent feature of many classic neurological and neurodegenerative diseases. 5,6 The need to develop new therapeutics for CNS disorders such as spinal cord and brain trauma and the current lack of specific therapy underscores the importance of identifying and characterizing novel neuroprotective compounds. Anti-inflammatory mechanisms may be triggered in parallel and serve to terminate neuroinflammation and reduce pathological outcomes. Treatments and interventions may be targeted at various levels to inhibit the neuroinflammatory processes, or to promote the resolution of inflammation. In this regard, PEA-OXA was able to decrease all inflammatory and behavioral parameters associated with the SCI and TBI models used here. Thus, the modulation of intracellular NAAA by PEA-OXA treatment could represent a novel mechanistic approach to control pathological conditions associated with neuroinflammation. Additional experimentation will be needed to better explain the mechanism of action of this new compound.
Footnotes
Acknowledgments
The authors would like to thank Francesco Soraci and Antonietta Medici for their excellent technical assistance during this study and Valentina Malvagni for editorial assistance with the manuscript.
This study was supported by grant PON01-02512.
Author Disclosure Statement
Dr. Salvatore Cuzzocrea, researcher on the study team, is co-inventor on patent WO2013121449 A8 (application number PCT/IT2012/000050, Italy) (Epitech Group SpA), which deals with compositions and methods for the modulation of amidases capable of hydrolysing N-acylethanolamines useable in the therapy of inflammatory diseases. Moreover, Dr. Cuzzocrea is also a co-inventor with Epitech group on the following patents: EP 2 821 083, MI2014 A001495, and 102015000067344.
No other authors have conflicts of interests.