
Abstract
Traumatic brain injury (TBI) is associated with the development of numerous psychiatric diseases. Of particular concern for TBI patients is the impact of chronic impulsivity on daily functioning. Despite the scope of the human problem, little has been done to address impulsivity in animal models of brain injury. In the current study, we examined the effects of either a severe or a milder bilateral frontal controlled cortical impact injury on impulsivity using the Delay Discounting Task (DDT), in which preference for smaller-sooner over larger-later rewards is indicative of greater impulsive choice. Both milder and severe TBI caused a significant, chronic increase in impulsive decision making. Despite these pronounced changes in performance of the DDT, memory function, as assessed by the Morris Water Maze, was not impaired in more mildly injured rats and only transiently impacted in the severe TBI group. Whereas a significant lesion was only evident in severely injured rats, analysis of cytokine levels within the frontal cortex revealed a selective increase in interleukin (IL)-12 that was associated with the magnitude of the change in impulsive choice caused by both milder and severe TBI. These findings suggest that tissue loss alone cannot explain the increased impulsivity observed, and that inflammatory pathways mediated by IL-12 may be a contributing factor. The findings from this study highlight the sensitivity of sophisticated behavioral measures designed to assess neuropsychiatric dysfunction in the detection of TBI-induced cognitive impairments and their utility in identifying potential mechanistic pathways and therapeutic targets.
Introduction
T
Given that impulsivity is a complex construct, understanding its manifestation post-TBI requires the combination of animal models of injury and assays of impulsive behavior with high translational validity. In the field of experimental TBI, numerous injury methods have been developed that replicate various aspects of acute and chronic pathology associated with brain injury. 7 –9 Despite the sophistication of many studies examining pathophysiological changes caused by these injury models, the sensitivity of behavioral assessments has lagged behind other techniques used in the field. In particular, there is a strong reliance on the Morris Water Maze (MWM) as the sole measure of cognitive function, which ignores crucial aspects of cognition, such as decision making and impulsivity. In recent years, interest in improving functional assessments, particularly those relating to the psychiatric aspects of brain injury, has increased. 10 –13
Fortunately, there exist a number of well-validated behavioral tasks in the fields of behavioral neuroscience and the experimental analysis of behavior that can be applied to questions of complex cognitive function. Specifically, with respect to impulsive choice, the Delay Discounting Task (DDT) has been developed as a particularly robust method. 14,15 In the task, subjects are asked to make a choice between a small reinforcer delivered immediately or a large reinforcer that is made available after a delay. Choices of the smaller option have lower utility and provide an index of intolerance to delayed gratification, one of the hallmarks of impulsive decision making. By comparing choice patterns across several delays, researchers can plot the discounting function of the individual (subjective value of a reinforcer at different delays) and evaluate the impulsivity of the subject. The slope of this function is typically represented by the variable k and provides a unitary measure for comparisons within and between subjects. 14 This behavioral assessment has been used in many species, including mice, rats, pigeons, monkeys, and humans, because of its highly robust cross-species validity. 16 It is particularly useful for evaluating impulsivity in patient populations, such as individuals with substance use disorders, psychiatric disease, and even persons with brain injury. 17 –19
Although studies are limited, TBI patients display increased discounting of delayed reinforcers, similar to those with diagnosed psychiatric conditions. 19 –21 However, given the current state of literature on impulsivity in TBI, it is difficult to reach strong conclusions regarding potential mechanisms for observed changes in impulsive choice. In particular, whereas impulsivity has been readily identified after severe injury, particularly in the form of impulsive aggression, 22 it has also been reported to emerge long after the injury event 22,23 and has been suggested to occur following even mild brain injuries. 5 Despite these complications, several key points are known about the neurobiology of impulsivity and may offer insight into the specific case of TBI. In particular, increased impulsive choice is associated with loss of frontal lobe function. 24 Thus, brain injury causing direct damage to this region may result in increased impulsivity, and disruption of frontal projections could provide an explanation for the delayed occurrence of impulsivity or its emergence following milder events. In addition, there is the potential for neuroinflammation to play a role in the development of impulsivity. Markers of increased neuroinflammation have been observed in several psychiatric disorders with strong impulsive components, such as substance dependence, bipolar disorder, and suicidality. 25 –27 Further, we have recently shown an association between motor impulsivity and neuroinflammation in rodents post-TBI. 28
The current study was designed to test the hypothesis that increased impulsive choice could be caused by both milder and more severe frontal TBI, even in the absence of memory deficits as assessed by the MWM, and that neuroinflammation could be associated with such behavioral change. We therefore trained rats to perform a DDT and then delivered either a milder or severe TBI using the controlled cortical impact (CCI) model bilaterally over the frontal cortex. We reassessed rats' performance on the DDT 5 days per week until 2 months post-injury and also measured spatial memory using the MWM. Measurements of both tissue loss and cytokine expression within the frontal cortex were obtained post-mortem and associations with impulsivity analyzed statistically.
Methods
Animals
Subjects were male Long-Evans rats (N = 30), 3 months of age when training began. Rats were food restricted to 85% free-feeding weight (14–20 g of maintenance chow daily); water was available ad libitum. Rats were pair-housed until surgery, after which they were singly housed in standard cages on a (12:12) reverse light cycle, with a plastic hut and shredded paper towel available as enrichment. Housing and testing were performed in accord with the Canadian Council on Animal Care, and all procedures were approved by the University of British Columbia Animal Care Committee.
Apparatus
Behavior was conducted in a bank of 12 standard operant chambers (Med Associates, St Albans, VT) equipped on one side with a five-hole array and on the other with a tone generator, two retractable levers, two lights above the levers, a sucrose pellet dispenser (45-mg pellets; Bio-Serv, Flemington, NJ), and a houselight. The left, center, and right (1, 3, 5) holes in the five-choice panel and the pellet dispenser were used in this experiment.
Behavioral training and baseline
Training
Rats were trained similar to previously published parameters. 29 Rats were habituated to the chamber with sugar pellets placed in the food hopper, and in holes 1, 3, and 5 for one to two sessions. In the first stage of training, rats learned to respond to an illuminated hole 3 to receive one pellet until they successfully completed 30 trials in a single session. In stage two of training, rats responded first to the illuminated hole 3, which turned on a light in either hole 1 or 5 (pseudorandomly), and correct responses were reinforced with a single sugar pellet. Finally, in the last stage, a differential was introduced, with hole 1 delivering four pellets and hole 5 delivering a single pellet. The location of the larger reward hole (hole 1 or 5) was kept constant for each rat throughout training and testing, but counterbalanced between subjects.
Delay Discounting Task
After the above training, the delay discounting task began. A task schematic is provided in Figure 1. Sessions lasted 40 min and consisted of four blocks of 12 trials, each of which had a fixed length of 40 sec. A trial began with the illumination of hole 3 and a nose poke would illuminate both choice holes 1 and 5. Failure to respond within 10 sec was scored as an omission and activated the intertrial interval (ITI). Rats chose between the large (four-pellet) and small (one-pellet) option (counterbalanced as above). The small option was always delivered immediately, whereas the delay to the large option increased in a step-wise fashion across successive blocks of trials, from 0 to 5, 10, and finally 20 sec. Every block began with two forced-choice trials to ensure rats were exposed to the current delay contingency. After a choice was made and the reinforcer delivered, there was an ITI equal to the remaining trial time (40 sec minus the delay to reinforcement). Rats were trained until a statistically stable choice baseline emerged (approximately 30 sessions).
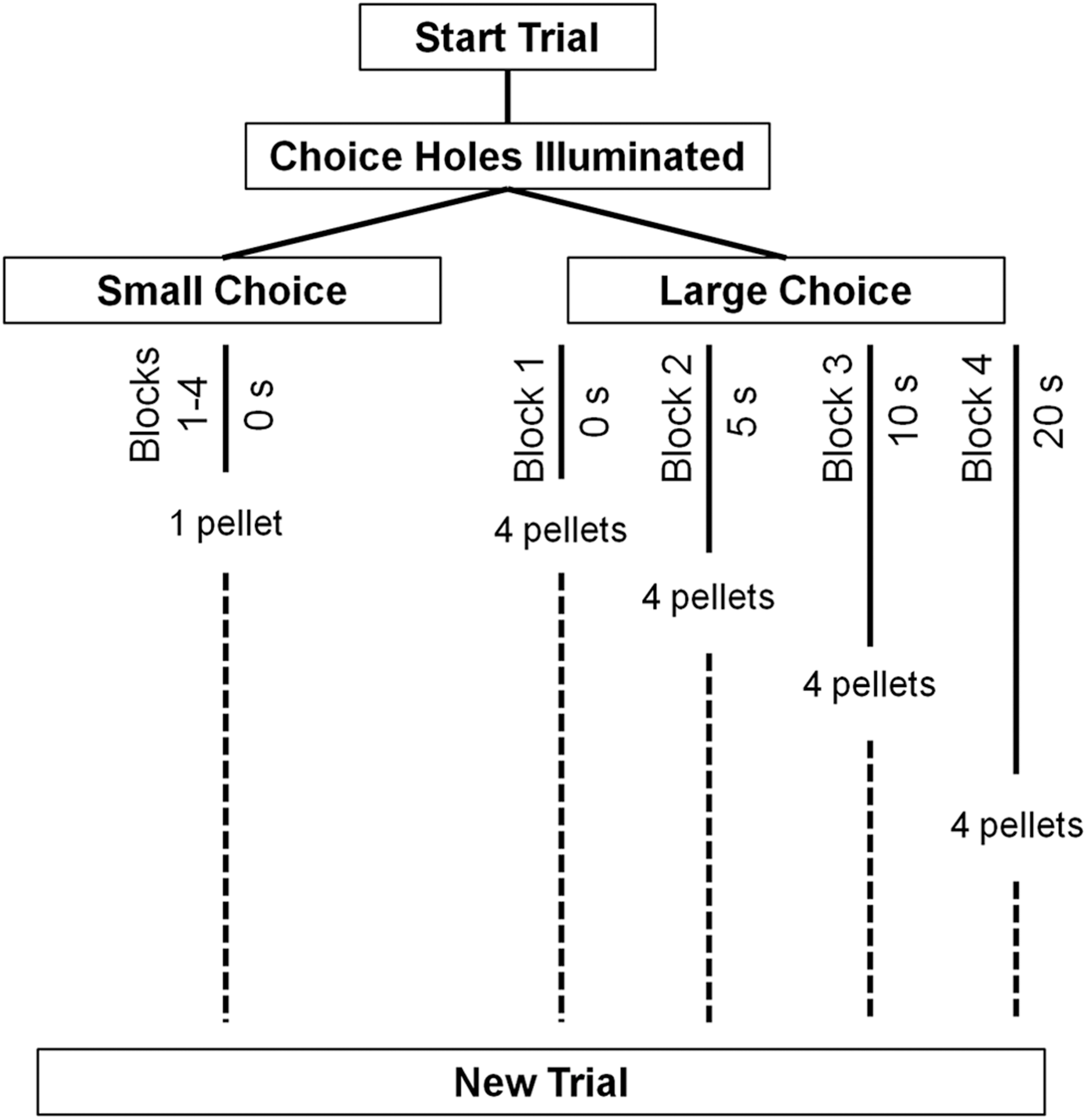
Schematic diagram of the delay discounting task. Rats started a trial by nose poking the illuminated center hole. They then chose between a small (1 pellet) and large (4 pellets) option. The small option was delivered immediately, whereas the large option was associated with some level of delay to reinforcement. Across four blocks of 12 trials each, the delay to the large reinforcer was increased from 0 to 5 to 10 to 20 sec. The total trial time was held constant across all blocks. Each block began with two forced choice trials to demonstrate the delay.
Surgery
Rats were matched for task performance and then assigned to severe TBI (n = 10), milder TBI (n = 10), or sham (n = 10) groups. TBI rats were given a bilateral frontal CCI, as previously described, with either severe or milder settings. 13,28 Although a truly mild injury is typically induced by closed-skull techniques, these milder settings allow a direct comparison between a severe and much less severe form of injury within the CCI model. In brief, rats were anesthetized and placed in a stereotaxic frame. Ketoprofen (5 mg/kg, subcutaneously [s.c.]), lactated ringer solution (8 mL, s.c.), and bupivicane (0.1 mL of 0.5% solution, s.c. at incision site) were administered. Under aseptic conditions, a mid-line incision was made in the scalp and the fascia retracted. A 6.0-mm-diameter circular craniotomy was performed centered at anteroposterior +3.0, mediolateral 0.0 mm from bregma. A TBI was then induced using an electromagnetic CCI device (Leica Biosystems, Buffalo Grove, IL) with a circular, flat-faced, 5-mm-diameter tip, at a rate of 3 m/s to a depth of −2.5 mm for 0.5 sec (severe) or a rate of 1 m/s to a depth of −0.8 mm for 0.5 sec (milder, ∼11% of severe force). Post-injury, bleeding was stopped with sterile gauze and the incision sutured. Sham procedures included everything above with the exception of craniotomy and impact. Ketoprofen (5 mg/kg) was administered for pain management 24 h post-surgery.
Behavioral assessment
Delay Discounting Task
After 1 week of recovery, testing resumed 5 days per week on the DDT until week 8 post-injury.
Morris Water Maze
At week 9 post-injury, rats were assessed in acquisition for 4 days using a fixed-platform MWM paradigm, followed by 4 days using a repeated acquisition paradigm, with a novel platform location each day. 12 For the standard acquisition paradigm, rats were given four trials daily with a 15-min ITI. Each trial began with a pseudorandomized start point at the four cardinal directions on the maze (N, E, S, W), and the platform remained located in the interior of the SE quadrant. If a rat failed to locate the platform within 90 sec, they were guided by hand to the platform. Rats were allowed to remain on the platform for 10 sec and then were removed to a heated cage. On the fifth day, rats were given a 30-sec probe trial in which the platform was removed. The repeated acquisition paradigm began after the probe trial on the fifth day, conducted as specified above, except that the platform location was randomized at the start of each day (NW, NE, SW, SE), and the first trial was considered a learning trial and not included in the data analysis. Latency and path data were recorded using AnyMaze (Stoelting, Wood Dale, IL).
Enzyme-linked immunosorbent assay analysis of cytokine levels
After MWM testing completed, at approximately 10 weeks post-injury, one half of the animals (n = 15) were rapidly decapitated, the orbitofrontal and ventral aspects of medial pre-frontal cortex extracted, rapidly frozen on dry ice, and stored at −80°C. Tissue was lysed in radioimmunoprecipitation assay buffer (pH 8.0) with protease and phosphotase inhibitors. Samples were spun at 13,000 RPM, supernatant extracted, and measured for protein content. Samples were then analyzed for interleukin (IL)-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12, tumor necrosis factor alpha (TNFα), and interferon-gamma IFNγ by multi-plex enzyme-linked immunosorbent assay (Quansys Q-plex, Logan, UT). In brief, with washes between each step, standards and samples were placed in antibody pre-coated plate wells and incubated under agitation for 90 min, then incubated with a detection mixture (secondary antibody) for 60 min, incubated with a streptavidin/horseradish peroxidase solution for 15 min, and incubated with a coloring reagent until color gradient appeared in standard wells. Optical density was then read using a Q-view imager. Protein concentrations were calculated using the standard curve.
Histology and lesion analysis
The other half of subjects (n = 15) were transcardially perfused with saline, followed by 3.7% formalin. Brains were removed and post-fixed in formalin for 24 h and then placed in 30% sucrose solution. Following 3 days in sucrose, brains were sliced at −20° on a cryostat at 50 μm and mounted to gelatin-subbed slides.
Lesion analysis
Slides were stained with cresyl-violet and imaged on a Zeiss microscope (Carl Zeiss, jena, Germany). The total remaining area, lesion area, and ventricular area for five brain sections, evenly spaced through the injury location (+6, +4.5, +3, +1.5, and 0 mm from bregma) were measured using ImageJ software (National Institutes of Health, Bethesda, MD) and the volume estimated using the Cavalieri method. 30
Statistical analysis
The percent choice of the large option in the DDT was used to estimate the slope of the hyperbolic discounting function (k) using the following equation: V = 1 / (1 + k*D), where V represents the subjective value (preference for the large option expressed as percent choice of large) and D represents the delay to reinforcement, as outlined in previous work. 14 This measure provides a single value for impulsivity to be compared within and across subjects over repeated testing.
Statistical tests were conducted using R statistical software (
Results
Delay discounting
The k values, or slopes of the discounting function, representing choice impulsivity, were compared across groups and over the testing period using a mixed-effects regression that included baseline impulsivity as a covariate (k ∼ Group*Week + Baseline). Both milder and severe TBI groups had a significant interaction of impulsivity and time (milder vs. sham, β = 0.06; t = 2.44; p = 0.015; severe vs. sham, β = −0.37; t = −14.53; p < 0.001; see Fig. 2), suggesting increased impulsivity across time relative to the sham group. Whereas animals with milder TBI showed higher impulsivity early on in testing that did not change across time, impulsivity both increased and then decreased over time in the severe group, resulting in a significant difference between the trajectory of behavioral change for the two TBI severities (milder vs. severe, β = −0.30; t = −16.72; p < 0.001). Only the severe TBI animals showed a significant overall effect of group (milder, β = 0.53; t = 1.85; p = 0.076; severe, β = 0.77; t = 2.68; p = 0.012) attributed to the large initial increase in impulsivity.
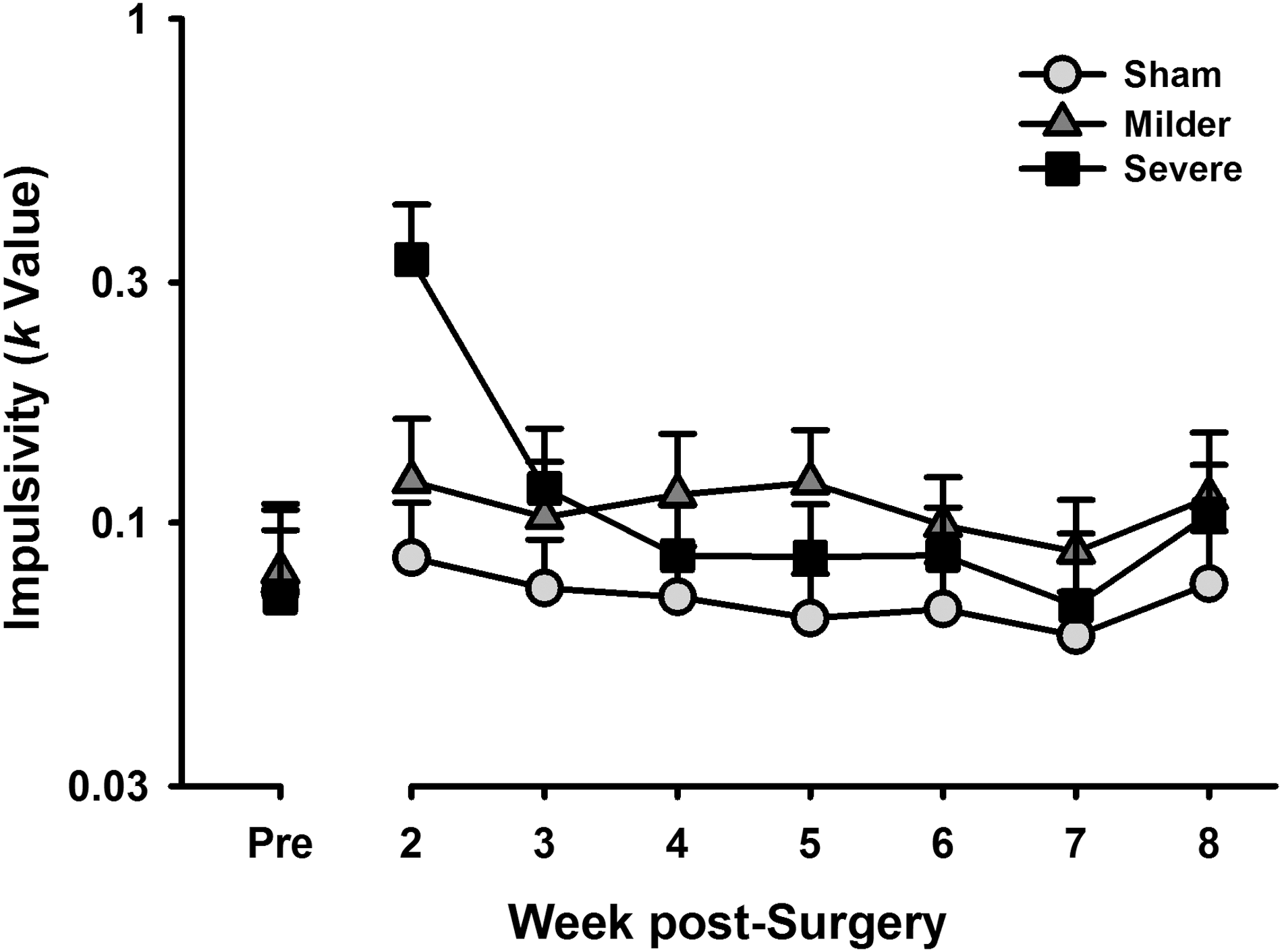
Impulsivity over time. Both the severe and milder traumatic brain injury groups showed a significant interaction of impulsivity and time relative to sham animals (p < 0.001; p = 0.015), with levels of impulsivity peaking early and returning to basal for the severe-injured group, and a small, but permanent increase in impulsivity for the mild-injured group. Data are mean + standard error of the mean.
To follow up the interaction of impulsivity across time, ANOVAs were performed on the raw choice values (% Choice ∼ Group*Delay) at time points of pre-surgery, 4 weeks post-surgery, and 8 weeks post-surgery. There were no group differences before the injury (F (2,588) = 0.32; p = 0.727; Fig. 3A). At 4 weeks, there was a significant group by delay interaction (F (2,708) = 21.91; p < 0.001; Fig. 3B); follow-up ANOVAs and post-hoc tests revealed decreased choice of the large reinforcer (increased impulsivity) relative to sham at the 0-sec delay for the severe TBI group (HSD = −1.75; p < 0.001), at the 5-sec delay for the milder and severe TBI group (milder, HSD = −1.02; p = 0.025; severe, HSD = −2.03; p < 0.001), at the 10-sec delay for the milder TBI group (HSD = −1.27; p = 0.021), and at the 20-sec delay for the milder TBI group (HSD = −0.87; p = 0.043). At 8 weeks, there was a significant effect of group (F (2,340) = 3.77; p = 0.024; Fig. 3C), and post-hoc analyses revealed that the milder TBI group had higher levels of impulsivity relative to the sham group (HSD = −0.74; p = 0.026), whereas the severe TBI group did not (HSD = −0.59; p = 0.098).
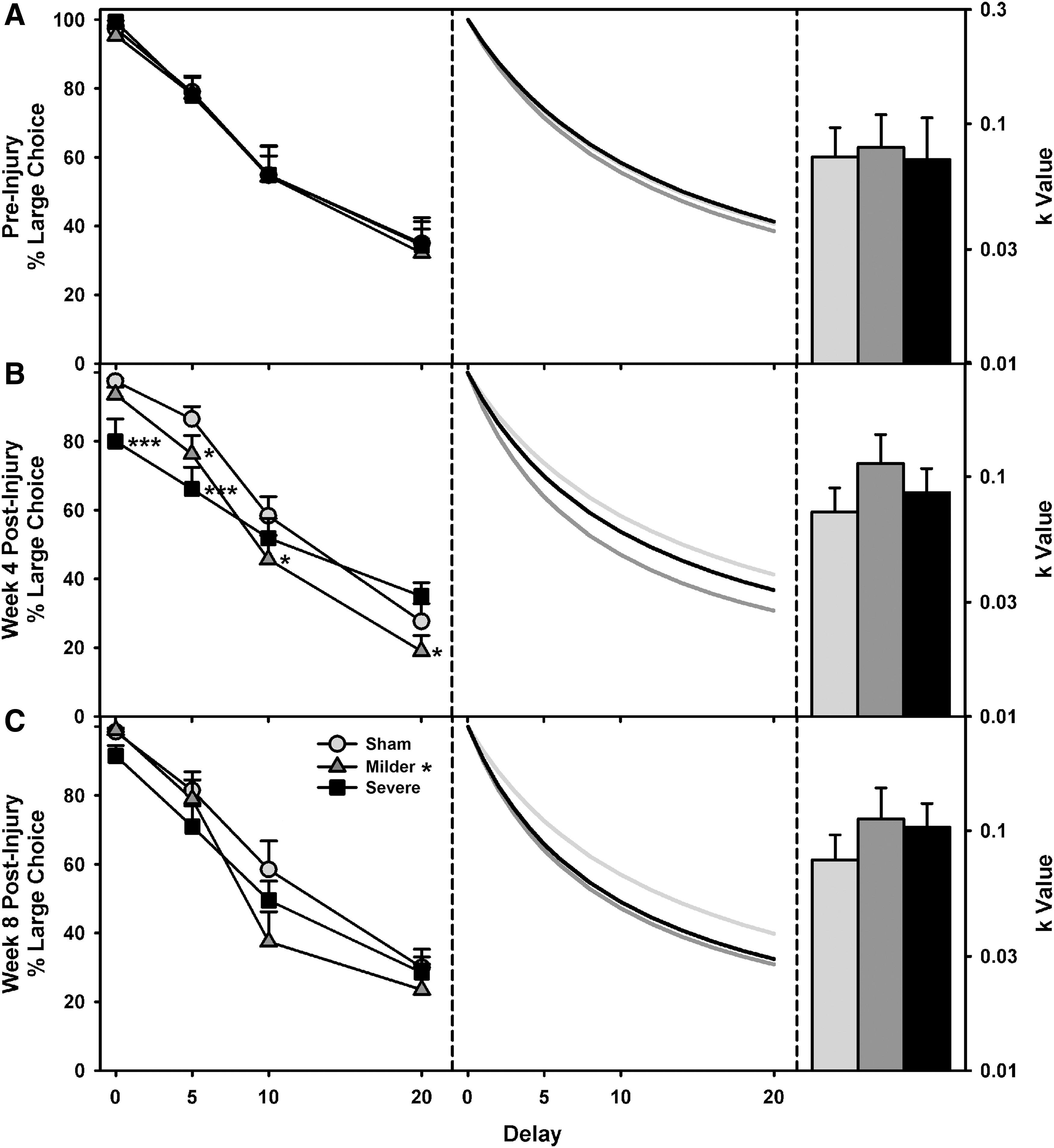
Delay discounting data, fitted curves, and k-values. (
Morris Water Maze
The latencies to reach the platform for the standard and repeat acquisition paradigm of the MWM were analyzed in a linear mixed-effects regression (Latency ∼ Group*Day). On the standard acquisition paradigm, severely injured animals were slower than both the milder TBI and sham controls to find the hidden platform, but there were no differences between the mild-injured and sham animals (Fig. 4A; severe vs. sham, β = 0.76; t = 4.05; p < 0.001; milder vs. sham, β = 0.25; t = 1.36; p = 0.186; milder vs. severe, β = 0.50; t = 2.70; p = 0.012). However, the rate at which the latency to find the hidden platform decreased over successive training, indicative of the speed of learning, was identical across all animals (severe vs. sham, β = −0.02; t = −0.20; p = 0.839; severe vs. milder, β = 0.01; t = 0.06; p = 0.949; milder vs. severe, β = −0.03; t = −0.27; p = 0.790). As such, although the severely lesioned animals may have been initially worse at locating the platform, it is hard to conclude that they showed any impairment in learning, particularly given the comparable latencies to find the platform on the final test day. During the probe test, although the severe TBI group appeared to spend less time in the quadrant where the platform had previously been hidden, this only approached significance (Fig. 4B; Group, F (2, 27) = 3.31; p = 0.056).
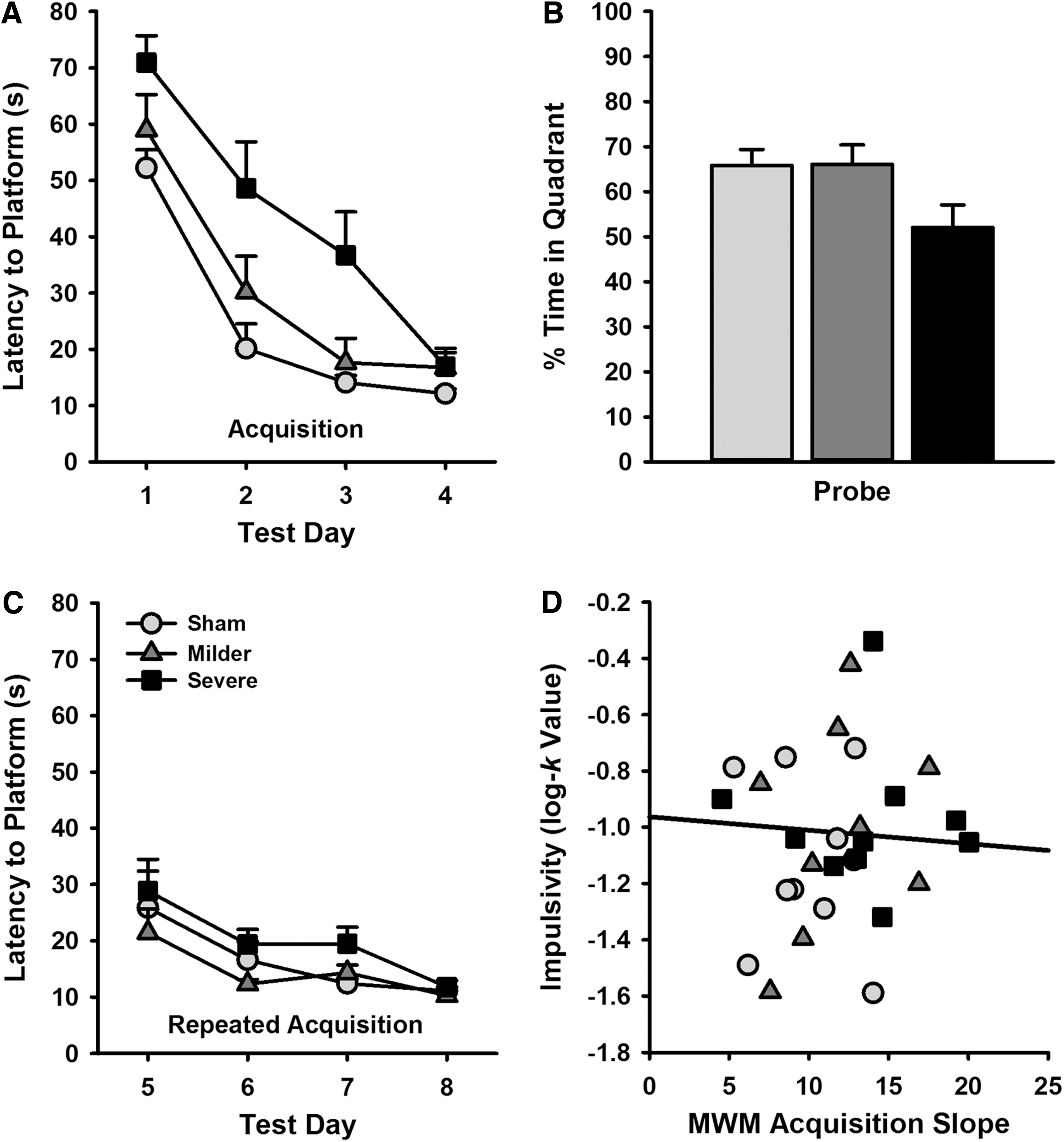
MWM performance at 9 weeks post-TBI. (
When tested in the repeat acquisition paradigm, latencies were low across all groups and there were no significant differences in the latency of either the severe or milder TBI group to locate the submerged platform as compared to sham controls (Fig. 4C; severe vs. sham, β = 0.41; t = 1.78; p = 0.087; milder vs. sham, β = −0.12; t = −0.50; p = 0.618), although the milder group performed slightly better compared to the severe group (β = 0.52; t = 2.28; p = 0.031). As per the initial acquisition of the MWM, there were no differences in learning rates between any of the groups (severe vs. sham, β = 0.07; t = 0.43; p = 0.671; milder vs. sham, β = −0.06; t = −0.38; p = 0.706; milder vs. severe, β = −0.13; t = −0.80; p = 0.423).
Behavioral task correlation
If deficits in impulsive choice and MWM performance were correlated post-TBI, this would provide indirect evidence to suggest that any such behavioral changes could be subserved by a common mechanism. However, there was no significant correlation between acquisition MWM learning rate and the k parameter estimate of impulsivity (r = −0.07; t = −0.36; p = 0.723; see Fig. 4).
Lesion analysis
The lesion and ventricle volumes were analyzed across brain position in a mixed-effects regression (Volume ∼ Group*Slice). The severe group had a significant increase in lesion volume (β = 2.11; t = 4.36; p < 0.001) compared to the sham group, whereas the milder group approached significance (β = 0.89; t = 1.86; p = 0.070). The severe group also had significantly larger lesions than the milder group (β = 1.22; t = 2.51; p = 0.017). Despite fluctuations in lesion size across slice position, there were no significant interactions between the groups in amount of tissue lost in the anterior versus posterior portion of the lesioned area (milder vs. sham, β = −0.03; t = −0.25; p = 0.807; severe vs. sham, β = −0.13; t = −1.14; p = 0.260; milder vs. severe, β = −0.10; t = −0.90; p = 0.371; Fig. 5).
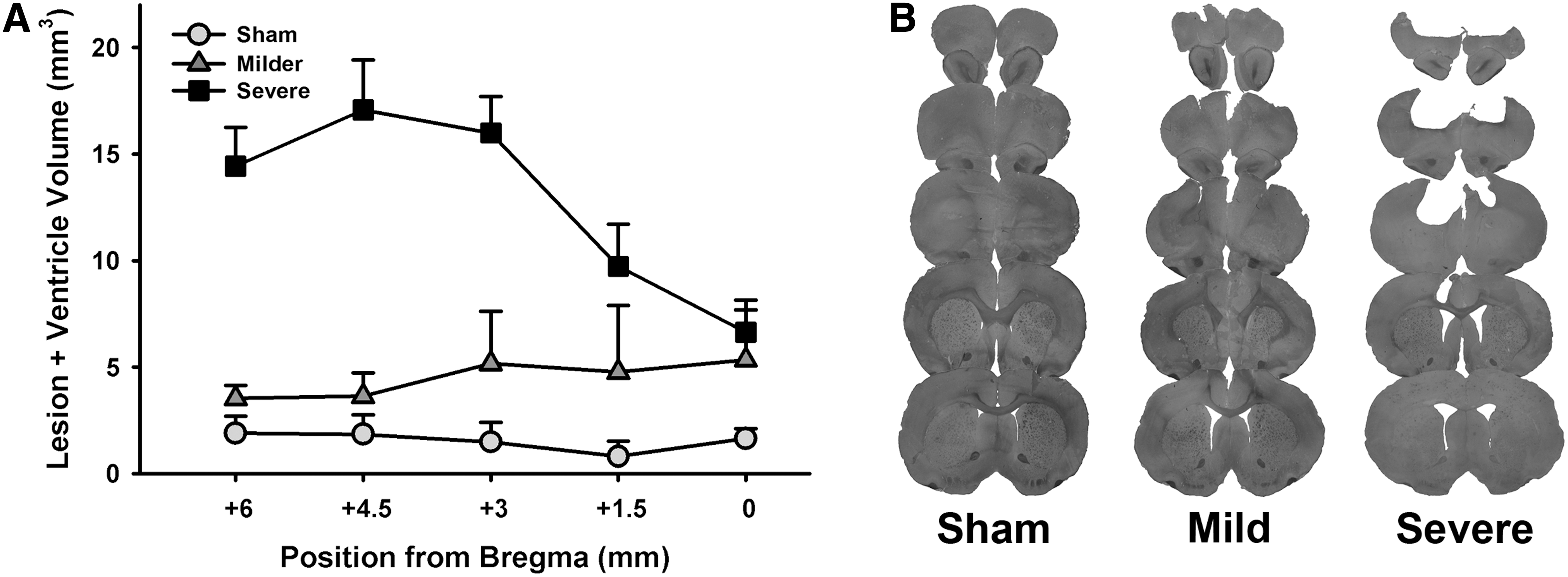
Lesion quantification. (
Cortical cytokine levels
ANOVAs were performed on all measured cytokines (Cytokine ∼ Group). There were no group differences in IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, TNFα, or IFNγ (Fs(2, 12) < 1.10; ps > 0.365; see Fig. 6). However, IL-12 levels were significantly increased at comparable levels in both the milder and severe TBI groups (Group, F (2, 12) = 9.81; p = 0.003; severe vs. sham, HSD = 2.54; p = 0.003; milder vs. sham, HSD = 2.02; p = 0.015; milder vs. severe, HSD = 0.51; p = 0.681; Fig. 6).
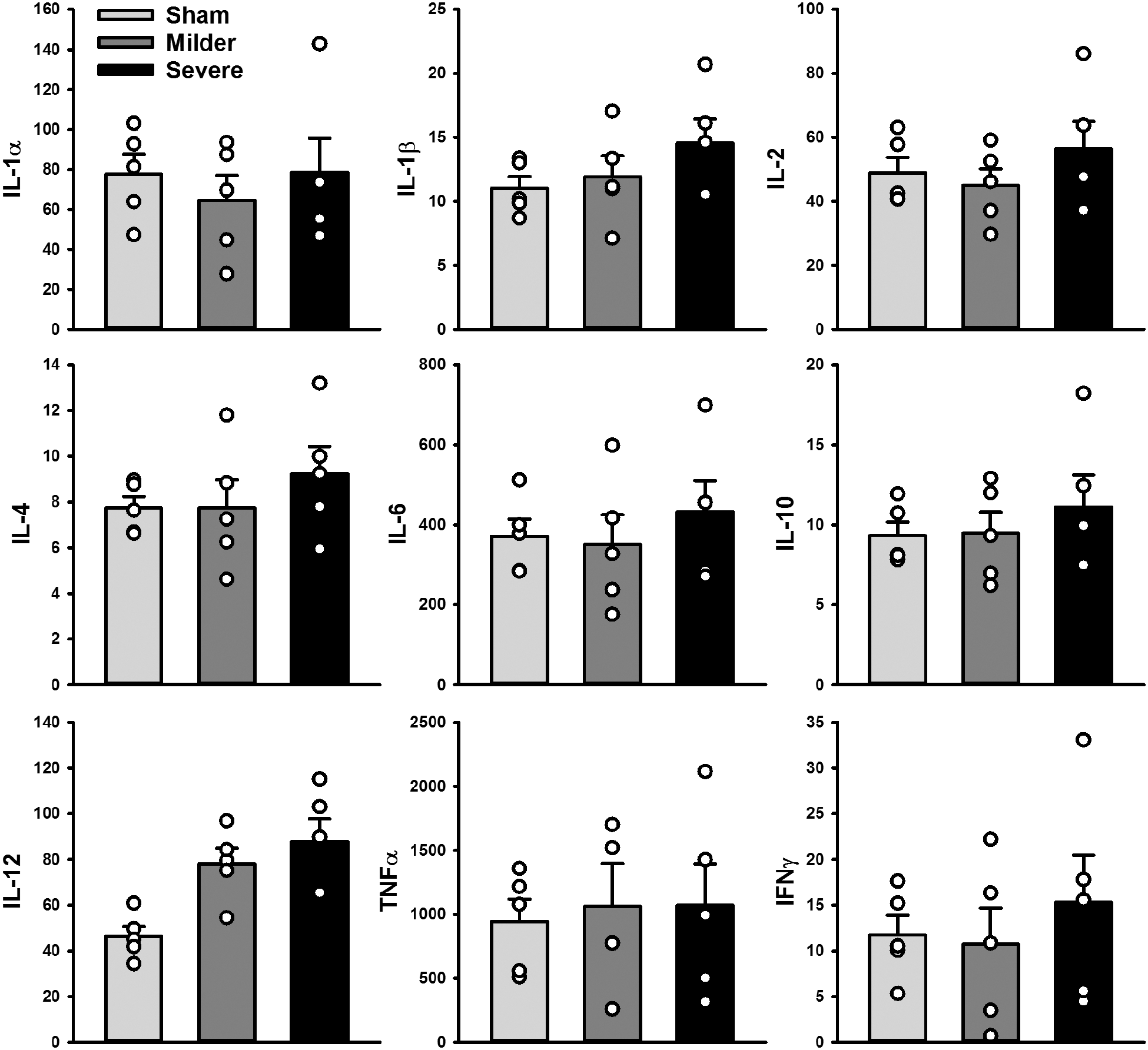
Cortical cytokine levels. IL-12 (bottom left) was the only cytokine with significantly increased levels in both the milder (p = 0.015) and severe groups (p = 0.003). Data are mean + standard error of the mean, with individual data points superimposed in circles. IFNγ, interferon-gamma; IL, interleukin; TNFα, tumor necrosis factor alpha.
Because the relationship between cytokines is complex, a PCA was also performed to account for any interactions between cytokines. Two components were identified that accounted for 89.68% of the variance in the data set. The first component represented 79.64% of the variance and featured relatively equal component loadings across all cytokines. The second component represented 10.04% of the variance and was heavily dominated by IL-12 (see Fig. 7).
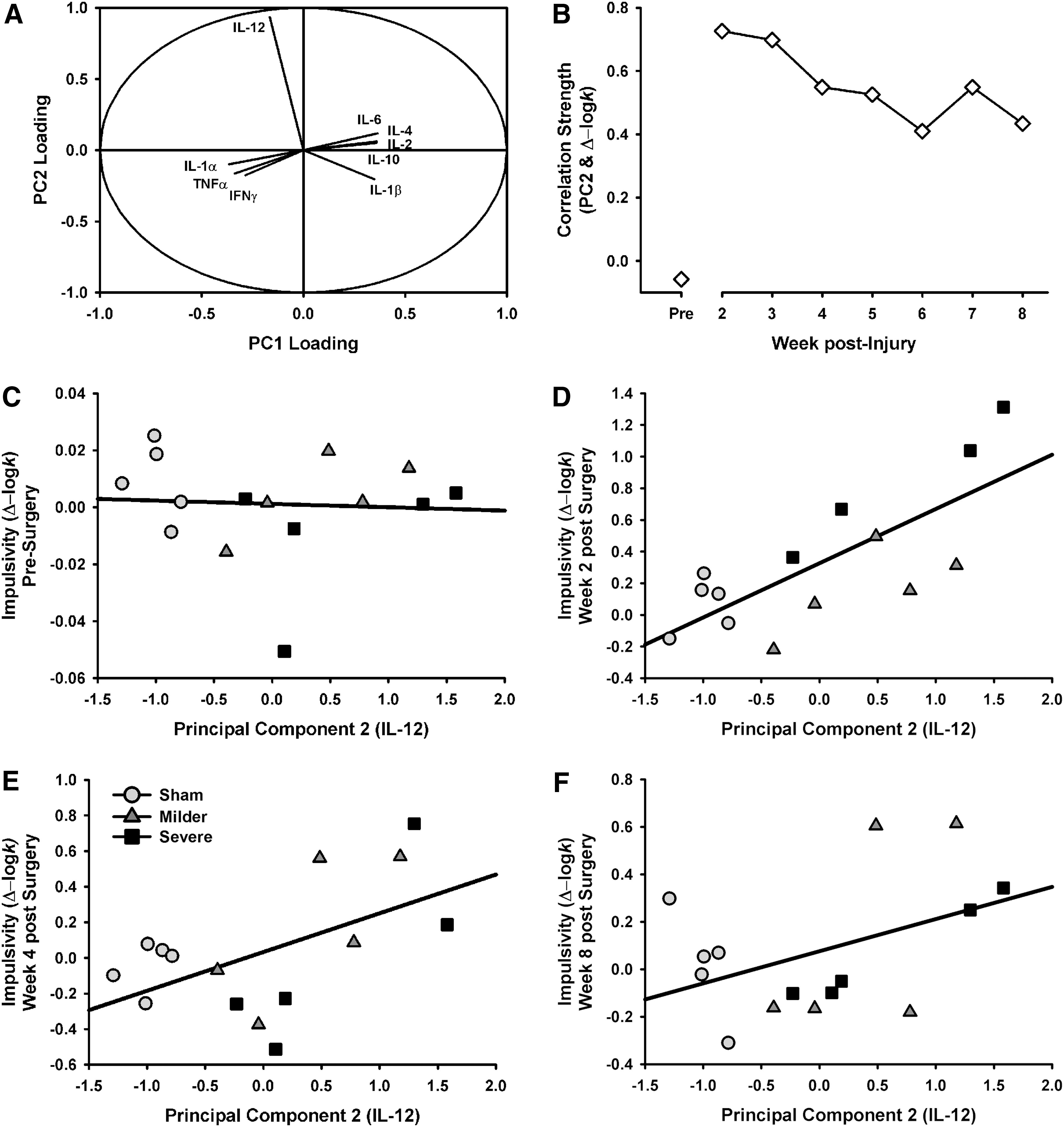
Relationship between cytokines and impulsivity. (
To determine to what degree the various cytokines accounted for impulsivity, multiple regression was performed to examine the relationship of principal component (PC) 1 and PC2 across the testing period with baseline impulsivity as a covariate (k ∼ PC1*Week + PC2*Week + Baseline). Model selection was performed and indicated that neither PC1 nor its interaction with time were significant contributors and thus were dropped from the model, yielding a final model of k ∼ PC2*Week + Baseline. The IL-12-dominated PC2 was significantly related to impulsivity level (β = 0.97; t = 4.56; p < 0.001), but over the testing period, impulsivity generally declined from its peak at week 2 post-injury (β = −0.10; t = −3.45; p < 0.001) and the strength of the PC2 effect was similarly moderated by time (β = −0.09; t = −2.85; p = 0.005; see Fig. 7). This pattern of data suggests that early changes in function may be associated with long-term IL-12 levels.
The baseline level of impulsivity exhibited by any subject can understandably determine, at least to some degree, the final level of impulsive choice observed following any manipulation, given that it is the starting value from which any alteration must deviate and as such is important to include in predictive models. Interestingly, PC2 (IL-12 dominated) accounted for impulsivity almost as well as baseline levels of performance (PC2, β = 0.97; Baseline, β = 1.32), whereas PC1 (all other cytokine activity) and tissue loss were not significantly associated with behavioral function.
Discussion
Impulsivity following brain injury is a pervasive problem that chronically affects a large number of patients. 2,4,5 In the current study, we observed persistent increases in choice impulsivity in rats following a frontal TBI, which continued to 8 weeks post-injury, replicating long-term symptoms reported in humans. Further, impulsivity was increased regardless of injury severity, suggesting that TBI-induced impulsivity occurs by mechanisms independent of tissue loss. Additionally, our other measure of cognitive function, the MWM, failed to capture the long-term deficits associated with injury and showed no functional overlap with the impulsivity measure. Finally, when samples of frontal cortex tissue were analyzed at 10 weeks post-injury, we did not observe elevation in numerous cytokines, as would have been expected from a general long-lasting inflammatory response. Instead, we found a selective and substantial increase in levels of IL-12 across both milder and severe TBI groups that was significantly associated with levels of impulsivity.
TBI has been associated with a multitude of psychiatric disorders, 2,3 many of which have impulsivity as a major component. However, dissecting whether high impulsivity contributes to a behavioral phenotype resulting in greater likelihood of TBI, or whether the TBI itself causes elevated impulsivity, is difficult to resolve from clinical data sets and is a recognized issue with respect to other psychiatric conditions in which impulsivity is a prominent symptom, such as addiction. 31 Recently, the National Institutes for Mental Health instituted the Research Domain Criteria initiative to redefine neuropsychiatric disease by symptom clusters, 32 of which impulsivity constitutes an important component of the Cognitive Control domain. Importantly, understanding the pathology of TBI in relation to the development of impulse control deficits could reveal common pathways by which these disorders occur in otherwise healthy individuals.
This study is the first to demonstrate increased impulsive choice in an animal model of TBI. Our observation that impulsive choice was altered regardless of injury severity is of particular interest. Although the milder TBI group did not show a significant increase in lesion volume, minor damage was evident from histological examination (see Fig. 5). In contrast, almost the entire medial pre-frontal cortex was ablated in the severe TBI group. These data suggest that gross loss of pre-frontal cortex tissue is not sufficient to explain changes in behavioral function following a brain injury. An extensive literature has characterized the role of the prefrontal cortex in impulsive choice in both rodents and humans (for review, see Winstanley 2010), 24 and our findings do not map perfectly onto changes in behavior caused by selective excitotoxic lesions to either the anterior cingulate, pre-limbic, or infralimbic regions of the medial pre-frontal cortex in the rat. If anything, the increase in impulsive choice observed here is most reminiscent of a subset of studies in which the ventrolateral orbitofrontal cortex is silenced, 29,33 but this region was largely spared even in severely lesioned animals. Collectively, these data suggest that, at least for impulsive choice, disruption of cortical circuits may be as detrimental, if not more so, than their complete destruction.
Considerable data suggest that, in the case of human brain injury, such disruption of cortical function may be mediated by mechanisms other than regional cell death. Injury causes many corollary actions, such as axonal damage, and alters the activity of key neurochemical systems that regulate higher-order cognitive processes. 34 –36 Numerous studies indicate that the monoamines, dopamine and serotonin, play a significant role in regulating impulsivity, 37 –39 and these neurotransmitter systems may be compromised following brain injury, 40,41 which may impair frontocortical connectivity. 42,43 However, TBI has also been associated with long-term changes in inflammatory status. 36,44 Inflammation can affect neurotransmission through a variety of mechanisms, such as alterations to glutamatergic signaling, degradation of synaptic connections, and increases in membrane excitability. 45 –47 Given the large-scale changes in neuroinflammation post-TBI, particularly increases in proinflammatory cytokines, these represent potential mechanisms by which disruption of frontal circuits could lead to increased impulsivity. However, in the current study, we found relatively few cytokines that were still elevated at 10 weeks post-injury in the frontal cortex, with the notable exception of IL-12. This is more surprising when the collection method (of specific frontal regions: orbitofrontal cortex and ventral pre-frontal cortex) is considered. It would be expected that the severely injured animals might show higher cytokine levels given the closer proximity to the lesion. Further, when all inflammatory markers were examined in a PCA, IL-12 was markedly different than all other cytokines (see Fig. 7). Further, we observed a significant association between the IL-12-dominated PC2 and impulsive choice (see Fig. 7). This relationship was strongest in the early post-injury phase, and weakened over the testing period, being driven at the end primarily by a subset of rats with elevated levels of IL-12 and high impulsive choice. These data are in keeping with previous work showing long-lasting increases in frontal IL-12 post-injury. 28
Although our studies indicate that IL-12 is clearly elevated long after injury, the time course of IL-12 expression is still unknown. As such, we cannot ascertain whether IL-12 levels peak before or after elevations in impulsivity. Further, we have yet to determine whether IL-12 is itself a mechanistic driver of impulse control deficits or whether its expression increases in parallel to, but is independent from, changes in impulsivity. Although other studies have noted that IL-12 levels can be elevated initially after neural injury, 48,49 some observed improved outcomes with reductions of IL-12, 50,51 whereas others saw recovery associated with increased IL-12 levels. 52,53 As such, the role played by IL-12 in the cognitive sequelae of TBI is likely a complex process, with both potential benefits and impairments associated with acute versus chronic expression respectively.
With emerging awareness of problems related to psychiatric disease and brain injury, the current study underscores the need for more-advanced behavioral assessments in the field of experimental TBI. The most commonly used task for assessing cognitive dysfunction is the MWM, 54 but an over-reliance on one behavioral measure provides an undifferentiated view of cognition. Although this task is effective in measuring hippocampal, and to a lesser extent, working memory, the fact that it is rapidly acquired, and not particularly amenable to repeat testing, decreases its utility in the assessment of long-lasting behavioral deficits that may wax or wane over time. Although the MWM can be adapted (typically through radial arm or t-maze inserts) to assess simple decision-making behavior, these assessments still rely on spatial ability 55 and require labor-intensive preparations relative to a bank of operant chambers. When moving to examine behaviors beyond spatial discrimination, such as impulsivity, other tasks are needed. Finally, it may also be worth considering the fundamental type of behavior measured in the MWM versus many operant tasks. The MWM operates on escape from an aversive stimulus (negative reinforcement), whereas operant tasks such as the DDT utilize delivery of a rewarding stimulus (positive reinforcement). This may be an important consideration for the use of both behaviors in assessing brain injury, given that studies have noted both increased stress and emotional responses, as well as anhedonia, in TBI populations. 56,57
Previously, we have shown chronic problems with behavioral disinhibition lasting 4 months post-injury, 28 and others have demonstrated inhibitory deficits after juvenile TBI that continued through adulthood, 58,59 even with milder injury parameters. In the current study, we observed increases in impulsive choice lasting 2 months post-injury, which showed no signs of resolving, and that were unrelated to MWM performance. Indeed, had we only tested brain function using the MWM, we would have concluded that these rats were not cognitively impaired. This is of particular note, given that reports with mild injury in the MWM have focused on the relatively early post-acute period (1–2 weeks post-injury), whereas later assessments have observed spontaneous recovery on this behavioral measure. 60,61 Given the increasing realization that impulse control deficits significantly contribute to poor mental health post-TBI, it is important for the research community to consider incorporating behavioral paradigms with strong translational validity for the measurement of impulsivity into programs of TBI assessment, such as the DDT used here.
Treatments directed at the post-acute and chronic phases of TBI have historically been neglected, potentially attributed to a lack of functional assessments that are resistant to endogenous recovery in animals. Stable, long-term deficits such as those observed here could aid in identifying the physiological factors that contribute to enduring dysfunction and susceptibility to psychiatric disease, such as the contribution of generalized neuroinflammation to motor impulsivity, which we have demonstrated previously, 28 and the identification of IL-12 as a potential contributor to choice impulsivity as shown in the current study. Given the complexity of neuroinflammation, it may seem unlikely that modulating a single cytokine could improve cognitive performance post-TBI, but the selective modulation of other cytokines, such as IL-6, has been shown to have highly specific behavioral effects. 62 Although we have assessed an important component of cognitive dysfunction, namely impulsive decision making, there are numerous other higher-order cognitive processes, which are compromised in both psychiatric and brain-injured populations, and that are also urgently in need of study, such as behavioral flexibility, working memory, and risk-based decision making. 2,3 Nevertheless, this study demonstrates the utility of incorporating behavioral assessments into pre-clinical models of brain injury that have greater relevance for patients living with chronic neuropsychiatric deficits resulting from TBI.
Footnotes
Acknowledgments
Funding for this project was provided by an internal grant from the Djavad Mowafaghian Centre for Brain Health to C.V., K.M., C.L.W., and C.A.W., operating grants from the Canadian Institutes of Health Research (CIHR) awarded to C.A.W. and to C.L.W., from the Weston Brain Institute to C.L.W., and the National Institute of Neurological Disorders and Stroke (R21NS087458) awarded to S.R. C.V. was supported by a (CIHR) post-doctoral fellowship. In the past 3 years, C.A.W. has received salary support from the CIHR New Investigator Award program and the Michael Smith Foundation for Health Research.
Author Disclosure Statement
In the past 3 years, C.A.W. has been a member of an Advisory Board for Shire Pharmaceuticals with reference to an unrelated matter. C.L.W. has a sponsored research project with AstraZeneca on an unrelated topic. The authors have no other financial disclosures or potential conflicts of interest to declare.